FeI₂ is an effective spin-1 material with strong single-ion anisotropy. Quadrupolar fluctuations give rise to a single-ion bound state that cannot be described by a dipole-only model. This tutorial illustrates how to use the linear spin wave theory of SU(3) coherent states (i.e. 2-flavor bosons) to model the magnetic behavior in FeI₂. The original study was performed in Bai et al., Nature Physics 17, 467–472 (2021).
The Fe atoms are arranged in stacked triangular layers. The effective spin Hamiltonian takes the form,
where the set of exchange matrices $J_{ij}$ between bonded sites $(i,j)$ includes competing ferromagnetic and antiferromagnetic interactions. This model also includes a strong easy axis anisotropy, $D > 0$.
We will formulate this Hamiltonian in Sunny and then calculate its dynamic structure factor.
Sunny is implemented in Julia. This is a relatively new programming language that allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users may wish to take a look at our Getting Started with Julia guide. Sunny requires Julia 1.9 or later.
From the Julia prompt, load Sunny. For plotting, one can choose either GLMakie (a pop-up window) or WGLMakie (inline plots for a Jupyter notebook).
using Sunny, GLMakie
If these packages are not yet installed, Julia should offer to install them using its built-in package management system. If old versions are installed, you may need to update them to run this tutorial.
A Crystal describes the crystallographic unit cell and will usually be loaded from a .cif file. Here, we instead build a crystal by listing all atoms and their types.
a = b = 4.05012 # Lattice constants for triangular lattice
+c = 6.75214 # Spacing in the z-direction
+
+latvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that
+ # define the conventional unit cell
+positions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions
+ # of lattice vectors
+types = ["Fe", "I", "I"]
+FeI2 = Crystal(latvecs, positions; types)
Importantly, cryst retains the spacegroup symmetry of the full FeI₂ crystal. This information will be used, for example, to propagate exchange interactions between symmetry-equivalent bonds.
In a running Julia environment, the crystal can be viewed interactively using view_crystal.
The command print_symmetry_table provides a list of all the symmetry-allowed interactions up to a cutoff distance.
print_symmetry_table(cryst, 8.0)
Atom 1
+Type 'Fe', position [0, 0, 0], multiplicity 1
+Allowed g-tensor: [A 0 0
+ 0 A 0
+ 0 0 B]
+Allowed anisotropy in Stevens operators:
+ c₁*𝒪[2,0] +
+ c₂*𝒪[4,-3] + c₃*𝒪[4,0] +
+ c₄*𝒪[6,-3] + c₅*𝒪[6,0] + c₆*𝒪[6,6]
+
+Bond(1, 1, [1, 0, 0])
+Distance 4.05012, coordination 6
+Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 0, 0]
+Allowed exchange matrix:[A 0 0
+ 0 B D
+ 0 D C]
+
+Bond(1, 1, [0, 0, 1])
+Distance 6.75214, coordination 2
+Connects 'Fe' at [0, 0, 0] to 'Fe' at [0, 0, 1]
+Allowed exchange matrix:[A 0 0
+ 0 A 0
+ 0 0 B]
+
+Bond(1, 1, [1, 2, 0])
+Distance 7.0150136167509, coordination 6
+Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 2, 0]
+Allowed exchange matrix:[A 0 0
+ 0 B D
+ 0 D C]
+
+Bond(1, 1, [1, 0, 1])
+Distance 7.8736818956572, coordination 12
+Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 0, 1]
+Allowed exchange matrix:[A F E
+ F B D
+ E D C]
The allowed $g$-tensor is expressed as a 3×3 matrix in the free coefficients A, B, ... The allowed single-ion anisotropy is expressed as a linear combination of Stevens operators. The latter correspond to polynomials of the spin operators, as we will describe below.
The allowed exchange interactions are given as a 3×3 matrix for representative bonds. The notation Bond(i, j, n) indicates a bond between atom indices i and j, with cell offset n. In the general case, it will be necessary to associate atom indices with their positions in the unit cell; these can be viewed with display(cryst). Note that the order of the pair $(i, j)$ is significant if the exchange tensor contains antisymmetric Dzyaloshinskii–Moriya (DM) interactions.
In the case of FeI₂, Bond(1, 1, [1,0,0]) is one of the 6 nearest-neighbor Fe-Fe bonds on a triangular lattice layer, and Bond(1, 1, [0,0,1]) is an Fe-Fe bond between layers.
System [SU(3)]
+Lattice (4×4×4)×1
+Energy per site 0
+
This system includes $4×4×4$ unit cells, i.e. 64 Fe atoms, each with spin $S=1$ and a $g$-factor of 2. Quantum mechanically, spin $S=1$ involves a superposition of $2S+1=3$ distinct angular momentum states. In :SUN mode, this superposition will be modeled explicitly using the formalism of SU(3) coherent states, which captures both dipolar and quadrupolar fluctuations. For the more traditional dipole dynamics, use :dipole mode instead.
Next we will use set_exchange! to assign interaction to bonds. Sunny will automatically propagate each interaction to all symmetry-equivalent bonds in the unit cell. The FeI₂ interactions below follow Bai et al.
The function set_onsite_coupling! assigns a single-ion anisotropy. The argument can be constructed using spin_matrices or stevens_matrices. Here we use Julia's anonymous function syntax to assign an easy-axis anisotropy along the direction $\hat{z}$.
D = 2.165
+set_onsite_coupling!(sys, S -> -D*S[3]^2, 1)
In the remainder of this tutorial, we will examine Sunny's tools for calculating the dynamical structure factor using a multi-boson generalization of linear spin wave theory (LSWT). This theory describes non-interacting quasi-particle excitations that hybridize dipolar and quadrupolar modes.
Begin with a random configuration and use minimize_energy! to find a configuration of the SU(3) coherent states (i.e. spin dipoles and quadrupoles) that locally minimizes energy.
randomize_spins!(sys)
+minimize_energy!(sys)
49
A positive number above indicates that the procedure has converged to a local energy minimum. The configuration, however, may still have defects. This can be checked by visualizing the spins, colored according to their $z$-components.
plot_spins(sys; color=[s[3] for s in sys.dipoles])
A different understanding of the magnetic ordering can be obtained by moving to Fourier space. The 'instant' structure factor $𝒮(𝐪)$ is an experimental observable. To investigate $𝒮(𝐪)$ as true 3D data, Sunny provides instant_correlations and related functions. Here, however, we will use print_wrapped_intensities, which gives average intensities for the individual Bravais sublattices (in effect, all wavevectors are wrapped to the first Brillouin zone).
The result will likely be approximately consistent with the known zero-field energy-minimizing magnetic structure of FeI₂, which is single-$Q$ (two-up, two-down antiferromagnetic order). Mathematically, spontaneous symmetry breaking should select one of $±Q = [0, -1/4, 1/4]$, $[1/4, 0, 1/4]$, or $[-1/4,1/4,1/4]$, associated with the three-fold rotational symmetry of the crystal spacegroup. In nature, however, one will frequently encounter competing "domains" associated with the three possible orientations of the ground state.
If the desired ground state is already known, as with FeI₂, it could be entered by hand using set_dipole!. Alternatively, in the case of FeI₂, we could repeatedly employ the above randomization and minimization procedure until a defect-free configuration is found. Some systems will have more complicated ground states, which can be much more challenging to find. For this, Sunny provides experimental support for powerful simulated annealing via parallel tempering, but that is outside the scope of this tutorial.
Here, let's break the three-fold symmetry of FeI₂ by hand. Given one or more desired $Q$ modes, Sunny can suggest a magnetic supercell with appropriate periodicity. Let's arbitrarily select one of the three possible ordering wavevectors, $Q = [0, -1/4, 1/4]$. Sunny suggests a corresponding magnetic supercell in units of the crystal lattice vectors.
suggest_magnetic_supercell([[0, -1/4, 1/4]])
Suggested magnetic supercell in multiples of lattice vectors:
+
+ [1 0 0; 0 2 1; 0 -2 1]
+
+for wavevectors [[0, -1/4, 1/4]].
The system returned by reshape_supercell is smaller, and is sheared relative to the original system. This makes it much easier to find the global energy minimum.
Now that we have found the ground state for a magnetic supercell, we can immediately proceed to perform zero-temperature calculations using linear spin wave theory. We begin by instantiating a SpinWaveTheory type using the supercell.
swt = SpinWaveTheory(sys_min)
SpinWaveTheory [Dipole correlations]
+Atoms in magnetic supercell: 4
+
Select a sequence of wavevectors that will define a piecewise linear interpolation in reciprocal lattice units (RLU).
The function reciprocal_space_path will linearly sample a path between the provided $q$-points with a given density. The xticks return value provides labels for use in plotting.
density = 50
+path, xticks = reciprocal_space_path(cryst, q_points, density);
The dispersion function defines the quasiparticle excitation energies $ω_i(𝐪)$ for each point $𝐪$ along the reciprocal space path.
disp = dispersion(swt, path);
In addition to the band energies $ω_i(𝐪)$, Sunny can calculate the inelastic neutron scattering intensity $I_i(𝐪)$ for each band $i$ according to an intensity_formula. We choose to apply a polarization correction $(1 - 𝐪⊗𝐪)$ by setting the mode argument to :perp. Selecting delta_function_kernel specifies that we want the energy and intensity of each band individually.
formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)
Quantum Scattering Intensity Formula
+At any Q and for each band ωᵢ = εᵢ(Q), with S = S(Q,ωᵢ):
+
+ Intensity(Q,ω) = ∑ᵢ δ(ω-ωᵢ) ∑_ij (I - Q⊗Q){i,j} S{i,j}
+
+ (i,j = Sx,Sy,Sz)
+
+BandStructure information (ωᵢ and intensity) reported for each band
+
The function intensities_bands uses linear spin wave theory to calculate both the dispersion and intensity data for the provided path.
A real FeI₂ sample will exhibit competing magnetic domains associated with spontaneous symmetry breaking of the 6-fold rotational symmetry of the triangular lattice. Note that the wavevectors $𝐪$ and $-𝐪$ are equivalent in the structure factor, which leaves three distinct domain orientations, which are related by 120° rotations about the $ẑ$-axis. Rather than rotating the spin configuration directly, on can rotate the $𝐪$-space path. Below, we use rotation_in_rlu to average the intensities over all three possible orientations.
This result can be directly compared to experimental neutron scattering data from Bai et al.
(The publication figure accidentally used a non-standard coordinate system to label the wave vectors.)
To get this agreement, the use of SU(3) coherent states is essential. In other words, we needed a theory of multi-flavored bosons. The lower band has large quadrupolar character, and arises from the strong easy-axis anisotropy of FeI₂. By setting mode = :SUN, the calculation captures this coupled dipole-quadrupole dynamics.
An interesting exercise is to repeat the same study, but using mode = :dipole instead of :SUN. That alternative choice would constrain the coherent state dynamics to the space of dipoles only.
The full dynamical spin structure factor (DSSF) can be retrieved as a $3×3$ matrix with the dssf function, for a given path of $𝐪$-vectors.
disp, is = dssf(swt, path);
The first output disp is identical to that obtained from dispersion. The second output is contains a list of $3×3$ matrix of intensities. For example, is[q,n][2,3] yields the $(ŷ,ẑ)$ component of the structure factor intensity for nth mode at the qth wavevector in the path.
The multi-boson linear spin wave theory, applied above, can be understood as the quantization of a certain generalization of the Landau-Lifshitz spin dynamics. Rather than dipoles, this dynamics takes places on the space of SU(N) coherent states.
The full SU(N) coherent state dynamics, with appropriate quantum correction factors, can be useful to model finite temperature scattering data. In particular, it captures certain anharmonic effects due to thermal fluctuations. See our generalized spin dynamics tutorial.
The classical dynamics is also a good starting point to study non-equilibrium phenomena. Empirical noise and damping terms can be used to model coupling to a thermal bath. This yields a Langevin dynamics of SU(N) coherent states. Our dynamical SU(N) quench tutorial illustrates how a temperature quench can give rise to novel liquid phase of CP² skyrmions.
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
diff --git a/dev/examples/02_LSWT_CoRh2O4-7eb4b415.png b/dev/examples/02_LSWT_CoRh2O4-7eb4b415.png
new file mode 100644
index 000000000..862569f3e
Binary files /dev/null and b/dev/examples/02_LSWT_CoRh2O4-7eb4b415.png differ
diff --git a/dev/examples/02_LSWT_CoRh2O4-822521c1.png b/dev/examples/02_LSWT_CoRh2O4-822521c1.png
new file mode 100644
index 000000000..1928a2877
Binary files /dev/null and b/dev/examples/02_LSWT_CoRh2O4-822521c1.png differ
diff --git a/dev/examples/powder_averaging-a4701496.png b/dev/examples/02_LSWT_CoRh2O4-a4701496.png
similarity index 100%
rename from dev/examples/powder_averaging-a4701496.png
rename to dev/examples/02_LSWT_CoRh2O4-a4701496.png
diff --git a/dev/examples/02_LSWT_CoRh2O4-d8c9a147.png b/dev/examples/02_LSWT_CoRh2O4-d8c9a147.png
new file mode 100644
index 000000000..e42041ad6
Binary files /dev/null and b/dev/examples/02_LSWT_CoRh2O4-d8c9a147.png differ
diff --git a/dev/examples/02_LSWT_CoRh2O4.html b/dev/examples/02_LSWT_CoRh2O4.html
new file mode 100644
index 000000000..1330740eb
--- /dev/null
+++ b/dev/examples/02_LSWT_CoRh2O4.html
@@ -0,0 +1,60 @@
+
+2. Spin wave simulations of CoRh₂O₄ · Sunny documentation
This tutorial illustrates the conventional spin wave theory of dipoles. We consider a simple model of the diamond-cubic crystal CoRh₂O₄, with parameters extracted from Ge et al., Phys. Rev. B 96, 064413.
using Sunny, GLMakie
Construct a diamond Crystal in the conventional (non-primitive) cubic unit cell. Sunny will populate all eight symmetry-equivalent sites when given the international spacegroup number 227 ("Fd-3m") and the appropriate setting. For this spacegroup, there are two conventional translations of the unit cell, and it is necessary to disambiguate through the setting keyword argument. (On your own: what happens if setting is omitted?)
a = 8.5031 # (Å)
+latvecs = lattice_vectors(a, a, a, 90, 90, 90)
+cryst = Crystal(latvecs, [[0,0,0]], 227, setting="1")
In a running Julia environment, the crystal can be viewed interactively using view_crystal.
view_crystal(cryst, 8.0)
Construct a System with quantum spin $S=3/2$ constrained to the space of dipoles. Including an antiferromagnetic nearest neighbor interaction J will favor Néel order. To optimize this magnetic structure, it is sufficient to employ a magnetic lattice consisting of a single crystal unit cell, latsize=(1,1,1). Passing an explicit random number seed will ensure repeatable results.
In the ground state, each spin is exactly anti-aligned with its 4 nearest-neighbors. Because every bond contributes an energy of $-JS^2$, the energy per site is $-2JS^2$. In this calculation, a factor of 1/2 avoids double-counting the bonds. Due to lack of frustration, direct energy minimization is successful in finding the ground state.
Plotting the spins confirms the expected Néel order. Note that the overall, global rotation of dipoles is arbitrary.
s0 = sys.dipoles[1,1,1,1]
+plot_spins(sys; color=[s'*s0 for s in sys.dipoles])
For numerical efficiency, it will be helpful to work with the smallest possible magnetic supercell. Here, it is the primitive unit cell, which contains just two sites. The variable shape below defines the primitive lattice vectors cryst.prim_latvecs in units of the conventional lattice vectors. This result is used as input to reshape_supercell. The energy per site remains the same, which verifies that the magnetic supercell is valid.
Now estimate $𝒮(𝐪,ω)$ with SpinWaveTheory and an intensity_formula. The mode :perp contracts with a dipole factor to return the unpolarized intensity. The formula also employs lorentzian broadening. The isotropic FormFactor for Cobalt(2+) dampens intensities at large $𝐪$.
Quantum Scattering Intensity Formula
+At any (Q,ω), with S = S(Q,ωᵢ):
+
+ Intensity(Q,ω) = ∑ᵢ Kernel(ω-ωᵢ) ∑_ij (I - Q⊗Q){i,j} S{i,j}
+
+ (i,j = Sx,Sy,Sz)
+
+Intensity(ω) reported
+
For the "single crystal" result, we may use reciprocal_space_path to construct a path that connects high-symmetry points in reciprocal space. The intensities_broadened function collects intensities along this path for the given set of energy values.
A powder measurement effectively involves an average over all possible crystal orientations. We use the function reciprocal_space_shell to sample n wavevectors on a sphere of a given radius (inverse angstroms), and then calculate the spherically-averaged intensity.
Use the stochastic Landau-Lifshitz dynamics to thermalize system into equilibrium at finite temperature. This is a Langevin equation, which includes damping and noise terms. The strength of the noise term is automatically fixed according to the damping time scale λ and the target temperature, according to a fluctuation-dissipation theorem.
Δt = 0.05/abs(J*S) # Time step
+λ = 0.1 # Dimensionless damping time-scale
+kT = 16 * meV_per_K # 16K, a temperature slightly below ordering
+langevin = Langevin(Δt; λ, kT);
Because the magnetic order has been initialized correctly, relatively few additional Langevin time-steps are required to reach thermal equilibrium.
To visualize the instantaneous (equal-time) structure factor, create an object instant_correlations and use add_sample! to accumulated data for the equilibrated spin configuration.
sc = instant_correlations(sys)
+add_sample!(sc, sys) # Accumulate the newly sampled structure factor into `sf`
Collect 20 additional decorrelated samples. For each sample, about 100 Langevin time-steps is sufficient to collect approximately uncorrelated statistics.
for _ in 1:20
+ for _ in 1:100
+ step!(sys, langevin)
+ end
+ add_sample!(sc, sys)
+end
Define a slice of momentum space. Wavevectors are specified in reciprocal lattice units (RLU). The notation q1s in -10:0.1:10 indicates that the first $q$-component ranges from -10 to 10 in intervals of 0.1. That is, $q$ spans over 20 Brillouin zones. To convert to absolute momentum units, each component of $q$ would need to be scaled by a reciprocal lattice vector.
q1s = -10:0.1:10
+q2s = -10:0.1:10
+qs = [[q1, q2, 0.0] for q1 in q1s, q2 in q2s];
Plot the instantaneous structure factor for the given $q$-slice. We employ the appropriate FormFactor for Co2⁺. An intensity_formula defines how dynamical correlations correspond to the observable structure factor. The function instant_intensities_interpolated calculates intensities at the target qs by interpolating over the data available at discrete reciprocal-space lattice points.
To collect statistics for the dynamical structure factor intensities $I(𝐪,ω)$ at finite temperature, use dynamical_correlations. Now, each call to add_sample! will run a classical spin dynamics trajectory. Longer-time trajectories will be required to achieve greater energy resolution, as controlled by nω. Here, we pick a moderate number of energies, nω = 50, which will make the simulation run quickly.
ωmax = 6.0 # Maximum energy to resolve (meV)
+nω = 50 # Number of energies to resolve
+sc = dynamical_correlations(sys; Δt, nω, ωmax, process_trajectory=:symmetrize)
Define spherical shells in reciprocal space via their radii, in absolute units of 1/Å. For each shell, calculate and average the intensities at 100 $𝐪$-points, sampled approximately uniformly.
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
diff --git a/dev/examples/fei2_classical-6b435b86.png b/dev/examples/04_GSD_FeI2-6b435b86.png
similarity index 100%
rename from dev/examples/fei2_classical-6b435b86.png
rename to dev/examples/04_GSD_FeI2-6b435b86.png
diff --git a/dev/examples/fei2_classical-9c3ace5d.png b/dev/examples/04_GSD_FeI2-9c3ace5d.png
similarity index 100%
rename from dev/examples/fei2_classical-9c3ace5d.png
rename to dev/examples/04_GSD_FeI2-9c3ace5d.png
diff --git a/dev/examples/fei2_classical-9fe52114.png b/dev/examples/04_GSD_FeI2-9fe52114.png
similarity index 100%
rename from dev/examples/fei2_classical-9fe52114.png
rename to dev/examples/04_GSD_FeI2-9fe52114.png
diff --git a/dev/examples/fei2_classical-a20d933e.png b/dev/examples/04_GSD_FeI2-a20d933e.png
similarity index 100%
rename from dev/examples/fei2_classical-a20d933e.png
rename to dev/examples/04_GSD_FeI2-a20d933e.png
diff --git a/dev/examples/fei2_classical-de3c2258.png b/dev/examples/04_GSD_FeI2-de3c2258.png
similarity index 100%
rename from dev/examples/fei2_classical-de3c2258.png
rename to dev/examples/04_GSD_FeI2-de3c2258.png
diff --git a/dev/examples/fei2_classical-ef051221.png b/dev/examples/04_GSD_FeI2-ef051221.png
similarity index 100%
rename from dev/examples/fei2_classical-ef051221.png
rename to dev/examples/04_GSD_FeI2-ef051221.png
diff --git a/dev/examples/fei2_classical-f49e7f7e.png b/dev/examples/04_GSD_FeI2-f49e7f7e.png
similarity index 100%
rename from dev/examples/fei2_classical-f49e7f7e.png
rename to dev/examples/04_GSD_FeI2-f49e7f7e.png
diff --git a/dev/examples/fei2_classical-ffcf4af4.png b/dev/examples/04_GSD_FeI2-ffcf4af4.png
similarity index 100%
rename from dev/examples/fei2_classical-ffcf4af4.png
rename to dev/examples/04_GSD_FeI2-ffcf4af4.png
diff --git a/dev/examples/fei2_classical.html b/dev/examples/04_GSD_FeI2.html
similarity index 50%
rename from dev/examples/fei2_classical.html
rename to dev/examples/04_GSD_FeI2.html
index e97cc6ca3..615cb23b0 100644
--- a/dev/examples/fei2_classical.html
+++ b/dev/examples/04_GSD_FeI2.html
@@ -1,5 +1,5 @@
-FeI₂ at Finite Temperature · Sunny documentation
In our previous Case Study: FeI₂, we used linear spin wave theory (LSWT) to calculate the dynamical structure factor. Here, we perform a similar calculation using classical spin dynamics. Because we are interested in the coupled dynamics of spin dipoles and quadrupoles, we employ a classical dynamics of SU(3) coherent states that generalizes the Landau-Lifshitz equation.
Compared to LSWT, simulations using classical dynamics are much slower, and are limited in $k$-space resolution. However, they make it is possible to capture nonlinear effects associated with finite temperature fluctuations. Classical dynamics are also appealing for studying out-of-equilibrium systems (e.g., relaxation of spin glasses), or systems with quenched inhomogeneities that require large simulation volumes.
In this tutorial, we show how to study the finite temperature dynamics of FeI₂ using the classical approach. It is important to stress that the estimation of $S(𝐪,ω)$ with classical dynamics is fundamentally a Monte Carlo calculation: sample spin configurations are drawn from thermal equilibrium and used as initial conditions for generating dissipationless trajectories. The correlations of these trajectories are then averaged and used to calculate scattering intensities. It is therefore important to ensure that the initial spin configurations are sampled appropriately and that sufficient statistics are collected. We will demonstrate one approach here.
As an overview, we will:
Identify the ground state
Measure correlation data describing the excitations around that ground state
Use the correlation data to compute scattering intensities
As the implementation of the FeI₂ model is already covered in detail in the LSWT tutorial, we will not repeat it below. Instead, we will assume that you already have defined a sys in the same way with lattice dimensions $4×4×4$.
sys
System [SU(3)]
+4. Generalized spin dynamics of FeI₂ at finite T · Sunny documentation
In the previous FeI₂ tutorial, we used multi-flavor spin wave theory to calculate the dynamical structure factor. Here, we perform a similar calculation using classical spin dynamics at finite temperature. Because we are interested in the coupled dynamics of spin dipoles and quadrupoles, we employ a classical dynamics of SU(3) coherent states that generalizes the Landau-Lifshitz equation.
Compared to LSWT, simulations using classical dynamics are much slower, and are limited in $k$-space resolution. However, they make it is possible to capture nonlinear effects associated with finite temperature fluctuations. Classical dynamics are also appealing for studying out-of-equilibrium systems (e.g., relaxation of spin glasses), or systems with quenched inhomogeneities that require large simulation volumes.
In this tutorial, we show how to study the finite temperature dynamics of FeI₂ using the classical approach. It is important to stress that the estimation of $S(𝐪,ω)$ with classical dynamics is fundamentally a Monte Carlo calculation: sample spin configurations are drawn from thermal equilibrium and used as initial conditions for generating dissipationless trajectories. The correlations of these trajectories are then averaged and used to calculate scattering intensities. It is therefore important to ensure that the initial spin configurations are sampled appropriately and that sufficient statistics are collected. We will demonstrate one approach here.
As an overview, we will:
Identify the ground state
Measure correlation data describing the excitations around that ground state
Use the correlation data to compute scattering intensities
As the implementation of the FeI₂ model is already covered in detail in the LSWT tutorial, we will not repeat it below. Instead, we will assume that you already have defined a sys in the same way with lattice dimensions $4×4×4$.
sys
System [SU(3)]
Lattice (4×4×4)×1
Energy per site -353/250
Sunny uses the Langevin dynamics of SU(N) coherent states to sample spin configurations from the thermal equlibrium. One first constructs a Langevin integrator. This requires a time step, temperature, and a phenomenological damping parameter $λ$ that sets the coupling to the thermal bath.
Δt = 0.05/D # Should be inversely proportional to the largest energy scale
@@ -13,8 +13,8 @@
langevin = Langevin(Δt; kT, λ);
Langevin dynamics can be used to search for a magnetically ordered state. For this, the temperature kT must be below the ordering temperature, but large enough that the dynamical sampling procedure can overcome local energy barriers and eliminate defects.
randomize_spins!(sys)
for _ in 1:20_000
step!(sys, langevin)
-end
Although thermal fluctuations are present, the correct antiferromagnetic order (2 up, 2 down) is apparent.
plot_spins(sys; color=[s[3] for s in sys.dipoles])
For other systems, it can be much harder to find the magnetic ordering in an unbiased way, and more complicated sampling procedures may be necessary.
Our aim is to study the classical spin dynamics for states sampled in thermal equilibrium. To minimize finite size effects, and achieve sufficient momentum space resolution, we should significantly enlarge the system volume. The function resize_supercell takes new dimensions as multiples of the unit cell lattice vectors.
sys_large = resize_supercell(sys, (16,16,4)) # 16x16x4 copies of the original unit cell
-plot_spins(sys_large; color=[s[3] for s in sys_large.dipoles])
Now we will re-thermalize the system to a configuration just above the ordering temperature. Sunny expects energies in meV by default, so we use meV_per_K to convert from kelvin.
kT = 3.5 * meV_per_K # 3.5K ≈ 0.30 meV
+end
Although thermal fluctuations are present, the correct antiferromagnetic order (2 up, 2 down) is apparent.
plot_spins(sys; color=[s[3] for s in sys.dipoles])
For other systems, it can be much harder to find the magnetic ordering in an unbiased way, and more complicated sampling procedures may be necessary.
Our aim is to study the classical spin dynamics for states sampled in thermal equilibrium. To minimize finite size effects, and achieve sufficient momentum space resolution, we should significantly enlarge the system volume. The function resize_supercell takes new dimensions as multiples of the unit cell lattice vectors.
sys_large = resize_supercell(sys, (16,16,4)) # 16x16x4 copies of the original unit cell
+plot_spins(sys_large; color=[s[3] for s in sys_large.dipoles])
Now we will re-thermalize the system to a configuration just above the ordering temperature. Sunny expects energies in meV by default, so we use meV_per_K to convert from kelvin.
The resolution in energy can be improved by increasing nω (and decreasing Δt), and the general accuracy can be improved by collecting additional samples from the thermal equilibrium.
For real calculations, one often wants to apply further corrections and more accurate formulas. Here, we apply FormFactor corrections appropriate for Fe2 magnetic ions, and a dipole polarization correction :perp.
The resolution in energy can be improved by increasing nω (and decreasing Δt), and the general accuracy can be improved by collecting additional samples from the thermal equilibrium.
For real calculations, one often wants to apply further corrections and more accurate formulas. Here, we apply FormFactor corrections appropriate for Fe2 magnetic ions, and a dipole polarization correction :perp.
Often it is useful to plot cuts across multiple wave vectors but at a single energy. We'll pick an energy,
ωidx = 60
target_ω = ωs[ωidx]
3.7312072301840273
and take a constant-energy cut at that energy. The most straightforward way is to make a plot whose axes are aligned with the conventional reciprocal lattice of the crystal. This is accomplished using unit_resolution_binning_parameters:
In the above plot, the dashed-line (direct) lattice vectors are clearly orthogonal. However, we know that in real space, the lattice vectors $a$ and $b$ are not orthogonal, but rather point along the edges of a hexagon (see lower left corner):
Thus, plotting the direct lattice vectors as orthogonal (even in reciprocal space) is somewhat misleading. Worse yet, the [H,0,0] by [0,K,0] plot apparently loses the 6-fold symmetry of the crystal! Lastly, if one works out the components of the real-space metric with respect to the axes of the plot, one finds that there are non-zero off-diagonal entries,
latvecs = sys.crystal.latvecs
+fig
In the above plot, the dashed-line (direct) lattice vectors are clearly orthogonal. However, we know that in real space, the lattice vectors $a$ and $b$ are not orthogonal, but rather point along the edges of a hexagon (see lower left corner):
Thus, plotting the direct lattice vectors as orthogonal (even in reciprocal space) is somewhat misleading. Worse yet, the [H,0,0] by [0,K,0] plot apparently loses the 6-fold symmetry of the crystal! Lastly, if one works out the components of the real-space metric with respect to the axes of the plot, one finds that there are non-zero off-diagonal entries,
so real-space rotations and angles map into reciprocal space rotations angles in a complicated way.
To resolve these important issues, we want to use axes which are orthogonal (i.e. they diagonalize the metric and solve all of the problems just mentioned). The canonical choice is to use the combination $\frac{1}{2}a + b$ of lattice vectors (equiv. $a^* - \frac{1}{2}b^*$), which is orthogonal to $a$:
(latvecs * [1/2,1,0]) ⋅ (latvecs * [1,0,0]) == 0
true
This new vector $\frac{1}{2}a+b$ is visibly orthogonal to $a$ in real space:
To use "projection onto the new vector" as a histogram axis, only a single change is needed to the binning parameters. The second covector (previously $b$) must be swapped out for $\frac{1}{2}a + b$ (recall that reciprocal space covectors, such as those used in BinningParameters correspond to direct space vectors).
params.covectors[2,1:3] = [1/2,1,0] # [1/2,1,0] times [a;b;c] is (a/2 + b)
Binning Parameters
+ 0.0 0.0 45.5914
so real-space rotations and angles map into reciprocal space rotations angles in a complicated way.
To resolve these important issues, we want to use axes which are orthogonal (i.e. they diagonalize the metric and solve all of the problems just mentioned). The canonical choice is to use the combination $\frac{1}{2}a + b$ of lattice vectors (equiv. $a^* - \frac{1}{2}b^*$), which is orthogonal to $a$:
(latvecs * [1/2,1,0]) ⋅ (latvecs * [1,0,0]) == 0
true
This new vector $\frac{1}{2}a+b$ is visibly orthogonal to $a$ in real space:
To use "projection onto the new vector" as a histogram axis, only a single change is needed to the binning parameters. The second covector (previously $b$) must be swapped out for $\frac{1}{2}a + b$ (recall that reciprocal space covectors, such as those used in BinningParameters correspond to direct space vectors).
params.covectors[2,1:3] = [1/2,1,0] # [1/2,1,0] times [a;b;c] is (a/2 + b)
Binning Parameters⊡ 31 bins from -1.000 to +0.969 along [+1.00 dx] (Δ = 0.064)
⊡ 31 bins from -1.000 to +0.969 along [+0.50 dx +1.00 dy] (Δ = 0.057)
∫ Integrated from -0.125 to +1.625 along [+1.00 dz] (Δ = 1.750)
@@ -161,7 +161,7 @@
is = intensities_interpolated(sc, qs, new_formula; interpolation=:linear)
add_lines!(ax_left,params)
-fig
Now, not only are the dashed-line lattice vectors no longer misleadingly orthogonal, but the six-fold symmetry has been restored as well! Further, the metric has been diagonalized:
Now, not only are the dashed-line lattice vectors no longer misleadingly orthogonal, but the six-fold symmetry has been restored as well! Further, the metric has been diagonalized:
Finally, we note that instantaneous structure factor data, $𝒮(𝐪)$, can be obtained from a dynamic structure factor with instant_intensities_interpolated. Here we'll reuse the grid of wave vectors we generated above.
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
+hm
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
diff --git a/dev/examples/ising2d-f39f34dd.png b/dev/examples/05_MC_Ising-f39f34dd.png
similarity index 100%
rename from dev/examples/ising2d-f39f34dd.png
rename to dev/examples/05_MC_Ising-f39f34dd.png
diff --git a/dev/examples/05_MC_Ising.html b/dev/examples/05_MC_Ising.html
new file mode 100644
index 000000000..e7bfedf6f
--- /dev/null
+++ b/dev/examples/05_MC_Ising.html
@@ -0,0 +1,17 @@
+
+5. Monte Carlo sampling of the Ising model · Sunny documentation
This tutorial illustrates simulation of the classical 2D Ising model.
using Sunny, GLMakie
Sunny expects a 3D Crystal unit cell. To model a square lattice, we create an orthogonal unit cell where the $z$-spacing is distinct from the $x$ and $y$ spacing.
a = 1
+latvecs = lattice_vectors(a,a,10a,90,90,90)
+crystal = Crystal(latvecs, [[0,0,0]])
Create a System of spins with linear size L in the $x$ and $y$ directions, and only one layer in the $z$ direction. The option :dipole means that the system will store Heisenberg spins, as opposed to SU($N$) coherent states. Polarize the initial spin configuration using polarize_spins!. Following the Ising convention, we will restrict these spins to the $z$-axis and give them magnitude $S=1$.
By default, Sunny uses physical units, e.g. magnetic field in tesla. Here we specify an alternative Units system, so that the Zeeman coupling between the spin dipole $𝐬$ and an external field $𝐁$ has the dimensionless form $-𝐁⋅𝐬$.
Use set_exchange! to include a ferromagnetic Heisenberg interaction along nearest-neighbor bonds. The Bond below connects two spins displaced by one lattice constant in the $x$-direction. This interaction will be propagated to all nearest-neighbors bonds in the system, consistent with the symmetries of the square lattice.
The critical temperature for the Ising model is known analytically.
Tc = 2/log(1+√2)
2.269185314213022
Use a LocalSampler to perform nsweeps Monte Carlo sweeps. A sweep consists of, on average, one trial update per spin in the system. Each proposed update is accepted or rejected according to the Metropolis acceptance probability. As its name suggests, the propose_flip function will only propose pure spin flips, $𝐬 \rightarrow -𝐬$.
nsweeps = 4000
+sampler = LocalSampler(kT=Tc, propose=propose_flip)
+for i in 1:nsweeps
+ step!(sys, sampler)
+end
Plot the Ising spins by extracting the $z$-component of the dipoles
heatmap(reshape([s.z for s in sys.dipoles], (L,L)))
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
diff --git a/dev/examples/out_of_equilibrium-c31de6b4.png b/dev/examples/06_CP2_Skyrmions-c31de6b4.png
similarity index 100%
rename from dev/examples/out_of_equilibrium-c31de6b4.png
rename to dev/examples/06_CP2_Skyrmions-c31de6b4.png
diff --git a/dev/examples/06_CP2_Skyrmions.html b/dev/examples/06_CP2_Skyrmions.html
new file mode 100644
index 000000000..ef5246f0c
--- /dev/null
+++ b/dev/examples/06_CP2_Skyrmions.html
@@ -0,0 +1,55 @@
+
+6. Dynamical quench into CP² skyrmion liquid · Sunny documentation
This example demonstrates Sunny's ability to simulate the out-of-equilibrium dynamics of generalized spin systems. We will implement the model Hamiltonian of Zhang et al., Nature Communications 14, 3626 (2023), which supports a novel type of topological defect, a CP² skyrmion, that involves both the dipolar and quadrupolar parts of a quantum spin.
Beginning from an initial high-temperature state, a disordered gas of CP² skyrmions can be formed by rapidly quenching to low temperature. To model the coupled dynamics of dipoles and quadrupoles, Sunny uses a recently developed generalization of the Landau-Lifshitz spin dynamics, Dahlbom et al., Phys. Rev. B 106, 235154 (2022).
contains competing ferromagnetic nearest-neightbor and antiferromagnetic next-nearest-neighbor exchange terms on a triangular lattice. Both exchanges exhibit anisotropy on the z-term. Additionally, there is an external magnetic field, $h$, and easy-plane single-ion anisotropy, $D > 0$. We begin by implementing the Crystal.
The crystal is then used to create a spin System. All parameters in this model system are dimensionless, so we select "theory" units and set the g-factor to one.
System [SU(3)]
+Lattice (40×40×1)×1
+Energy per site 0
+
We proceed to implement each term of the Hamiltonian, selecting our parameters so that the system occupies a region of the phase diagram that supports skyrmions. The exchange interactions are set as follows.
h = 15.5
+field = set_external_field!(sys, [0.0 0.0 h])
and finally an easy-plane single-ion anisotropy,
D = 19.0
+set_onsite_coupling!(sys, S -> D*S[3]^2, 1)
Initialize system to an infinite temperature (fully randomized) initial condition.
randomize_spins!(sys)
We are now ready to simulate the quenching process using a generalized Langevin spin dynamics. If we were working with spin dipoles only, then Langevin dynamics would be the usual Landau-Lifshitz spin dynamics, augmented with damping and noise terms. In the present study, we are instead working with quantum spin-1 (an ($N=3$)-level system that includes both dipoles and quadrupoles). Here, Langevin captures the coupled dipole-quadrupole dynamics using the formalism of SU($N$) coherent states.
Selecting kT = 0 in the Langevin dynamics will effective disable the noise term. Then the parameter λ effectively determines the damping time-scale.
Δt = 0.2/D # Integration time step (inverse meV). Typically this will be
+ # inversely proportional to the largest energy scale in the
+ # system. We can use a fairly large time-step here because
+ # accuracy isn't critical.
+kT = 0 # Target equilibrium temperature (meV)
+λ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)
+integrator = Langevin(Δt; kT, λ)
Langevin(0.010526315789473684, 0.1, 0.0)
Finally we run the dynamics. We will record the state of the system at three different times during the quenching process by copying the coherents field of the System.
τs = [4., 16, 256] # Times to record snapshots
+frames = [] # Empty array to store snapshots
+for i in eachindex(τs)
+ dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate
+ numsteps = round(Int, dur/Δt)
+ for _ in 1:numsteps # Perform the integration
+ step!(sys, integrator)
+ end
+ push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration
+end
To visualize the state of the system contained in each snapshot, we will calculate and plot the skyrmion density on each plaquette of our lattice. The function plot_triangular_plaquettes is not part of the core Sunny package, but rather something you could define yourself. We are using the definition in plotting2d.jl from the Sunny examples/extra directory.
The times are given in $\hbar/|J_1|$. The white background corresponds to a quantum paramagnetic state, where the local spin exhibits a strong quadrupole moment and little or no dipole moment. Observe that the process has generated a number of well-formed skyrmions of both positive (red) and negative (blue) charge in addition to a number of other metastable spin configurations. A full-sized version of this figure is available in Dahlbom et al..
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
diff --git a/dev/examples/contributed/CoRh2O4-tutorial.html b/dev/examples/contributed/CoRh2O4-tutorial.html
index 53eec38ca..511ef21c9 100644
--- a/dev/examples/contributed/CoRh2O4-tutorial.html
+++ b/dev/examples/contributed/CoRh2O4-tutorial.html
@@ -1,5 +1,5 @@
-Powder Averaged CoRh₂O₄ at Finite Temperature · Sunny documentation
Goal: This script is to calculate the temperature dependence of the magnon excitations in the spin-3/2 Heisenberg Diamond Antiferromagnet and compare to powder-averaged results obtained for the compound CoRh₂O₄
Goal: This script is to calculate the temperature dependence of the magnon excitations in the spin-3/2 Heisenberg Diamond Antiferromagnet and compare to powder-averaged results obtained for the compound CoRh₂O₄
The function quench! randomizes the spins of a given System, fixes a target temperature, and lets the system relax at this temperature for nrelax integration steps.
Author: Martin Mourigal <br> Date: September 9, 2022 (Updated by October 28, 2023 using Sunny 0.5.5)
In this tutorial, we will walk through an example in Sunny and calculate the spin dynamical properties of the Heisenberg pyrochlore antiferromagnet and apply this knowledge to MgCr2O4 and ZnCr2O4, which are known to approximate this model. Relevant publications include:
To run the examples in the tutorial, you will need a working installation of the Julia programming language and the Sunny package. Some useful references for getting started are:
Author: Martin Mourigal <br> Date: September 9, 2022 (Updated by October 28, 2023 using Sunny 0.5.5)
In this tutorial, we will walk through an example in Sunny and calculate the spin dynamical properties of the Heisenberg pyrochlore antiferromagnet and apply this knowledge to MgCr2O4 and ZnCr2O4, which are known to approximate this model. Relevant publications include:
To run the examples in the tutorial, you will need a working installation of the Julia programming language and the Sunny package. Some useful references for getting started are:
Before specifying the interactions of our system, we first must set up the crystal. We will begin by specifying the pyrochlore lattice (illustrated below) in the manner that is typical of theorists.
Picture Credits: Theory of Quantum Matter Unit, OIST
FeI₂ is an effective spin-1 material with strong single-ion anisotropy. Quadrupolar fluctuations give rise to a single-ion bound state that cannot be described by a dipole-only model. This tutorial illustrates how to use the linear spin wave theory of SU(3) coherent states (i.e. 2-flavor bosons) to model the magnetic behavior in FeI₂. The original study was performed in Bai et al., Nature Physics 17, 467–472 (2021).
The Fe atoms are arranged in stacked triangular layers. The effective spin Hamiltonian takes the form,
where the set of exchange matrices $J_{ij}$ between bonded sites $(i,j)$ includes competing ferromagnetic and antiferromagnetic interactions. This model also includes a strong easy axis anisotropy, $D > 0$.
We will formulate this Hamiltonian in Sunny and then calculate its dynamic structure factor.
Sunny is implemented in Julia. This is a relatively new programming language that allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users may wish to take a look at our Getting Started with Julia guide. Sunny requires Julia 1.9 or later.
From the Julia prompt, load Sunny. For plotting, one can choose either GLMakie (a pop-up window) or WGLMakie (inline plots for a Jupyter notebook or VSCode).
using Sunny, GLMakie
If these packages are not yet installed, Julia should offer to install them using its built-in package management system. If old versions are installed, you may need to update them to run this tutorial.
A Crystal describes the crystallographic unit cell and will usually be loaded from a .cif file. Here, we instead build a crystal by listing all atoms and their types.
a = b = 4.05012 # Lattice constants for triangular lattice
-c = 6.75214 # Spacing in the z-direction
-
-latvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that
- # define the conventional unit cell
-positions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions
- # of lattice vectors
-types = ["Fe", "I", "I"]
-FeI2 = Crystal(latvecs, positions; types)
Observe that cryst retains the spacegroup symmetry of the full FeI₂ crystal. This information will be used, for example, to propagate exchange interactions between symmetry-equivalent bonds.
In a running Julia environment, the crystal can be viewed interactively using view_crystal.
The command print_symmetry_table provides a list of all the symmetry-allowed interactions up to a cutoff distance.
print_symmetry_table(cryst, 8.0)
Atom 1
-Type 'Fe', position [0, 0, 0], multiplicity 1
-Allowed g-tensor: [A 0 0
- 0 A 0
- 0 0 B]
-Allowed anisotropy in Stevens operators:
- c₁*𝒪[2,0] +
- c₂*𝒪[4,-3] + c₃*𝒪[4,0] +
- c₄*𝒪[6,-3] + c₅*𝒪[6,0] + c₆*𝒪[6,6]
-
-Bond(1, 1, [1, 0, 0])
-Distance 4.05012, coordination 6
-Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 0, 0]
-Allowed exchange matrix:[A 0 0
- 0 B D
- 0 D C]
-
-Bond(1, 1, [0, 0, 1])
-Distance 6.75214, coordination 2
-Connects 'Fe' at [0, 0, 0] to 'Fe' at [0, 0, 1]
-Allowed exchange matrix:[A 0 0
- 0 A 0
- 0 0 B]
-
-Bond(1, 1, [1, 2, 0])
-Distance 7.0150136167509, coordination 6
-Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 2, 0]
-Allowed exchange matrix:[A 0 0
- 0 B D
- 0 D C]
-
-Bond(1, 1, [1, 0, 1])
-Distance 7.8736818956572, coordination 12
-Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 0, 1]
-Allowed exchange matrix:[A F E
- F B D
- E D C]
The allowed $g$-tensor is expressed as a 3×3 matrix in the free coefficients A, B, ... The allowed single-ion anisotropy is expressed as a linear combination of Stevens operators. The latter correspond to polynomials of the spin operators, as we will describe below.
The allowed exchange interactions are given as a 3×3 matrix for representative bonds. The notation Bond(i, j, n) indicates a bond between atom indices i and j, with cell offset n. In the general case, it will be necessary to associate atom indices with their positions in the unit cell; these can be viewed with display(cryst). Note that the order of the pair $(i, j)$ is significant if the exchange tensor contains antisymmetric Dzyaloshinskii–Moriya (DM) interactions.
In the case of FeI₂, Bond(1, 1, [1,0,0]) is one of the 6 nearest-neighbor Fe-Fe bonds on a triangular lattice layer, and Bond(1, 1, [0,0,1]) is an Fe-Fe bond between layers.
System [SU(3)]
-Lattice (4×4×4)×1
-Energy per site 0
-
This system includes $4×4×4$ unit cells, i.e. 64 Fe atoms, each with spin $S=1$ and a $g$-factor of 2. Quantum mechanically, spin $S=1$ involves a superposition of $2S+1=3$ distinct angular momentum states. In :SUN mode, this superposition will be modeled explicitly using the formalism of SU(3) coherent states, which captures both dipolar and quadrupolar fluctuations. For the more traditional dipole dynamics, use :dipole mode instead.
Next we will use set_exchange! to assign interaction to bonds. Sunny will automatically propagate each interaction to all symmetry-equivalent bonds in the unit cell. The FeI₂ interactions below follow Bai et al.
The function set_onsite_coupling! assigns a single-ion anisotropy. The argument can be constructed using spin_matrices or stevens_matrices. Here we use Julia's anonymous function syntax to assign an easy-axis anisotropy along the direction $\hat{z}$.
D = 2.165
-set_onsite_coupling!(sys, S -> -D*S[3]^2, 1)
In the remainder of this tutorial, we will examine Sunny's tools for calculating the dynamical structure factor using a multi-boson generalization of linear spin wave theory (LSWT). This theory describes non-interacting quasi-particle excitations that hybridize dipolar and quadrupolar modes.
Begin with a random configuration and use minimize_energy! to find a configuration of the SU(3) coherent states (i.e. spin dipoles and quadrupoles) that locally minimizes energy.
randomize_spins!(sys)
-minimize_energy!(sys)
49
A positive number above indicates that the procedure has converged to a local energy minimum. The configuration, however, may still have defects. This can be checked by visualizing the spins, colored according to their $z$-components.
plot_spins(sys; color=[s[3] for s in sys.dipoles])
A different understanding of the magnetic ordering can be obtained by moving to Fourier space. The 'instant' structure factor $𝒮(𝐪)$ is an experimental observable. To investigate $𝒮(𝐪)$ as true 3D data, Sunny provides instant_correlations and related functions. Here, however, we will use print_wrapped_intensities, which gives average intensities for the individual Bravais sublattices (in effect, all wavevectors are wrapped to the first Brillouin zone).
The result will likely be approximately consistent with the known zero-field energy-minimizing magnetic structure of FeI₂, which is single-$Q$ (two-up, two-down antiferromagnetic order). Mathematically, spontaneous symmetry breaking should select one of $±Q = [0, -1/4, 1/4]$, $[1/4, 0, 1/4]$, or $[-1/4,1/4,1/4]$, associated with the three-fold rotational symmetry of the crystal spacegroup. In nature, however, one will frequently encounter competing "domains" associated with the three possible orientations of the ground state.
If the desired ground state is already known, as with FeI₂, it could be entered by hand using set_dipole!. Alternatively, in the case of FeI₂, we could repeatedly employ the above randomization and minimization procedure until a defect-free configuration is found. Some systems will have more complicated ground states, which can be much more challenging to find. For this, Sunny provides experimental support for powerful simulated annealing via parallel tempering, but that is outside the scope of this tutorial.
Here, let's break the three-fold symmetry of FeI₂ by hand. Given one or more desired $Q$ modes, Sunny can suggest a magnetic supercell with appropriate periodicity. Let's arbitrarily select one of the three possible ordering wavevectors, $Q = [0, -1/4, 1/4]$. Sunny suggests a corresponding magnetic supercell in units of the crystal lattice vectors.
suggest_magnetic_supercell([[0, -1/4, 1/4]])
Suggested magnetic supercell in multiples of lattice vectors:
-
- [1 0 0; 0 2 1; 0 -2 1]
-
-for wavevectors [[0, -1/4, 1/4]].
The system returned by reshape_supercell is smaller, and is sheared relative to the original system. This makes it much easier to find the global energy minimum.
Now that we have found the ground state for a magnetic supercell, we can immediately proceed to perform zero-temperature calculations using linear spin wave theory. We begin by instantiating a SpinWaveTheory type using the supercell.
swt = SpinWaveTheory(sys_min)
SpinWaveTheory [Dipole correlations]
-Atoms in magnetic supercell: 4
-
Select a sequence of wavevectors that will define a piecewise linear interpolation in reciprocal lattice units (RLU).
The function reciprocal_space_path will linearly sample a path between the provided $q$-points with a given density. The xticks return value provides labels for use in plotting.
density = 50
-path, xticks = reciprocal_space_path(cryst, q_points, density);
The dispersion function defines the quasiparticle excitation energies $ω_i(𝐪)$ for each point $𝐪$ along the reciprocal space path.
disp = dispersion(swt, path);
In addition to the band energies $ω_i(𝐪)$, Sunny can calculate the inelastic neutron scattering intensity $I_i(𝐪)$ for each band $i$ according to an intensity_formula. We choose to apply a polarization correction $(1 - 𝐪⊗𝐪)$ by setting the mode argument to :perp. Selecting delta_function_kernel specifies that we want the energy and intensity of each band individually.
formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)
Quantum Scattering Intensity Formula
-At any Q and for each band ωᵢ = εᵢ(Q), with S = S(Q,ωᵢ):
-
- Intensity(Q,ω) = ∑ᵢ δ(ω-ωᵢ) ∑_ij (I - Q⊗Q){i,j} S{i,j}
-
- (i,j = Sx,Sy,Sz)
-
-BandStructure information (ωᵢ and intensity) reported for each band
-
The function intensities_bands uses linear spin wave theory to calculate both the dispersion and intensity data for the provided path.
A real FeI₂ sample will exhibit competing magnetic domains associated with spontaneous symmetry breaking of the 6-fold rotational symmetry of the triangular lattice. Note that the wavevectors $𝐪$ and $-𝐪$ are equivalent in the structure factor, which leaves three distinct domain orientations, which are related by 120° rotations about the $ẑ$-axis. Rather than rotating the spin configuration directly, on can rotate the $𝐪$-space path. Below, we use rotation_in_rlu to average the intensities over all three possible orientations.
This result can be directly compared to experimental neutron scattering data from Bai et al.
(The publication figure accidentally used a non-standard coordinate system to label the wave vectors.)
To get this agreement, the use of SU(3) coherent states is essential. In other words, we needed a theory of multi-flavored bosons. The lower band has large quadrupolar character, and arises from the strong easy-axis anisotropy of FeI₂. By setting mode = :SUN, the calculation captures this coupled dipole-quadrupole dynamics.
An interesting exercise is to repeat the same study, but using mode = :dipole instead of :SUN. That alternative choice would constrain the coherent state dynamics to the space of dipoles only.
The full dynamical spin structure factor (DSSF) can be retrieved as a $3×3$ matrix with the dssf function, for a given path of $𝐪$-vectors.
disp, is = dssf(swt, path);
The first output disp is identical to that obtained from dispersion. The second output is contains a list of $3×3$ matrix of intensities. For example, is[q,n][2,3] yields the $(ŷ,ẑ)$ component of the structure factor intensity for nth mode at the qth wavevector in the path.
The multi-boson linear spin wave theory, applied above, can be understood as the quantization of a certain generalization of the Landau-Lifshitz spin dynamics. Rather than dipoles, this dynamics takes places on the space of SU(N) coherent states.
The full SU(N) coherent state dynamics, with appropriate quantum correction factors, can be useful to model finite temperature scattering data. In particular, it captures certain anharmonic effects due to thermal fluctuations. This is the subject of our FeI₂ at Finite Temperature tutorial.
The classical dynamics is also a good starting point to study non-equilibrium phenomena. Empirical noise and damping terms can be used to model coupling to a thermal bath. This yields a Langevin dynamics of SU(N) coherent states. Our CP² Skyrmion Quench tutorial shows how this dynamics gives rise to the formation of novel topological defects in a temperature quench.
Relative to LSWT calculations, it can take much more time to estimate $\mathcal{S}(𝐪,ω)$ intensities using classical dynamics simulation. See the SunnyTutorials notebooks for examples of "production-scale" simulations.
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
This tutorial illustrates simulation of the classical 2D Ising model.
using Sunny, GLMakie
Sunny expects a 3D Crystal unit cell. To model a square lattice, we create an orthogonal unit cell where the $z$-spacing is distinct from the $x$ and $y$ spacing.
a = 1
-latvecs = lattice_vectors(a,a,10a,90,90,90)
-crystal = Crystal(latvecs, [[0,0,0]])
Create a System of spins with linear size L in the $x$ and $y$ directions, and only one layer in the $z$ direction. The option :dipole means that the system will store Heisenberg spins, as opposed to SU($N$) coherent states. Polarize the initial spin configuration using polarize_spins!. Following the Ising convention, we will restrict these spins to the $z$-axis and give them magnitude $S=1$.
By default, Sunny uses physical units, e.g. magnetic field in tesla. Here we specify an alternative Units system, so that the Zeeman coupling between the spin dipole $𝐬$ and an external field $𝐁$ has the dimensionless form $-𝐁⋅𝐬$.
Use set_exchange! to include a ferromagnetic Heisenberg interaction along nearest-neighbor bonds. The Bond below connects two spins displaced by one lattice constant in the $x$-direction. This interaction will be propagated to all nearest-neighbors bonds in the system, consistent with the symmetries of the square lattice.
The critical temperature for the Ising model is known analytically.
Tc = 2/log(1+√2)
2.269185314213022
Use a LocalSampler to perform nsweeps Monte Carlo sweeps. A sweep consists of, on average, one trial update per spin in the system. Each proposed update is accepted or rejected according to the Metropolis acceptance probability. As its name suggests, the propose_flip function will only propose pure spin flips, $𝐬 \rightarrow -𝐬$.
nsweeps = 4000
-sampler = LocalSampler(kT=Tc, propose=propose_flip)
-for i in 1:nsweeps
- step!(sys, sampler)
-end
Plot the Ising spins by extracting the $z$-component of the dipoles
heatmap(reshape([s.z for s in sys.dipoles], (L,L)))
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
This example demonstrates Sunny's ability to simulate the out-of-equilibrium dynamics of generalized spin systems. We will implement the model Hamiltonian of Zhang et al., Nature Communications 14, 3626 (2023), which supports a novel type of topological defect, a CP² skyrmion, that involves both the dipolar and quadrupolar parts of a quantum spin.
Beginning from an initial high-temperature state, a disordered gas of CP² skyrmions can be formed by rapidly quenching to low temperature. To model the coupled dynamics of dipoles and quadrupoles, Sunny uses a recently developed generalization of the Landau-Lifshitz spin dynamics, Dahlbom et al., Phys. Rev. B 106, 235154 (2022).
contains competing ferromagnetic nearest-neightbor and antiferromagnetic next-nearest-neighbor exchange terms on a triangular lattice. Both exchanges exhibit anisotropy on the z-term. Additionally, there is an external magnetic field, $h$, and easy-plane single-ion anisotropy, $D > 0$. We begin by implementing the Crystal.
The crystal is then used to create a spin System. All parameters in this model system are dimensionless, so we select "theory" units and set the g-factor to one.
System [SU(3)]
-Lattice (40×40×1)×1
-Energy per site 0
-
We proceed to implement each term of the Hamiltonian, selecting our parameters so that the system occupies a region of the phase diagram that supports skyrmions. The exchange interactions are set as follows.
h = 15.5
-field = set_external_field!(sys, [0.0 0.0 h])
and finally an easy-plane single-ion anisotropy,
D = 19.0
-set_onsite_coupling!(sys, S -> D*S[3]^2, 1)
Initialize system to an infinite temperature (fully randomized) initial condition.
randomize_spins!(sys)
We are now ready to simulate the quenching process using a generalized Langevin spin dynamics. If we were working with spin dipoles only, then Langevin dynamics would be the usual Landau-Lifshitz spin dynamics, augmented with damping and noise terms. In the present study, we are instead working with quantum spin-1 (an ($N=3$)-level system that includes both dipoles and quadrupoles). Here, Langevin captures the coupled dipole-quadrupole dynamics using the formalism of SU($N$) coherent states.
Selecting kT = 0 in the Langevin dynamics will effective disable the noise term. Then the parameter λ effectively determines the damping time-scale.
Δt = 0.2/D # Integration time step (inverse meV). Typically this will be
- # inversely proportional to the largest energy scale in the
- # system. We can use a fairly large time-step here because
- # accuracy isn't critical.
-kT = 0 # Target equilibrium temperature (meV)
-λ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)
-integrator = Langevin(Δt; kT, λ)
Langevin(0.010526315789473684, 0.1, 0.0)
Finally we run the dynamics. We will record the state of the system at three different times during the quenching process by copying the coherents field of the System.
τs = [4., 16, 256] # Times to record snapshots
-frames = [] # Empty array to store snapshots
-for i in eachindex(τs)
- dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate
- numsteps = round(Int, dur/Δt)
- for _ in 1:numsteps # Perform the integration
- step!(sys, integrator)
- end
- push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration
-end
To visualize the state of the system contained in each snapshot, we will calculate and plot the skyrmion density on each plaquette of our lattice. The function plot_triangular_plaquettes is not part of the core Sunny package, but rather something you could define yourself. We are using the definition in plotting2d.jl from the Sunny examples/extra directory.
The times are given in $\hbar/|J_1|$. The white background corresponds to a quantum paramagnetic state, where the local spin exhibits a strong quadrupole moment and little or no dipole moment. Observe that the process has generated a number of well-formed skyrmions of both positive (red) and negative (blue) charge in addition to a number of other metastable spin configurations. A full-sized version of this figure is available in Dahlbom et al..
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
This tutorial illustrates the calculation of the powder-averaged structure factor by performing an orientational average. We consider a simple model of the diamond-cubic crystal CoRh₂O₄, with parameters extracted from Ge et al., Phys. Rev. B 96, 064413.
using Sunny, GLMakie
Construct a diamond Crystal in the conventional (non-primitive) cubic unit cell. Sunny will populate all eight symmetry-equivalent sites when given the international spacegroup number 227 ("Fd-3m") and the appropriate setting. For this spacegroup, there are two conventional translations of the unit cell, and it is necessary to disambiguate through the setting keyword argument. (On your own: what happens if setting is omitted?)
a = 8.5031 # (Å)
-latvecs = lattice_vectors(a, a, a, 90, 90, 90)
-cryst = Crystal(latvecs, [[0,0,0]], 227, setting="1")
In a running Julia environment, the crystal can be viewed interactively using view_crystal.
view_crystal(cryst, 8.0)
Construct a System with an antiferromagnetic nearest neighbor interaction J. Because the diamond crystal is bipartite, the ground state will have unfrustrated Néel order. Selecting latsize=(1,1,1) is sufficient because the ground state is periodic over each cubic unit cell. By passing an explicit seed, the system's random number generator will give repeatable results.
In the ground state, each spin is exactly anti-aligned with its 4 nearest-neighbors. Because every bond contributes an energy of $-JS^2$, the energy per site is $-2JS^2$. In this calculation, a factor of 1/2 avoids double-counting the bonds. Due to lack of frustration, direct energy minimization is successful in finding the ground state.
Plotting the spins confirms the expected Néel order. Note that the overall, global rotation of dipoles is arbitrary.
s0 = sys.dipoles[1,1,1,1]
-plot_spins(sys; color=[s'*s0 for s in sys.dipoles])
For numerical efficiency, it will be helpful to work with the smallest possible magnetic supercell. Here, it is the primitive unit cell, which contains just two sites. The variable shape below defines the primitive lattice vectors cryst.prim_latvecs in units of the conventional lattice vectors. This result is used as input to reshape_supercell. The energy per site remains the same, which verifies that the magnetic supercell is valid.
Now estimate $𝒮(𝐪,ω)$ with SpinWaveTheory and an intensity_formula. The mode :perp contracts with a dipole factor to return the unpolarized intensity. The formula also employs lorentzian broadening. The isotropic FormFactor for Cobalt(2+) dampens intensities at large $𝐪$.
Quantum Scattering Intensity Formula
-At any (Q,ω), with S = S(Q,ωᵢ):
-
- Intensity(Q,ω) = ∑ᵢ Kernel(ω-ωᵢ) ∑_ij (I - Q⊗Q){i,j} S{i,j}
-
- (i,j = Sx,Sy,Sz)
-
-Intensity(ω) reported
-
For the "single crystal" result, we may use reciprocal_space_path to construct a path that connects high-symmetry points in reciprocal space. The intensities_broadened function collects intensities along this path for the given set of energy values.
A powder measurement effectively involves an average over all possible crystal orientations. We use the function reciprocal_space_shell to sample n wavevectors on a sphere of a given radius (inverse angstroms), and then calculate the spherically-averaged intensity.
This is a Sunny port of SpinW Tutorial 8, originally authored by Bjorn Fak and Sandor Toth. The goal is to calculate the linear spin wave theory spectrum for the $\sqrt{3} \times \sqrt{3}$ order of a Kagome antiferromagnet.
This is a Sunny port of SpinW Tutorial 15, originally authored by Sandor Toth. The goal is to calculate the linear spin wave theory spectrum for Ba₃NbFe₃Si₂O₁₄.
Create a System with a lattice size of $(1,1,7)$. The magnetic structure of Ba₃NbFe₃Si₂O₁₄ was determined to have the ordering wavevector $𝐐=(0,0,1/7)$ and hence the magnetic unit cell has 7 sites.
The final two exchanges define the chirality of the magnetic structure. The crystal chirality, $\epsilon_T$, the chirality of each triangle, $ϵ_D$ and the sense of rotation of the spin helices along $c$, $ϵ_{H}$. The three chiralities are related by $ϵ_T=ϵ_D ϵ_H$. We now assign $J_3$ and $J_5$ according to the crystal chirality.
Whilst Sunny provides tools to optimize the ground state automatically, in this case we already know the model ground state. Set the spiral magnetic order using set_spiral_order_on_sublattice!. It takes an ordering wavevector q, an axis of rotation for the spins axis, and the initial spin S0 for each sublattice.
q = [0, 0, 1/7]
-axis = [0,0,1]
-set_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[1, 0, 0])
-set_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[-1/2, -sqrt(3)/2, 0])
-set_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[-1/2, +sqrt(3)/2, 0])
-
-plot_spins(sys; color=[s[1] for s in sys.dipoles])
Define a path in reciprocal space, $[0,1,-1+\xi]$ for $\xi = 0 \dots 3$.
This is a Sunny port of SpinW Tutorial 8, originally authored by Bjorn Fak and Sandor Toth. The goal is to calculate the linear spin wave theory spectrum for the $\sqrt{3} \times \sqrt{3}$ order of a Kagome antiferromagnet.
This is a Sunny port of SpinW Tutorial 15, originally authored by Sandor Toth. The goal is to calculate the linear spin wave theory spectrum for Ba₃NbFe₃Si₂O₁₄.
Create a System with a lattice size of $(1,1,7)$. The magnetic structure of Ba₃NbFe₃Si₂O₁₄ was determined to have the ordering wavevector $𝐐=(0,0,1/7)$ and hence the magnetic unit cell has 7 sites.
The final two exchanges define the chirality of the magnetic structure. The crystal chirality, $\epsilon_T$, the chirality of each triangle, $ϵ_D$ and the sense of rotation of the spin helices along $c$, $ϵ_{H}$. The three chiralities are related by $ϵ_T=ϵ_D ϵ_H$. We now assign $J_3$ and $J_5$ according to the crystal chirality.
Whilst Sunny provides tools to optimize the ground state automatically, in this case we already know the model ground state. Set the spiral magnetic order using set_spiral_order_on_sublattice!. It takes an ordering wavevector q, an axis of rotation for the spins axis, and the initial spin S0 for each sublattice.
q = [0, 0, 1/7]
+axis = [0,0,1]
+set_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[1, 0, 0])
+set_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[-1/2, -sqrt(3)/2, 0])
+set_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[-1/2, +sqrt(3)/2, 0])
+
+plot_spins(sys; color=[s[1] for s in sys.dipoles])
Define a path in reciprocal space, $[0,1,-1+\xi]$ for $\xi = 0 \dots 3$.
Sunny is a Julia package for modeling atomic-scale magnetism. It provides powerful tools to study equilibrium and non-equilibrium magnetic phenomena. In particular, it allows estimation of dynamical structure factor intensities, $\mathcal{S}(𝐪,ω)$, to support quantitative modeling of experimental scattering data.
Ability to specify a crystal from a .cif file or its spacegroup symmetry.
Interactive visualizations of the 3D crystals and magnetic ordering.
Symmetry analysis to classify allowed interaction terms, and to propagate them by symmetry.
Single-ion anisotropy at arbitrary order, which can be specified using Stevens operators or as a polynomial of spin operators.
Monte Carlo sampling of spin configurations in thermal equilibrium, and optimization tools.
Measurements of dynamical correlations. At low temperature, one can use linear spin wave theory and its multi-boson generalization. This generalizes to finite temperatures using the classical dynamics, which allows for strongly nonlinear effects.
Long-range dipole-dipole interactions accelerated with the fast Fourier transform (FFT).
Support for comparison with experimental data: form factor, dipole factor, temperature-dependent classical-to-quantum factors, intensity binning, etc.
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
Sunny is a Julia package for modeling atomic-scale magnetism. It provides powerful tools to study equilibrium and non-equilibrium magnetic phenomena. In particular, it allows estimation of dynamical structure factor intensities, $\mathcal{S}(𝐪,ω)$, to support quantitative modeling of experimental scattering data.
Ability to specify a crystal from a .cif file or its spacegroup symmetry.
Interactive visualizations of the 3D crystals and magnetic ordering.
Symmetry analysis to classify allowed interaction terms, and to propagate them by symmetry.
Single-ion anisotropy at arbitrary order, which can be specified using Stevens operators or as a polynomial of spin operators.
Monte Carlo sampling of spin configurations in thermal equilibrium, and optimization tools.
Measurements of dynamical correlations. At low temperature, one can use linear spin wave theory and its multi-boson generalization. This generalizes to finite temperatures using the classical dynamics, which allows for strongly nonlinear effects.
Long-range dipole-dipole interactions accelerated with the fast Fourier transform (FFT).
Support for comparison with experimental data: form factor, dipole factor, temperature-dependent classical-to-quantum factors, intensity binning, etc.
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
diff --git a/dev/library.html b/dev/library.html
index 5008e0ebb..845bc8d3c 100644
--- a/dev/library.html
+++ b/dev/library.html
@@ -1,8 +1,8 @@
-Library API · Sunny documentation
This page describes the public types and functions exported by Sunny. This documentation can be also be accessed using the Julia help system (enter ? at the Julia command prompt).
Four indices identifying a single site in a System. The first three indices select the lattice cell and the last selects the sublattice (i.e., the atom within the unit cell).
This object can be used to index dipoles and coherents fields of a System. A Site is also required to specify inhomogeneous interactions via functions such as set_external_field_at! or set_exchange_at!.
Note that the definition of a cell may change when a system is reshaped. In this case, it is convenient to construct the Site using position_to_site, which always takes a position in fractional coordinates of the original lattice vectors.
The unit system is implicitly determined by the definition of two physical constants: the vacuum permeability $μ₀$ and the Bohr magneton $μ_B$. Temperatures are effectively measured in units of energy ($k_B = 1$) and time is effectively measured in units of inverse energy ($ħ = 1$). The default unit system, Units.meV, employs (meV, Å, tesla). Select alternatively Units.theory for a units system defined so that $μ₀ = μ_B = 1$.
This page describes the public types and functions exported by Sunny. This documentation can be also be accessed using the Julia help system (enter ? at the Julia command prompt).
Four indices identifying a single site in a System. The first three indices select the lattice cell and the last selects the sublattice (i.e., the atom within the unit cell).
This object can be used to index dipoles and coherents fields of a System. A Site is also required to specify inhomogeneous interactions via functions such as set_external_field_at! or set_exchange_at!.
Note that the definition of a cell may change when a system is reshaped. In this case, it is convenient to construct the Site using position_to_site, which always takes a position in fractional coordinates of the original lattice vectors.
The unit system is implicitly determined by the definition of two physical constants: the vacuum permeability $μ₀$ and the Bohr magneton $μ_B$. Temperatures are effectively measured in units of energy ($k_B = 1$) and time is effectively measured in units of inverse energy ($ħ = 1$). The default unit system, Units.meV, employs (meV, Å, tesla). Select alternatively Units.theory for a units system defined so that $μ₀ = μ_B = 1$.
The coordinates of the histogram axes are specified by multiplication of (q,ω) with each row of the covectors matrix, with q given in [R.L.U.]. Since the default covectors matrix is the identity matrix, the default axes are (qx,qy,qz,ω) in absolute units.
The convention for the binning scheme is that:
The left edge of the first bin starts at binstart
The bin width is binwidth
The last bin contains binend
There are no "partial bins;" the last bin may contain values greater than binend. C.f. count_bins.
An object describing a crystallographic unit cell and its space group symmetry. Constructors are as follows:
Crystal(filename; symprec=1e-5)
Reads the crystal from a .cif file located at the path filename. The optional parameter symprec controls the precision tolerance for spacegroup symmetries.
Constructs a crystal from the complete list of atom positions positions, with coordinates (between 0 and 1) in units of lattice vectors latvecs. Spacegroup symmetry information is automatically inferred. The optional parameter types is a list of strings, one for each atom, and can be used to break symmetry-equivalence between atoms.
Builds a crystal by applying symmetry operators for a given international spacegroup number. For certain spacegroups, there are multiple possible unit cell settings; in this case, a warning message will be printed, and a list of crystals will be returned, one for every possible setting. Alternatively, the optional setting string will disambiguate between unit cell conventions.
Currently, crystals built using only the spacegroup number will be missing some symmetry information. It is generally preferred to build a crystal from a .cif file or from the full specification of the unit cell.
Examples
# Read a Crystal from a .cif file
+bin_ix = 1 .+ floor.(Int64,(coords .- binstart) ./ binwidth)
An object describing a crystallographic unit cell and its space group symmetry. Constructors are as follows:
Crystal(filename; symprec=1e-5)
Reads the crystal from a .cif file located at the path filename. The optional parameter symprec controls the precision tolerance for spacegroup symmetries.
Constructs a crystal from the complete list of atom positions positions, with coordinates (between 0 and 1) in units of lattice vectors latvecs. Spacegroup symmetry information is automatically inferred. The optional parameter types is a list of strings, one for each atom, and can be used to break symmetry-equivalence between atoms.
Builds a crystal by applying symmetry operators for a given international spacegroup number. For certain spacegroups, there are multiple possible unit cell settings; in this case, a warning message will be printed, and a list of crystals will be returned, one for every possible setting. Alternatively, the optional setting string will disambiguate between unit cell conventions.
Currently, crystals built using only the spacegroup number will be missing some symmetry information. It is generally preferred to build a crystal from a .cif file or from the full specification of the unit cell.
Examples
# Read a Crystal from a .cif file
Crystal("filename.cif")
# Build an FCC crystal using the primitive unit cell. The spacegroup number
@@ -25,7 +25,7 @@
# overall unit cell translation.
latvecs = lattice_vectors(1, 1, 1, 90, 90, 90)
positions = [[1, 1, 1] / 4]
-cryst = Crystal(latvecs, positions, 227; setting="1")
The magnetic form factor for a given magnetic ion and charge state. When passed to an intensity_formula, determines a $|𝐪|$-dependent scaling of the structure factor.
The parameter ion must be one of the following strings:
The magnetic form factor for a given magnetic ion and charge state. When passed to an intensity_formula, determines a $|𝐪|$-dependent scaling of the structure factor.
The parameter ion must be one of the following strings:
involving the Landé $g$-factor. The $⟨j_l(s)⟩$ are radial integrals associated with the $l$th Bessel function of the magnetic dipole, where $s = |k|/4π$, and $|k|$ is the magnitude of momentum transfer.
The radial integrals have been calculated using Hartree-Fock for transition metals, or Dirac-Fock for the rare earths and actinide series [1–3]. Sunny uses approximate fits as a sum of Gaussians,
\[⟨j_0(s)⟩ = A e^{-as^2} + B e^{-bs^2} + C e^{-cs^2} + D e^{-ds^2} + E \
-⟨j_l(s)⟩ = (A e^{-as^2} + B e^{-bs^2} + C e^{-cs^2} + D e^{-ds^2} + E) s^2\]
Energy-conserving spin dynamics. One call to the step! function will advance a System by Δt units of time.
Uses the spherical midpoint integration scheme for dipole systems and the Schrödinger midpoint integration scheme for SU(N) spin systems. Both integration schemes are symplectic, and therefore avoid energy drift over long periods of simulation time.
Spin dynamics with coupling to a Langevin thermostat, which includes damping and noise terms. One call to the step! function will advance a System by Δt units of time.
Assuming ergodicity, the Langevin dynamics will sample from thermal equilibrium for the target temperature kT. The empirical parameter λ determines the strength of the coupling to the thermal bath. In other words, 1/λ is the decorrelation time-scale. If $λ = 0$, then the spin dynamics coincides with ImplicitMidpoint.
An alternative approach to sampling is LocalSampler, which may be preferred when the allowed spin values become effective discrete (e.g. Ising spins).
Monte Carlo simulation involving Metropolis updates to individual spins. One call to the step! function will perform nsweeps of MCMC sampling for a provided System. The default value of 1.0 means that step! performs, on average, one trial update per spin.
Assuming ergodicity, the LocalSampler will sample from thermal equilibrium for the target temperature kT.
The returned object stores fields ΔE and Δs, which represent the cumulative change to the net energy and dipole, respectively.
An alternative approach to sampling is Langevin, which may be preferred for simulating continuous spins, especially in the presence of long-range dipole-dipole interactions (cf. enable_dipole_dipole!).
Characterizes the spin at a given atom index within the crystal unit cell. S is an integer multiple of 1/2 and gives the spin angular momentum in units of ħ. g is the g-factor or tensor, such that an angular momentum dipole $s$ produces a magnetic moment $g s$ in units of the Bohr magneton.
Constructs an object to perform linear spin wave theory. Use it with dispersion and dssf functions.
The optional parameter energy_ϵ adds a small positive shift to the diagonal of the dynamical matrix $D$ to avoid numerical issues with zero-energy quasi-particle modes.
Construct a System of spins for a given Crystal symmetry. The latsize parameter determines the number of unit cells in each lattice vector direction. The infos parameter is a list of SpinInfo objects, which determine the magnitude $S$ and $g$-tensor of each spin.
The two primary options for mode are :SUN and :dipole. In the former, each spin-$S$ degree of freedom is described as an SU(N) coherent state, i.e. a quantum superposition of $N = 2S + 1$ levels. This formalism can be useful to capture multipolar spin fluctuations or local entanglement effects.
Mode :dipole projects the SU(N) dynamics onto the restricted space of pure dipoles. In practice this means that Sunny will simulate Landau-Lifshitz dynamics, but single-ion anisotropy and biquadratic exchange interactions will be renormalized to improve accuracy. To disable this renormalization, use the mode :dipole_large_S which applies the $S → ∞$ classical limit. For details, see the documentation page: Interaction Strength Renormalization.
The default units system of (meV, Å, tesla) can be overridden by with the units parameter; see Units.
An optional seed may be provided to achieve reproducible random number generation.
All spins are initially polarized in the $z$-direction.
add_trajectory uses the spin configuration contained in the System to generate a correlation data and accumulate it into sc. For static structure factors, this involves analyzing the spin-spin correlations of the spin configuration provided. For a dynamic structure factor, a trajectory is calculated using the given spin configuration as an initial condition. The spin-spin correlations are then calculating in time and accumulated into sc.
This function will change the state of sys when calculating dynamical structure factor data. To preserve the initial state of sys, it must be saved separately prior to calling add_sample!. Alternatively, the initial spin configuration may be copied into a new System and this new System can be passed to add_sample!.
Return the ω values for the energy index of a SampledCorrelations. By default, only returns values for non-negative energies, which corresponds to the default output of intensities. Set negative_energies to true to retrieve all ω values.
Performs a real-space convolution along the energy axis of an array of intensities. Assumes the format of the intensities array corresponds to what would be returned by intensities_interpolated. kernel must be a function that takes two numbers: kernel(ω, ω₀), where ω is a frequency, and ω₀ is the center frequency of the kernel. Sunny provides lorentzian for the most common use case:
Returns the number of bins in the binning scheme implied by binstart, binend, and binwidth. To count the bins in a BinningParameters, use params.numbins.
This function defines how partial bins are handled, so it should be used preferentially over computing the number of bins manually.
Computes the spin excitation energy dispersion relations given a SpinWaveTheory and an array of wave vectors qs. Each element $q$ of qs must be a 3-vector in units of reciprocal lattice units. I.e., $qᵢ$ is given in $2π/|aᵢ|$ with $|aᵢ|$ the lattice constant of the original chemical lattice.
The first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of the array is an energy.
Energy-conserving spin dynamics. One call to the step! function will advance a System by Δt units of time.
Uses the spherical midpoint integration scheme for dipole systems and the Schrödinger midpoint integration scheme for SU(N) spin systems. Both integration schemes are symplectic, and therefore avoid energy drift over long periods of simulation time.
Spin dynamics with coupling to a Langevin thermostat, which includes damping and noise terms. One call to the step! function will advance a System by Δt units of time.
Assuming ergodicity, the Langevin dynamics will sample from thermal equilibrium for the target temperature kT. The empirical parameter λ determines the strength of the coupling to the thermal bath. In other words, 1/λ is the decorrelation time-scale. If $λ = 0$, then the spin dynamics coincides with ImplicitMidpoint.
An alternative approach to sampling is LocalSampler, which may be preferred when the allowed spin values become effective discrete (e.g. Ising spins).
Monte Carlo simulation involving Metropolis updates to individual spins. One call to the step! function will perform nsweeps of MCMC sampling for a provided System. The default value of 1.0 means that step! performs, on average, one trial update per spin.
Assuming ergodicity, the LocalSampler will sample from thermal equilibrium for the target temperature kT.
The returned object stores fields ΔE and Δs, which represent the cumulative change to the net energy and dipole, respectively.
An alternative approach to sampling is Langevin, which may be preferred for simulating continuous spins, especially in the presence of long-range dipole-dipole interactions (cf. enable_dipole_dipole!).
Characterizes the spin at a given atom index within the crystal unit cell. S is an integer multiple of 1/2 and gives the spin angular momentum in units of ħ. g is the g-factor or tensor, such that an angular momentum dipole $s$ produces a magnetic moment $g s$ in units of the Bohr magneton.
Constructs an object to perform linear spin wave theory. Use it with dispersion and dssf functions.
The optional parameter energy_ϵ adds a small positive shift to the diagonal of the dynamical matrix $D$ to avoid numerical issues with zero-energy quasi-particle modes.
Construct a System of spins for a given Crystal symmetry. The latsize parameter determines the number of unit cells in each lattice vector direction. The infos parameter is a list of SpinInfo objects, which determine the magnitude $S$ and $g$-tensor of each spin.
The two primary options for mode are :SUN and :dipole. In the former, each spin-$S$ degree of freedom is described as an SU(N) coherent state, i.e. a quantum superposition of $N = 2S + 1$ levels. This formalism can be useful to capture multipolar spin fluctuations or local entanglement effects.
Mode :dipole projects the SU(N) dynamics onto the restricted space of pure dipoles. In practice this means that Sunny will simulate Landau-Lifshitz dynamics, but single-ion anisotropy and biquadratic exchange interactions will be renormalized to improve accuracy. To disable this renormalization, use the mode :dipole_large_S which applies the $S → ∞$ classical limit. For details, see the documentation page: Interaction Strength Renormalization.
The default units system of (meV, Å, tesla) can be overridden by with the units parameter; see Units.
An optional seed may be provided to achieve reproducible random number generation.
All spins are initially polarized in the $z$-direction.
add_trajectory uses the spin configuration contained in the System to generate a correlation data and accumulate it into sc. For static structure factors, this involves analyzing the spin-spin correlations of the spin configuration provided. For a dynamic structure factor, a trajectory is calculated using the given spin configuration as an initial condition. The spin-spin correlations are then calculating in time and accumulated into sc.
This function will change the state of sys when calculating dynamical structure factor data. To preserve the initial state of sys, it must be saved separately prior to calling add_sample!. Alternatively, the initial spin configuration may be copied into a new System and this new System can be passed to add_sample!.
Return the ω values for the energy index of a SampledCorrelations. By default, only returns values for non-negative energies, which corresponds to the default output of intensities. Set negative_energies to true to retrieve all ω values.
Performs a real-space convolution along the energy axis of an array of intensities. Assumes the format of the intensities array corresponds to what would be returned by intensities_interpolated. kernel must be a function that takes two numbers: kernel(ω, ω₀), where ω is a frequency, and ω₀ is the center frequency of the kernel. Sunny provides lorentzian for the most common use case:
Returns the number of bins in the binning scheme implied by binstart, binend, and binwidth. To count the bins in a BinningParameters, use params.numbins.
This function defines how partial bins are handled, so it should be used preferentially over computing the number of bins manually.
Computes the spin excitation energy dispersion relations given a SpinWaveTheory and an array of wave vectors qs. Each element $q$ of qs must be a 3-vector in units of reciprocal lattice units. I.e., $qᵢ$ is given in $2π/|aᵢ|$ with $|aᵢ|$ the lattice constant of the original chemical lattice.
The first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of the array is an energy.
qs is an array of wave vectors of arbitrary dimension. Each element $q$ of qs must be a 3-vector in reciprocal lattice units (RLU), i.e., in the basis of reciprocal lattice vectors.
The first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of this array is a tensor (3×3 matrix) corresponding to the indices $α$ and $β$.
Creates an empty SampledCorrelations object for calculating and storing dynamical structure factor intensities $𝒮(𝐪,ω)$. Call add_sample! to accumulate data for the given configuration of a spin system. Internally, this will run a dynamical trajectory and measure time correlations. The $𝒮(𝐪,ω)$ data can be retrieved by calling intensities_interpolated. Alternatively, instant_intensities_interpolated will integrate out $ω$ to obtain $𝒮(𝐪)$, optionally applying classical-to-quantum correction factors.
Three keywords are required to specify the dynamics used for the trajectory calculation.
Δt: The time step used for calculating the trajectory from which dynamic spin-spin correlations are calculated. The trajectories are calculated with an ImplicitMidpoint integrator.
ωmax: The maximum energy, $ω$, that will be resolved.
nω: The number of energy bins to calculated between 0 and ωmax.
Additional keyword options are the following:
observables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.
correlations: Specify which correlation functions are calculated, i.e. which matrix elements $αβ$ of $𝒮^{αβ}(q,ω)$ are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].
where the sum is over all pairs of spins (singly counted), including periodic images, regularized using the Ewald summation convention. The magnetic moments are $𝐌_i = μ_B g 𝐒_i$ where $g$ is the g-factor or g-tensor, and $𝐒_i$ is the spin angular momentum dipole in units of ħ. The Bohr magneton $μ_B$ and vacuum permeability $μ_0$ are physical constants, with numerical values determined by the unit system.
Generate a Mantid script which bins data according to the given BinningParameters.
Units
Take care to ensure the units are correct (R.L.U. or absolute). You may want to call Sunny.bin_rlu_as_absolute_units! or Sunny.bin_absolute_units_as_rlu! first.
Creates an empty SampledCorrelations object for calculating and storing instantaneous structure factor intensities $𝒮(𝐪)$. Call add_sample! to accumulate data for the given configuration of a spin system. Call instant_intensities_interpolated to retrieve averaged $𝒮(𝐪)$ data.
Important note: When dealing with continuous (non-Ising) spins, consider creating using dynamical_correlations instead of instant_correlations. The former will provide full $𝒮(𝐪,ω)$ data, from which $𝒮(𝐪)$ can be obtained by integrating out $ω$. During this integration step, Sunny can incorporate temperature- and $ω$-dependent classical-to-quantum correction factors to produce more accurate $𝒮(𝐪)$ estimates. See instant_intensities_interpolated for more information.
The following optional keywords are available:
observables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.
correlations: Specify which correlation functions are calculated, i.e. which matrix elements $αβ$ of $𝒮^{αβ}(q,ω)$ are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].
Return $𝒮(𝐪)$ intensities at wave vectors qs. The functionality is very similar to intensities_interpolated, except the returned array has dimensions identical to qs. If called on a SampledCorrelations with dynamical information, i.e., $𝒮(𝐪,ω)$, the $ω$ information is integrated out.
Computes the scattering intensities at each energy band for each momentum transfer k in ks, according to Linear Spin Wave Theory and the given intensity formula. The formula must have a delta-function kernel, e.g.:
formula = intensity_formula(swt, :perp, formula; kernel = delta_function_kernel)
or else the bands will be broadened, and their intensity can not be computed.
The outputs will be arrays with indices identical to ks, with the last index giving the band index. dispersions reports the energy of each band, while intensities reports the scattering intensity.
Given correlation data contained in a SampledCorrelations and BinningParameters describing the shape of a histogram, compute the intensity and normalization for each histogram bin using a given intensity_formula.
The BinningParameters are expected to accept (q,ω) in R.L.U. for the (possibly reshaped) crystal associated with sc.
If a function integrated_kernel(Δω) is passed, it will be used as the CDF of a kernel function for energy broadening. For example, integrated_kernel = Δω -> atan(Δω/η)/pi (c.f. integrated_lorentzian implements Lorentzian broadening with parameter η. Energy-dependent energy broadening can be achieved by providing an integrated_kernel(ω,Δω) whose first argument is the energy transfer ω.
Currently, energy broadening is only supported if the BinningParameters are such that the first three axes are purely spatial and the last (energy) axis is [0,0,0,1].
Computes the scattering intensities at each (Q,ω) according to Linear Spin Wave Theory and the given intensity formula. The required formula must have a non-delta-function kernel, e.g.:
formula = intensity_formula(swt, :perp; kernel = lorentzian(0.05))
or else the intensity at ωvals which are not exactly on the dispersion curve can not be calculated.
The intensity is computed at each wave vector in ks and each energy in ωvals. The output will be an array with indices identical to ks, with the last index matching ωvals.
Note that ks is an array of wave vectors of arbitrary dimension. Each element $k$ of ks must be a 3-wavevector in absolute units.
The basic function for retrieving $𝒮(𝐪,ω)$ information from a SampledCorrelations. Maps an array of wave vectors qs to an array of structure factor intensities, including an additional energy index. The values of $ω$ associated with the energy index can be retrieved by calling available_energies. The three coordinates of each wave vector are measured in reciprocal lattice units, i.e., multiples of the reciprocal lattice vectors.
interpolation: Since $𝒮(𝐪, ω)$ is calculated on a finite lattice, data is only available at discrete wave vectors. By default, Sunny will round a requested q to the nearest available wave vector. Linear interpolation can be applied by setting interpolation=:linear.
negative_energies: If set to true, Sunny will return the periodic extension of the energy axis. Most users will not want this.
formula = intensity_formula(sc::SampledCorrelations)
Establish a formula for computing the intensity of the discrete scattering modes (q,ω) using the correlation data $𝒮^{αβ}(q,ω)$ stored in the SampledCorrelations. The formula returned from intensity_formula can be passed to intensities_interpolated or intensities_binned.
There are keyword arguments providing temperature and form factor corrections:
kT: If a temperature is provided, the intensities will be rescaled by a temperature- and ω-dependent classical-to-quantum factor. kT should be specified when making comparisons with spin wave calculations or experimental data. If kT is not specified, infinite temperature (no correction) is assumed.
formfactors: To apply form factor corrections, provide this keyword with a list of FormFactors, one for each symmetry-distinct site in the crystal. The order of FormFactors must correspond to the order of site symmetry classes, e.g., as they appear when printed in display(crystal).
qs is an array of wave vectors of arbitrary dimension. Each element $q$ of qs must be a 3-vector in reciprocal lattice units (RLU), i.e., in the basis of reciprocal lattice vectors.
The first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of this array is a tensor (3×3 matrix) corresponding to the indices $α$ and $β$.
Creates an empty SampledCorrelations object for calculating and storing dynamical structure factor intensities $𝒮(𝐪,ω)$. Call add_sample! to accumulate data for the given configuration of a spin system. Internally, this will run a dynamical trajectory and measure time correlations. The $𝒮(𝐪,ω)$ data can be retrieved by calling intensities_interpolated. Alternatively, instant_intensities_interpolated will integrate out $ω$ to obtain $𝒮(𝐪)$, optionally applying classical-to-quantum correction factors.
Three keywords are required to specify the dynamics used for the trajectory calculation.
Δt: The time step used for calculating the trajectory from which dynamic spin-spin correlations are calculated. The trajectories are calculated with an ImplicitMidpoint integrator.
ωmax: The maximum energy, $ω$, that will be resolved.
nω: The number of energy bins to calculated between 0 and ωmax.
Additional keyword options are the following:
observables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.
correlations: Specify which correlation functions are calculated, i.e. which matrix elements $αβ$ of $𝒮^{αβ}(q,ω)$ are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].
where the sum is over all pairs of spins (singly counted), including periodic images, regularized using the Ewald summation convention. The magnetic moments are $𝐌_i = μ_B g 𝐒_i$ where $g$ is the g-factor or g-tensor, and $𝐒_i$ is the spin angular momentum dipole in units of ħ. The Bohr magneton $μ_B$ and vacuum permeability $μ_0$ are physical constants, with numerical values determined by the unit system.
Generate a Mantid script which bins data according to the given BinningParameters.
Units
Take care to ensure the units are correct (R.L.U. or absolute). You may want to call Sunny.bin_rlu_as_absolute_units! or Sunny.bin_absolute_units_as_rlu! first.
Creates an empty SampledCorrelations object for calculating and storing instantaneous structure factor intensities $𝒮(𝐪)$. Call add_sample! to accumulate data for the given configuration of a spin system. Call instant_intensities_interpolated to retrieve averaged $𝒮(𝐪)$ data.
Important note: When dealing with continuous (non-Ising) spins, consider creating using dynamical_correlations instead of instant_correlations. The former will provide full $𝒮(𝐪,ω)$ data, from which $𝒮(𝐪)$ can be obtained by integrating out $ω$. During this integration step, Sunny can incorporate temperature- and $ω$-dependent classical-to-quantum correction factors to produce more accurate $𝒮(𝐪)$ estimates. See instant_intensities_interpolated for more information.
The following optional keywords are available:
observables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.
correlations: Specify which correlation functions are calculated, i.e. which matrix elements $αβ$ of $𝒮^{αβ}(q,ω)$ are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].
Return $𝒮(𝐪)$ intensities at wave vectors qs. The functionality is very similar to intensities_interpolated, except the returned array has dimensions identical to qs. If called on a SampledCorrelations with dynamical information, i.e., $𝒮(𝐪,ω)$, the $ω$ information is integrated out.
Computes the scattering intensities at each energy band for each momentum transfer k in ks, according to Linear Spin Wave Theory and the given intensity formula. The formula must have a delta-function kernel, e.g.:
formula = intensity_formula(swt, :perp, formula; kernel = delta_function_kernel)
or else the bands will be broadened, and their intensity can not be computed.
The outputs will be arrays with indices identical to ks, with the last index giving the band index. dispersions reports the energy of each band, while intensities reports the scattering intensity.
Given correlation data contained in a SampledCorrelations and BinningParameters describing the shape of a histogram, compute the intensity and normalization for each histogram bin using a given intensity_formula.
The BinningParameters are expected to accept (q,ω) in R.L.U. for the (possibly reshaped) crystal associated with sc.
If a function integrated_kernel(Δω) is passed, it will be used as the CDF of a kernel function for energy broadening. For example, integrated_kernel = Δω -> atan(Δω/η)/pi (c.f. integrated_lorentzian implements Lorentzian broadening with parameter η. Energy-dependent energy broadening can be achieved by providing an integrated_kernel(ω,Δω) whose first argument is the energy transfer ω.
Currently, energy broadening is only supported if the BinningParameters are such that the first three axes are purely spatial and the last (energy) axis is [0,0,0,1].
Computes the scattering intensities at each (Q,ω) according to Linear Spin Wave Theory and the given intensity formula. The required formula must have a non-delta-function kernel, e.g.:
formula = intensity_formula(swt, :perp; kernel = lorentzian(0.05))
or else the intensity at ωvals which are not exactly on the dispersion curve can not be calculated.
The intensity is computed at each wave vector in ks and each energy in ωvals. The output will be an array with indices identical to ks, with the last index matching ωvals.
Note that ks is an array of wave vectors of arbitrary dimension. Each element $k$ of ks must be a 3-wavevector in absolute units.
The basic function for retrieving $𝒮(𝐪,ω)$ information from a SampledCorrelations. Maps an array of wave vectors qs to an array of structure factor intensities, including an additional energy index. The values of $ω$ associated with the energy index can be retrieved by calling available_energies. The three coordinates of each wave vector are measured in reciprocal lattice units, i.e., multiples of the reciprocal lattice vectors.
interpolation: Since $𝒮(𝐪, ω)$ is calculated on a finite lattice, data is only available at discrete wave vectors. By default, Sunny will round a requested q to the nearest available wave vector. Linear interpolation can be applied by setting interpolation=:linear.
negative_energies: If set to true, Sunny will return the periodic extension of the energy axis. Most users will not want this.
formula = intensity_formula(sc::SampledCorrelations)
Establish a formula for computing the intensity of the discrete scattering modes (q,ω) using the correlation data $𝒮^{αβ}(q,ω)$ stored in the SampledCorrelations. The formula returned from intensity_formula can be passed to intensities_interpolated or intensities_binned.
There are keyword arguments providing temperature and form factor corrections:
kT: If a temperature is provided, the intensities will be rescaled by a temperature- and ω-dependent classical-to-quantum factor. kT should be specified when making comparisons with spin wave calculations or experimental data. If kT is not specified, infinite temperature (no correction) is assumed.
formfactors: To apply form factor corrections, provide this keyword with a list of FormFactors, one for each symmetry-distinct site in the crystal. The order of FormFactors must correspond to the order of site symmetry classes, e.g., as they appear when printed in display(crystal).
formula = intensity_formula(swt::SpinWaveTheory; kernel = ...)
Establish a formula for computing the scattering intensity by diagonalizing the hamiltonian $H(q)$ using Linear Spin Wave Theory.
If kernel = delta_function_kernel, then the resulting formula can be used with intensities_bands.
If kernel is an energy broadening kernel function, then the resulting formula can be used with intensities_broadened. Energy broadening kernel functions can either be a function of Δω only, e.g.:
kernel = Δω -> ...
or a function of both the energy transfer ω and of Δω, e.g.:
kernel = (ω,Δω) -> ...
The integral of a properly normalized kernel function over all Δω is one.
Compute the lattice parameters $(a, b, c, α, β, γ)$ for the three lattice vectors provided as columns of latvecs. The inverse mapping is lattice_vectors.
Return the lattice vectors, as columns of the $3×3$ output matrix, that correspond to the conventional unit cell defined by the lattice constants $(a, b, c)$ and the angles $(α, β, γ)$ in degrees. The inverse mapping is lattice_params.
minimize_energy!(sys::System{N}; maxiters=100, subiters=20,
- method=Optim.ConjugateGradient(), kwargs...) where N
Optimizes the spin configuration in sys to minimize energy. A total of maxiters iterations will be attempted, with restarts after every subiters iterations. The remaining kwargs will be forwarded to the optimize method of the Optim.jl package.
Converts a position r to four indices of a Site. The coordinates of r are given in units of the lattice vectors for the original crystal. This function can be useful for working with systems that have been reshaped using reshape_supercell.
Example
# Find the `site` at the center of a unit cell which is displaced by four
+end
If your custom formula returns a type other than Float64, use the return_type keyword argument to flag this.
formula = intensity_formula(swt::SpinWaveTheory; kernel = ...)
Establish a formula for computing the scattering intensity by diagonalizing the hamiltonian $H(q)$ using Linear Spin Wave Theory.
If kernel = delta_function_kernel, then the resulting formula can be used with intensities_bands.
If kernel is an energy broadening kernel function, then the resulting formula can be used with intensities_broadened. Energy broadening kernel functions can either be a function of Δω only, e.g.:
kernel = Δω -> ...
or a function of both the energy transfer ω and of Δω, e.g.:
kernel = (ω,Δω) -> ...
The integral of a properly normalized kernel function over all Δω is one.
Compute the lattice parameters $(a, b, c, α, β, γ)$ for the three lattice vectors provided as columns of latvecs. The inverse mapping is lattice_vectors.
Return the lattice vectors, as columns of the $3×3$ output matrix, that correspond to the conventional unit cell defined by the lattice constants $(a, b, c)$ and the angles $(α, β, γ)$ in degrees. The inverse mapping is lattice_params.
minimize_energy!(sys::System{N}; maxiters=100, subiters=20,
+ method=Optim.ConjugateGradient(), kwargs...) where N
Optimizes the spin configuration in sys to minimize energy. A total of maxiters iterations will be attempted, with restarts after every subiters iterations. The remaining kwargs will be forwarded to the optimize method of the Optim.jl package.
Converts a position r to four indices of a Site. The coordinates of r are given in units of the lattice vectors for the original crystal. This function can be useful for working with systems that have been reshaped using reshape_supercell.
Example
# Find the `site` at the center of a unit cell which is displaced by four
# multiples of the first lattice vector
site = position_to_site(sys, [4.5, 0.5, 0.5])
# Print the dipole at this site
-println(sys.dipoles[site])
This function emulates the experimental situation of "powder averaging," where only the magnitude (and not the direction) of the momentum transfer is resolvable. The intensities are binned similarly to intensities_binned, but the histogram x-axis is |k| in absolute units, which is a nonlinear function of kx,ky,kz. The y-axis is energy.
Radial binning parameters are specified as tuples (start,end,bin_width), e.g. radial_binning_parameters = (0,6π,6π/55).
Energy broadening is supported in the same way as intensities_binned, and this function accepts the same kind of intensity_formula.
Returns the shape of the primitive cell as a 3×3 matrix, in fractional coordinates of the conventional lattice vectors. May be useful for constructing inputs to reshape_supercell.
Examples
# Valid if `cryst` has not been reshaped
-@assert cryst.prim_latvecs ≈ cryst.latvecs * primitive_cell_shape(cryst)
Prints symmetry information for bond bond. A symmetry-equivalent reference bond b_ref can optionally be provided to fix the meaning of the coefficients A, B, ...
Print symmetry information for the site i, including allowed g-tensor and allowed anisotropy operator. An optional rotation matrix R can be provided to define the reference frame for expression of the anisotropy.
Prints a local Hermitian operator as a linear combination of Stevens operators. The operator op may be a finite-dimensional matrix or an abstract spin polynomial in the large-$S$ limit.
This function emulates the experimental situation of "powder averaging," where only the magnitude (and not the direction) of the momentum transfer is resolvable. The intensities are binned similarly to intensities_binned, but the histogram x-axis is |k| in absolute units, which is a nonlinear function of kx,ky,kz. The y-axis is energy.
Radial binning parameters are specified as tuples (start,end,bin_width), e.g. radial_binning_parameters = (0,6π,6π/55).
Energy broadening is supported in the same way as intensities_binned, and this function accepts the same kind of intensity_formula.
Returns the shape of the primitive cell as a 3×3 matrix, in fractional coordinates of the conventional lattice vectors. May be useful for constructing inputs to reshape_supercell.
Examples
# Valid if `cryst` has not been reshaped
+@assert cryst.prim_latvecs ≈ cryst.latvecs * primitive_cell_shape(cryst)
Prints symmetry information for bond bond. A symmetry-equivalent reference bond b_ref can optionally be provided to fix the meaning of the coefficients A, B, ...
Print symmetry information for the site i, including allowed g-tensor and allowed anisotropy operator. An optional rotation matrix R can be provided to define the reference frame for expression of the anisotropy.
Prints a local Hermitian operator as a linear combination of Stevens operators. The operator op may be a finite-dimensional matrix or an abstract spin polynomial in the large-$S$ limit.
Print a suggested reference frame, as a rotation matrix R, that can be used as input to print_site(). The purpose is to simplify the description of allowed anisotropies.
Print symmetry information for all equivalence classes of sites and bonds, up to a maximum bond distance of max_dist. Equivalent to calling print_bond(cryst, b) for every bond b in reference_bonds(cryst, max_dist), where Bond(i, i, [0,0,0]) refers to a single site i.
For Bravais lattices: Prints up to nmax wavevectors according to their instantaneous (static) structure factor intensities, listed in descending order. For non-Bravais lattices: Performs the same analysis for each spin sublattice independently; the output weights are naïvely averaged over sublattices, without incorporating phase shift information. This procedure therefore wraps all wavevectors into the first Brillouin zone. Each wavevector coordinate is given between $-1/2$ and $1/2$ in reciprocal lattice units (RLU). The output from this function will typically be used as input to suggest_magnetic_supercell.
Because this function does not incorporate phase information in its averaging over sublattices, the printed weights are not directly comparable with experiment. For that purpose, use instant_correlations instead.
Generate a proposal function that adds a Gaussian perturbation to the existing spin state. In :dipole mode, the procedure is to first introduce a random three-vector perturbation $𝐬′ = 𝐬 + |𝐬| ξ$ and then return the properly normalized spin $|𝐬| (𝐬′/|𝐬′|)$. Each component of the random vector $ξ$ is Gaussian distributed with a standard deviation of magnitude; the latter is dimensionless and typically smaller than one.
In :SUN mode, the procedure is analogous, but now involving Gaussian perturbations to each of the $N$ complex components of an SU(N) coherent state.
In the limit of very large magnitude, this function coincides with propose_uniform.
Function to propose pure spin flip updates in the context of a LocalSampler. Dipoles are flipped as $𝐬 → -𝐬$. SU(N) coherent states are flipped using the time-reversal operator.
Function to propose a uniformly random spin update in the context of a LocalSampler. In :dipole mode, the result is a random three-vector with appropriate normalization. In :SUN mode, the result is a random SU(N) coherent state with appropriate normalization.
Returns a pair (path, xticks). The path return value is a list of wavevectors that samples linearly between the provided wavevectors qs. The xticks return value can be used to label the special $𝐪$ values on the x-axis of a plot.
Special note about units: the wavevectors qs must be provided in reciprocal lattice units (RLU) for the given crystal, but the sampling density must be specified in the global frame. Specifically, the density is given as number of sample points per unit of radian inverse length, where the unit of length is the same as that used to specify the lattice vectors of the Crystal. The path will therefore include more samples between q-points that are further apart in absolute Fourier distance.
Takes a list of wave vectors, qs in R.L.U., and builds a series of histogram BinningParameters whose first axis traces a path through the provided points. The second and third axes are integrated over according to the args and kwargs, which are passed through to slice_2D_binning_parameters.
Also returned is a list of marker indices corresponding to the input points, and a list of ranges giving the indices of each histogram x-axis within a concatenated histogram. The density parameter is given in samples per reciprocal lattice unit (R.L.U.).
Returns a full list of bonds, one for each symmetry equivalence class, up to distance max_dist. The reference bond b for each equivalence class is selected according to a scoring system that prioritizes simplification of the elements in basis_for_symmetry_allowed_couplings(cryst, b).
Maps an existing System to a new one that has the shape and periodicity of a requested supercell. The columns of the $3×3$ integer matrix shape represent the supercell lattice vectors measured in units of the original crystal lattice vectors.
resize_supercell(sys::System{N}, latsize::NTuple{3,Int}) where N
Creates a System with a given number of conventional unit cells in each lattice vector direction. Interactions and other settings will be inherited from sys.
Returns a $3×3$ matrix that rotates wavevectors in reciprocal lattice units (RLU). The axis vector is a real-space direction in absolute units (but arbitrary magnitude), and the angle is in radians.
Set a coherent spin state at a Site using the $N$ complex amplitudes in Z.
For a standard SpinInfo, these amplitudes will be interpreted in the eigenbasis of $𝒮̂ᶻ$. That is, Z[1] represents the amplitude for the basis state fully polarized along the $ẑ$-direction, and subsequent components represent states with decreasing angular momentum along this axis ($m = S, S-1, …, -S$).
Sets a 3×3 spin-exchange matrix J along bond, yielding a pairwise interaction energy $𝐒_i⋅J 𝐒_j$. This interaction will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells.
The parameter J may be scalar or matrix-valued. As a convenience, dmvec(D) can be used to construct the antisymmetric part of the exchange, where D is the Dzyaloshinskii-Moriya pseudo-vector. The resulting interaction will be $𝐃⋅(𝐒_i×𝐒_j)$.
For more general interactions, such as biquadratic, use set_pair_coupling! instead.
Print a suggested reference frame, as a rotation matrix R, that can be used as input to print_site(). The purpose is to simplify the description of allowed anisotropies.
Print symmetry information for all equivalence classes of sites and bonds, up to a maximum bond distance of max_dist. Equivalent to calling print_bond(cryst, b) for every bond b in reference_bonds(cryst, max_dist), where Bond(i, i, [0,0,0]) refers to a single site i.
For Bravais lattices: Prints up to nmax wavevectors according to their instantaneous (static) structure factor intensities, listed in descending order. For non-Bravais lattices: Performs the same analysis for each spin sublattice independently; the output weights are naïvely averaged over sublattices, without incorporating phase shift information. This procedure therefore wraps all wavevectors into the first Brillouin zone. Each wavevector coordinate is given between $-1/2$ and $1/2$ in reciprocal lattice units (RLU). The output from this function will typically be used as input to suggest_magnetic_supercell.
Because this function does not incorporate phase information in its averaging over sublattices, the printed weights are not directly comparable with experiment. For that purpose, use instant_correlations instead.
Generate a proposal function that adds a Gaussian perturbation to the existing spin state. In :dipole mode, the procedure is to first introduce a random three-vector perturbation $𝐬′ = 𝐬 + |𝐬| ξ$ and then return the properly normalized spin $|𝐬| (𝐬′/|𝐬′|)$. Each component of the random vector $ξ$ is Gaussian distributed with a standard deviation of magnitude; the latter is dimensionless and typically smaller than one.
In :SUN mode, the procedure is analogous, but now involving Gaussian perturbations to each of the $N$ complex components of an SU(N) coherent state.
In the limit of very large magnitude, this function coincides with propose_uniform.
Function to propose pure spin flip updates in the context of a LocalSampler. Dipoles are flipped as $𝐬 → -𝐬$. SU(N) coherent states are flipped using the time-reversal operator.
Function to propose a uniformly random spin update in the context of a LocalSampler. In :dipole mode, the result is a random three-vector with appropriate normalization. In :SUN mode, the result is a random SU(N) coherent state with appropriate normalization.
Returns a pair (path, xticks). The path return value is a list of wavevectors that samples linearly between the provided wavevectors qs. The xticks return value can be used to label the special $𝐪$ values on the x-axis of a plot.
Special note about units: the wavevectors qs must be provided in reciprocal lattice units (RLU) for the given crystal, but the sampling density must be specified in the global frame. Specifically, the density is given as number of sample points per unit of radian inverse length, where the unit of length is the same as that used to specify the lattice vectors of the Crystal. The path will therefore include more samples between q-points that are further apart in absolute Fourier distance.
Takes a list of wave vectors, qs in R.L.U., and builds a series of histogram BinningParameters whose first axis traces a path through the provided points. The second and third axes are integrated over according to the args and kwargs, which are passed through to slice_2D_binning_parameters.
Also returned is a list of marker indices corresponding to the input points, and a list of ranges giving the indices of each histogram x-axis within a concatenated histogram. The density parameter is given in samples per reciprocal lattice unit (R.L.U.).
Returns a full list of bonds, one for each symmetry equivalence class, up to distance max_dist. The reference bond b for each equivalence class is selected according to a scoring system that prioritizes simplification of the elements in basis_for_symmetry_allowed_couplings(cryst, b).
Maps an existing System to a new one that has the shape and periodicity of a requested supercell. The columns of the $3×3$ integer matrix shape represent the supercell lattice vectors measured in units of the original crystal lattice vectors.
resize_supercell(sys::System{N}, latsize::NTuple{3,Int}) where N
Creates a System with a given number of conventional unit cells in each lattice vector direction. Interactions and other settings will be inherited from sys.
Returns a $3×3$ matrix that rotates wavevectors in reciprocal lattice units (RLU). The axis vector is a real-space direction in absolute units (but arbitrary magnitude), and the angle is in radians.
Set a coherent spin state at a Site using the $N$ complex amplitudes in Z.
For a standard SpinInfo, these amplitudes will be interpreted in the eigenbasis of $𝒮̂ᶻ$. That is, Z[1] represents the amplitude for the basis state fully polarized along the $ẑ$-direction, and subsequent components represent states with decreasing angular momentum along this axis ($m = S, S-1, …, -S$).
Sets a 3×3 spin-exchange matrix J along bond, yielding a pairwise interaction energy $𝐒_i⋅J 𝐒_j$. This interaction will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells.
The parameter J may be scalar or matrix-valued. As a convenience, dmvec(D) can be used to construct the antisymmetric part of the exchange, where D is the Dzyaloshinskii-Moriya pseudo-vector. The resulting interaction will be $𝐃⋅(𝐒_i×𝐒_j)$.
For more general interactions, such as biquadratic, use set_pair_coupling! instead.
Sets an exchange interaction `𝐒_i⋅J 𝐒_j along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The system must support inhomogeneous interactions via to_inhomogeneous.
If the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.
For more general interactions, such as biquadratic, use set_pair_coupling_at! instead.
Set the single-ion anisotropy for the ith atom of every unit cell, as well as all symmetry-equivalent atoms. The operator op may be provided as an abstract function of the local spin operators, as a polynomial of spin_matrices, or as a linear combination of stevens_matrices.
Examples
# An easy axis anisotropy in the z-direction
+set_exchange!(sys, J2, bond)
Sets an exchange interaction `𝐒_i⋅J 𝐒_j along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The system must support inhomogeneous interactions via to_inhomogeneous.
If the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.
For more general interactions, such as biquadratic, use set_pair_coupling_at! instead.
Set the single-ion anisotropy for the ith atom of every unit cell, as well as all symmetry-equivalent atoms. The operator op may be provided as an abstract function of the local spin operators, as a polynomial of spin_matrices, or as a linear combination of stevens_matrices.
Examples
# An easy axis anisotropy in the z-direction
set_onsite_coupling!(sys, S -> -D*S[3]^3, i)
# The unique quartic single-ion anisotropy for a site with cubic point group
@@ -80,31 +80,31 @@
# An equivalent expression of this quartic anisotropy, up to a constant shift
O = stevens_matrices(spin_label(sys, i))
-set_onsite_coupling!(sys, O[4,0] + 5*O[4,4], i)
Sets the single-ion anisotropy operator op for a single Site, ignoring crystal symmetry. The system must support inhomogeneous interactions via to_inhomogeneous.
Sets an arbitrary coupling op along bond. This coupling will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites.
Sets the single-ion anisotropy operator op for a single Site, ignoring crystal symmetry. The system must support inhomogeneous interactions via to_inhomogeneous.
Sets an arbitrary coupling op along bond. This coupling will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites.
Examples
# Bilinear+biquadratic exchange involving 3×3 matrices J1 and J2
set_pair_coupling!(sys, (Si, Sj) -> Si'*J1*Sj + (Si'*J2*Sj)^2, bond)
# Equivalent expression using an appropriate fixed matrix representation
S = spin_matrices(1/2)
Si, Sj = to_product_space(S, S)
-set_pair_coupling!(sys, Si'*J1*Sj + (Si'*J2*Sj)^2, bond)
Sets an arbitrary coupling along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites. The documentation for set_pair_coupling! provides examples constructing op.
The system must support inhomogeneous interactions via to_inhomogeneous.
If the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.
Initializes the system with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0 at the real-space origin. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.
Example
# Spiral order for a wavevector propagating in the direction of the first
+set_pair_coupling!(sys, Si'*J1*Sj + (Si'*J2*Sj)^2, bond)
Sets an arbitrary coupling along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites. The documentation for set_pair_coupling! provides examples constructing op.
The system must support inhomogeneous interactions via to_inhomogeneous.
If the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.
Initializes the system with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0 at the real-space origin. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.
Example
# Spiral order for a wavevector propagating in the direction of the first
# reciprocal lattice vector (i.e., orthogonal to the lattice vectors ``𝐚_2``
# and ``𝐚_3``), repeating with a period of 10 lattice constants, and spiraling
# about the ``ẑ``-axis. The spin at the origin will point in the direction
# ``𝐒_0 = ŷ + ẑ``. Here, ``(x̂, ŷ, ẑ)`` are the axes of Cartesian coordinate
# system in the global frame.
-set_spiral_order!(sys; q=[1/10, 0, 0], axis=[0, 0, 1], S0=[0, 1, 1])
set_spiral_order_on_sublattice!(sys, i; q, axis, S0)
Initializes sublattice i with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0. The phase is selected such that the spin at sys.dipole[1,1,1,i] will point in the direction of S0. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.
This function is not available for systems with reshaped unit cells.
Creates BinningParameters which make a cut along one dimension of Q-space.
The x-axis of the resulting histogram consists of cut_bins-many bins ranging from cut_from_q to cut_to_q. The width of the bins in the transverse direciton is controlled by cut_width and cut_height.
The binning in the transverse directions is defined in the following way, which sets their normalization and orthogonality properties:
set_spiral_order_on_sublattice!(sys, i; q, axis, S0)
Initializes sublattice i with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0. The phase is selected such that the spin at sys.dipole[1,1,1,i] will point in the direction of S0. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.
This function is not available for systems with reshaped unit cells.
Creates BinningParameters which make a cut along one dimension of Q-space.
The x-axis of the resulting histogram consists of cut_bins-many bins ranging from cut_from_q to cut_to_q. The width of the bins in the transverse direciton is controlled by cut_width and cut_height.
The binning in the transverse directions is defined in the following way, which sets their normalization and orthogonality properties:
In other words, the axes are orthonormal with respect to the Euclidean metric.
If the cut is too narrow, there will be very few scattering vectors per bin, or the number per bin will vary substantially along the cut. If the output appears under-resolved, try increasing cut_width.
The four axes of the resulting histogram are:
Along the cut
Fist transverse Q direction
Second transverse Q direction
Energy
This function can be used without reference to a SampledCorrelations using this alternate syntax to manually specify the bin centers for the energy axis:
Returns a triple of $N×N$ spin matrices, where $N = 2S+1$. These are the generators of SU(2) in the spin-S representation.
If S == Inf, then the return values are abstract symbols denoting infinite-dimensional matrices that commute. These can be useful for repeating historical studies, or modeling micromagnetic systems. A technical discussion appears in the Sunny documentation page: Interaction Strength Renormalization.
Example
S = spin_matrices(3/2)
+cotransverse_covector = normalize(transverse_covector × cut_covector)
In other words, the axes are orthonormal with respect to the Euclidean metric.
If the cut is too narrow, there will be very few scattering vectors per bin, or the number per bin will vary substantially along the cut. If the output appears under-resolved, try increasing cut_width.
The four axes of the resulting histogram are:
Along the cut
Fist transverse Q direction
Second transverse Q direction
Energy
This function can be used without reference to a SampledCorrelations using this alternate syntax to manually specify the bin centers for the energy axis:
Returns a triple of $N×N$ spin matrices, where $N = 2S+1$. These are the generators of SU(2) in the spin-S representation.
If S == Inf, then the return values are abstract symbols denoting infinite-dimensional matrices that commute. These can be useful for repeating historical studies, or modeling micromagnetic systems. A technical discussion appears in the Sunny documentation page: Interaction Strength Renormalization.
Advance the spin configuration one dynamical time-step. The dynamics object may be a continuous spin dynamics, such as Langevin or ImplicitMidpoint, or it may be a discrete Monte Carlo sampling scheme such as LocalSampler.
Returns a generator of Stevens operators in the spin-S representation. The return value O can be indexed as O[k,q], where $0 ≤ k ≤ 6$ labels an irrep of SO(3) and $-k ≤ q ≤ k$. This will produce an $N×N$ matrix where ``N = 2S
1``. Linear combinations of Stevens operators can be used as a "physical
basis" for decomposing local observables. To see this decomposition, use print_stevens_expansion.
If S == Inf, then symbolic operators will be returned. In this infinite dimensional limit, the Stevens operators become homogeneous polynomials of commuting spin operators.
Example
O = stevens_matrices(2)
+@assert S[1]*S[2] - S[2]*S[1] == 0
Advance the spin configuration one dynamical time-step. The dynamics object may be a continuous spin dynamics, such as Langevin or ImplicitMidpoint, or it may be a discrete Monte Carlo sampling scheme such as LocalSampler.
Returns a generator of Stevens operators in the spin-S representation. The return value O can be indexed as O[k,q], where $0 ≤ k ≤ 6$ labels an irrep of SO(3) and $-k ≤ q ≤ k$. This will produce an $N×N$ matrix where ``N = 2S
1``. Linear combinations of Stevens operators can be used as a "physical
basis" for decomposing local observables. To see this decomposition, use print_stevens_expansion.
If S == Inf, then symbolic operators will be returned. In this infinite dimensional limit, the Stevens operators become homogeneous polynomials of commuting spin operators.
Example
O = stevens_matrices(2)
S = spin_matrices(2)
A = (1/20)O[4,0] + (1/4)O[4,4] + (102/5)I
B = S[1]^4 + S[2]^4 + S[3]^4
-@assert A ≈ B
Suggests a magnetic supercell, in units of the crystal lattice vectors, that is consistent with periodicity of the wavevectors qs in RLU. If the wavevectors are incommensurate (with respect to the maximum supercell size maxsize), one can select a larger error tolerance tol to find a supercell that is almost commensurate.
Prints a $3×3$ matrix of integers that is suitable for use in reshape_supercell.
Examples
# A magnetic supercell for a single-Q structure. Will print
+@assert A ≈ B
Suggests a magnetic supercell, in units of the crystal lattice vectors, that is consistent with periodicity of the wavevectors qs in RLU. If the wavevectors are incommensurate (with respect to the maximum supercell size maxsize), one can select a larger error tolerance tol to find a supercell that is almost commensurate.
Prints a $3×3$ matrix of integers that is suitable for use in reshape_supercell.
Examples
# A magnetic supercell for a single-Q structure. Will print
q1 = [0, -1/4, 1/4]
suggest_magnetic_supercell([q1]) # [1 0 0; 0 2 1; 0 -2 1]
@@ -117,11 +117,11 @@
suggest_magnetic_supercell([[0, 0, 1/√5], [0, 0, 1/√7]]; tol=1e-2)
# This prints [1 0 0; 0 1 0; 0 0 16], which becomes commensurate under the
-# approximations `1/√5 ≈ 7/16` and `1/√7 ≈ 3/8`.
Given a Bond for the original (unreshaped) crystal, return all symmetry equivalent bonds in the System. Each returned bond is represented as a pair of Sites, which may be used as input to set_exchange_at!. Reverse bonds are not included (no double counting).
Example
for (site1, site2, offset) in symmetry_equivalent_bonds(sys, bond)
+# approximations `1/√5 ≈ 7/16` and `1/√7 ≈ 3/8`.
Given a Bond for the original (unreshaped) crystal, return all symmetry equivalent bonds in the System. Each returned bond is represented as a pair of Sites, which may be used as input to set_exchange_at!. Reverse bonds are not included (no double counting).
Example
for (site1, site2, offset) in symmetry_equivalent_bonds(sys, bond)
@assert site1 < site2
set_exchange_at!(sys, J, site1, site2; offset)
-end
Given lists of operators acting on local Hilbert spaces individually, return the corresponding operators that act on the tensor product space. In typical usage, the inputs will represent local physical observables and the outputs will be used to define quantum couplings.
Create BinningParameters which place one histogram bin centered at each possible (q,ω) scattering vector of the crystal. This is the finest possible binning without creating bins with zero scattering vectors in them.
This function can be used without reference to a SampledCorrelations using an alternate syntax to manually specify the bin centers for the energy axis and the lattice size:
Macro to generate a proposal function that randomly selects among the provided functions according to the provided probability weights. For use with LocalSampler.
Example
# A proposal function that proposes a spin flip 40% of the time, and a
+end
Given lists of operators acting on local Hilbert spaces individually, return the corresponding operators that act on the tensor product space. In typical usage, the inputs will represent local physical observables and the outputs will be used to define quantum couplings.
Create BinningParameters which place one histogram bin centered at each possible (q,ω) scattering vector of the crystal. This is the finest possible binning without creating bins with zero scattering vectors in them.
This function can be used without reference to a SampledCorrelations using an alternate syntax to manually specify the bin centers for the energy axis and the lattice size:
Macro to generate a proposal function that randomly selects among the provided functions according to the provided probability weights. For use with LocalSampler.
Example
# A proposal function that proposes a spin flip 40% of the time, and a
# Gaussian perturbation 60% of the time.
-@mix_proposals 0.4 propose_flip 0.6 propose_delta(0.2)
Export a VTK-compatible file to filename (do not include file extension when specifying the file name) which contains the data as VTK Cell Data on a grid parameterized by params.
Export a VTK-compatible file to filename (do not include file extension when specifying the file name) which contains the data as VTK Cell Data on a grid parameterized by params.
Calculating structure factors with classical dynamics is computationally expensive, and Sunny does not currently parallelize these calculations at a fine-grained level. However, Julia provides facilities that allow users to run multiple simulations in parallel with only a little extra effort. We will look at two approaches to doing this: multithreading and Julia's Distributed package. We'll present these approaches in a series of code snippets that can be copied and pasted into your preferred Julia development environment.
The serial approach to calculating a structure factor, covered in FeI₂ at Finite Temperature, involves thermalizing a spin System and then calling add_sample!. add_sample! uses the state of the System as an initial condition for the calculation of a dynamical trajectory. The correlations of the trajectory are calculated and accumulated into a running average of the $𝒮(𝐪,ω)$. This sequence is repeated to generate additional samples.
To illustrate, we'll set up a a simple model: a spin-1 antiferromagnet on an FCC crystal.
using Sunny, GLMakie
+Parallelizing Calculations · Sunny documentation
Calculating structure factors with classical dynamics is computationally expensive, and Sunny does not currently parallelize these calculations at a fine-grained level. However, Julia provides facilities that allow users to run multiple simulations in parallel with only a little extra effort. We will look at two approaches to doing this: multithreading and Julia's Distributed package. We'll present these approaches in a series of code snippets that can be copied and pasted into your preferred Julia development environment.
The serial approach to calculating a structure factor, covered in the FeI₂ tutorial, involves thermalizing a spin System and then calling add_sample!. add_sample! uses the state of the System as an initial condition for the calculation of a dynamical trajectory. The correlations of the trajectory are calculated and accumulated into a running average of the $𝒮(𝐪,ω)$. This sequence is repeated to generate additional samples.
To illustrate, we'll set up a a simple model: a spin-1 antiferromagnet on an FCC crystal.
using Sunny, GLMakie
function make_system(cryst; J, dims, seed=nothing)
sys = System(cryst, dims, [SpinInfo(1, S=1, g=2)], :dipole; seed)
@@ -63,4 +63,4 @@
end
return sc
-end
Finally, merge the results into a summary SampledCorrelations.
sc = merge_correlations(scs)
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
+end
Finally, merge the results into a summary SampledCorrelations.
sc = merge_correlations(scs)
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
A unique feature of Sunny is its support for building classical models where quantum spin is represented as an $N$-level system, rather than just an expected dipole. This generalization can be important when modeling quantum spin Hamiltonians that include, e.g., a single-ion anisotropy, or a biquadratic coupling between sites. Sunny also supports constraining quantum spin to the space of pure dipoles; in this case, Sunny will automatically perform an interaction strength renormalization that maximizes accuracy.
A quantum spin-$S$ state has $N = 2S + 1$ levels. Each local spin operator $\hat{S}^{\{x,y,z\}}$ is faithfully represented as an $N×N$ matrix. These matrices can be accessed using spin_matrices for a given label $S$. For example, the Pauli matrices are associated with $S = 1/2$.
When $S > 1/2$, it is possible to construct multipole moments beyond the spin-dipole. For example,
S = spin_matrices(3/2)
+Interaction Strength Renormalization · Sunny documentation
A unique feature of Sunny is its support for building classical models where quantum spin is represented as an $N$-level system, rather than just an expected dipole. This generalization can be important when modeling quantum spin Hamiltonians that include, e.g., a single-ion anisotropy, or a biquadratic coupling between sites. Sunny also supports constraining quantum spin to the space of pure dipoles; in this case, Sunny will automatically perform an interaction strength renormalization that maximizes accuracy.
A quantum spin-$S$ state has $N = 2S + 1$ levels. Each local spin operator $\hat{S}^{\{x,y,z\}}$ is faithfully represented as an $N×N$ matrix. These matrices can be accessed using spin_matrices for a given label $S$. For example, the Pauli matrices are associated with $S = 1/2$.
When $S > 1/2$, it is possible to construct multipole moments beyond the spin-dipole. For example,
If the operator -S[3]^2 is passed to set_onsite_coupling!, it would set an easy-axis anisotropy in the $\hat{z}$ direction.
Any Hermitian operator can be expanded in the basis of Stevens operators $\hat{\mathcal{O}}_{k,q}$ up to a constant shift. To see this expansion, use print_stevens_expansion:
print_stevens_expansion((S[1]^2 + S[2]^2)) # Prints -(1/3)𝒪₂₀ + 5/2
Alternatively, the same operator could have been constructed directly from stevens_matrices:
Sunny will typically operate in one of two modes: :SUN or :dipole. The former faithfully represents quantum spin as an SU(N) coherent-state which, for our purposes, is an $N$-component complex vector. In contrast, :dipole mode constrains the coherent-state to the space of pure dipoles. Here, Sunny will automatically renormalize the magnitude of each Stevens operator to achieve maximal consistency with :SUN mode. This procedure was derived in D. Dahlbom et al., [arXiv:2304.03874].
By way of illustration, consider a quantum operator $\hat{\mathcal{H}}_{\mathrm{local}}$ giving a single-ion anisotropy for one site. In Stevens operators,
In :SUN mode, Sunny will faithfully represent $\hat{\mathcal H}_{\mathrm{local}}$ as an $N×N$ matrix. In :dipole mode, the expected energy $\langle \hat{\mathcal H}_{\mathrm{local}} \rangle$ must somehow be approximated as a function of the expected dipole $\mathbf{s}$.
One approach is to formally take $S \to \infty$, and this yields the traditional classical limit of a spin system. In this, limit spin operators commute, and expectation values of polynomials become polynomials of expectation values. For example, $\langle \hat{S}^\alpha \hat{S}^\beta\rangle \to \langle \hat{S}^\alpha \rangle \langle \hat{S}^\beta\rangle$, because any corrections are damped by the factor $S^{-1} \to 0$. The expectation of a Stevens operator $\langle \hat{\mathcal{O}}_{k,q} \rangle$ becomes a Stevens function $\mathcal{O}_{k,q}(\mathbf{s})$, i.e., a polynomial of expected dipole $\mathbf{s} = \langle \hat{\mathbf{S}} \rangle$.
In a real magnetic compound, however, the spin magnitude $S$ is not necessarily large. To obtain a better approximation, one should avoid the assumption $S \to \infty$. Our approach is to start with the full dynamics of SU(N) coherent states, and then constrain it to the space of pure dipole states $|\mathbf{s}\rangle$. The latter are defined such that expectation values,
For each $k$ value, the set of operators $\hat{\mathcal{O}}_{k,q'}$ for $q' = -k, \dots, k$ form an irreducible representation of the group of rotations O(3). That is, rotation will transform $\hat{\mathcal{O}}_{k,q}$ into a linear combination of $\hat{\mathcal{O}}_{k,q'}$ where $q'$ varies but $k$ remains fixed.
In taking the large-$S$ limit, each dipole operator is replaced by its expectation value $\mathbf{s} = \langle \hat{\mathbf{S}} \rangle$, and only leading-order terms are retained. The operator $\hat{\mathcal{O}}_{k,q}$ becomes a homogeneous polynomial $O_{k,q}(\mathbf{s})$ of order $k$ in the spin components. One can see these polynomials by constructing stevens_matrices with the argument S = Inf. Due to the normalization constraint, each dipole can be expressed in polar angles, $(\theta, \phi)$. Then the Stevens functions $O_{k,q}(\mathbf{s})$ correspond to the spherical harmonic functions $Y_l^m(\theta, \phi)$ where $l=k$ and $m=q$; this correspondence is valid up to $k$ and $q$-dependent rescaling factors.
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
For each $k$ value, the set of operators $\hat{\mathcal{O}}_{k,q'}$ for $q' = -k, \dots, k$ form an irreducible representation of the group of rotations O(3). That is, rotation will transform $\hat{\mathcal{O}}_{k,q}$ into a linear combination of $\hat{\mathcal{O}}_{k,q'}$ where $q'$ varies but $k$ remains fixed.
In taking the large-$S$ limit, each dipole operator is replaced by its expectation value $\mathbf{s} = \langle \hat{\mathbf{S}} \rangle$, and only leading-order terms are retained. The operator $\hat{\mathcal{O}}_{k,q}$ becomes a homogeneous polynomial $O_{k,q}(\mathbf{s})$ of order $k$ in the spin components. One can see these polynomials by constructing stevens_matrices with the argument S = Inf. Due to the normalization constraint, each dipole can be expressed in polar angles, $(\theta, \phi)$. Then the Stevens functions $O_{k,q}(\mathbf{s})$ correspond to the spherical harmonic functions $Y_l^m(\theta, \phi)$ where $l=k$ and $m=q$; this correspondence is valid up to $k$ and $q$-dependent rescaling factors.
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
diff --git a/dev/search_index.js b/dev/search_index.js
index 677263bf7..e3ec0f803 100644
--- a/dev/search_index.js
+++ b/dev/search_index.js
@@ -1,3 +1,3 @@
var documenterSearchIndex = {"docs":
-[{"location":"parallelism.html#Parallelizing-Calculations","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"","category":"section"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Calculating structure factors with classical dynamics is computationally expensive, and Sunny does not currently parallelize these calculations at a fine-grained level. However, Julia provides facilities that allow users to run multiple simulations in parallel with only a little extra effort. We will look at two approaches to doing this: multithreading and Julia's Distributed package. We'll present these approaches in a series of code snippets that can be copied and pasted into your preferred Julia development environment.","category":"page"},{"location":"parallelism.html#Review-of-the-serial-workflow","page":"Parallelizing Calculations","title":"Review of the serial workflow","text":"","category":"section"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"The serial approach to calculating a structure factor, covered in FeI₂ at Finite Temperature, involves thermalizing a spin System and then calling add_sample!. add_sample! uses the state of the System as an initial condition for the calculation of a dynamical trajectory. The correlations of the trajectory are calculated and accumulated into a running average of the 𝒮(𝐪ω). This sequence is repeated to generate additional samples.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"To illustrate, we'll set up a a simple model: a spin-1 antiferromagnet on an FCC crystal. ","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"using Sunny, GLMakie\n\nfunction make_system(cryst; J, dims, seed=nothing)\n sys = System(cryst, dims, [SpinInfo(1, S=1, g=2)], :dipole; seed)\n set_exchange!(sys, J, Bond(1,1,[1,0,0]))\n return sys\nend\n\nJ = 1.0 # meV \ncryst = Sunny.fcc_primitive_crystal()\nsys = make_system(cryst; J, dims=(10,10,2))","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"To thermalize and generate equilibrium samples, we'll need a Langevin integrator. ","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"kT = 0.5 # meV\nΔt = 0.05/J\nintegrator = Langevin(Δt; kT, λ=0.1)","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Finally, call dynamical_correlations to configure a SampledCorrelations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"sc = dynamical_correlations(sys; Δt=0.1, nω=100, ωmax=10.0)","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"The serial calculation can now be performed as follows:","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"nsamples = 10\n\n# Thermalize the system\nfor _ in 1:5000 # Sufficient number of steps to thermalize\n step!(sys, integrator)\nend\n\nfor _ in 1:nsamples\n # Generate new sample by running Langevin dynamics\n for _ in 1:1000 # Sufficient number of steps to decorrelate\n step!(sys, integrator)\n end\n # Add the sample to the correlation data\n add_sample!(sc, sys)\nend","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"This will take a second or two on a modern workstation, resulting in a single SampledCorrelations that contains 10 samples.","category":"page"},{"location":"parallelism.html#Multithreading-approach","page":"Parallelizing Calculations","title":"Multithreading approach","text":"","category":"section"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"To use threads in Julia, you must launch your Julia environment appropriately.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"From the command line, launch Julia with julia --threads=N, where N is the number of threads. If you don't know how many threads you'd like, you can let Julia decide with --threads=auto.\nJupyter notebook users will need to to set up a multithreaded Julia kernel and restart into this kernel. The kernel can be created inside Julia with the following:","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":" using IJulia\n IJulia.installkernel(\"Julia Multithreaded\",\n env=Dict(\"JULIA_NUM_THREADS\" => \"auto\"))","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"VSCode users should open their settings and search for Julia: Additional Args. There will be link called Edit in settings.json. Click on this, add \"--threads=auto\" to the list julia.additionalArgs, and start a new REPL.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Before going further, make sure that Threads.nthreads() returns a number greater than 1.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"We will use multithreading in a very simple way, essentially employing a distributed memory approach to avoid conflicts around memory access. First preallocate a number of systems and correlations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"npar = Threads.nthreads()\nsystems = [make_system(cryst; J, dims=(10,10,2), seed=i) for i in 1:npar]\nscs = [dynamical_correlations(sys; Δt=0.1, nω=100, ωmax=10.0) for _ in 1:npar]","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"warning: Dealing with memory constraints\nIf you have many threads available and are working with a large system, you may not have enough memory to store all these systems and correlations. In that case, simply reduce npar to a small enough value that you can make the necessary allocations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"When the Threads.@threads macro is applied before a for loop, the iterations of the loop will execute in parallel using the available threads. We will put the entire thermalization and sampling process inside the loop, with each thread acting on a unique System and SampledCorrelations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Threads.@threads for id in 1:npar\n integrator = Langevin(Δt; kT, λ=0.1)\n for _ in 1:5000\n step!(systems[id], integrator)\n end\n for _ in 1:nsamples\n for _ in 1:1000\n step!(systems[id], integrator)\n end\n add_sample!(scs[id], systems[id])\n end\nend","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"You may find this takes a little bit longer than the serial example, but, at the end of it, you will have generated npar correlation objects, each with 10 samples. We can merge these into a summary SampledCorrelations with 10*npar samples with merge_correlations:","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"sc = merge_correlations(scs)","category":"page"},{"location":"parallelism.html#Using-Distributed","page":"Parallelizing Calculations","title":"Using Distributed","text":"","category":"section"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Julia also provides a distributed memory approach to parallelism through the standard library package Distributed. This works by launching independent Julia environments on different \"processes.\" An advantage of this approach is that it scales naturally to clusters since the processes are easily distributed across many different compute nodes. A disadvantage, especially when working on a single computer, is the increased memory overhead associated with launching all these environments.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"We begin by importing the package,","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"using Distributed","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"and launching some new processes. It is often sensible to create as many processes as there are cores.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"ncores = length(Sys.cpu_info())\naddprocs(ncores)","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"You can think of each process as a separate computer running a fresh Julia environment, so we'll need to import Sunny and define our function in each of these environments. This is easily achieved with the @everywhere macro.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"@everywhere using Sunny\n\n@everywhere function make_system(cryst; J, dims, seed=nothing)\n sys = System(cryst, dims, [SpinInfo(1, S=1, g=2)], :dipole; seed)\n set_exchange!(sys, J, Bond(1,1,[1,0,0]))\n return sys\nend","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"A simple way to perform work on these processes is to use the parallel map function, pmap. This will apply a function to each element of some iterable, such as a list of numbers, and return a list of the results. It is a parallel map because these function calls may occur at the same time on different Julia processes. The pmap function takes care of distributing the work among the different processes and retrieving the results.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"In the example below, we give pmap a list of RNG seeds to iterate over, and we define the function that will be applied to each of these seeds in a do block. The contents of this block are essentially the same as what we put inside our parallel for loop in the multithreading example. The main difference is that the Systems and SampledCorrelations are not created in advance of the parallelization but are instead created inside each Julia process. The do block returns a SampledCorrelations, and the output of all the parallel computations are collected into list of SampledCorrelations called scs.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"scs = pmap(1:ncores) do seed\n sys = make_system(Sunny.fcc_primitive_crystal(); J=1.0, dims=(10,10,2), seed)\n sc = dynamical_correlations(sys; Δt=0.1, nω=100, ωmax=10.0)\n integrator = Langevin(Δt; kT, λ=0.1)\n\n for _ in 1:5000 # Thermalize\n step!(sys, integrator)\n end\n for _ in 1:nsamples \n for _ in 1:1000 \n step!(sys, integrator)\n end\n add_sample!(sc, sys)\n end\n\n return sc\nend","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Finally, merge the results into a summary SampledCorrelations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"sc = merge_correlations(scs)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"EditURL = \"../../../examples/fei2_tutorial.jl\"","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/fei2_tutorial.html#Case-Study:-FeI","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"FeI₂ is an effective spin-1 material with strong single-ion anisotropy. Quadrupolar fluctuations give rise to a single-ion bound state that cannot be described by a dipole-only model. This tutorial illustrates how to use the linear spin wave theory of SU(3) coherent states (i.e. 2-flavor bosons) to model the magnetic behavior in FeI₂. The original study was performed in Bai et al., Nature Physics 17, 467–472 (2021).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The Fe atoms are arranged in stacked triangular layers. The effective spin Hamiltonian takes the form,","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"mathcalH=sum_(ij) 𝐒_i J_ij 𝐒_j - Dsum_i left(S_i^zright)^2","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"where the set of exchange matrices J_ij between bonded sites (ij) includes competing ferromagnetic and antiferromagnetic interactions. This model also includes a strong easy axis anisotropy, D 0.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"We will formulate this Hamiltonian in Sunny and then calculate its dynamic structure factor.","category":"page"},{"location":"examples/fei2_tutorial.html#Get-Julia-and-Sunny","page":"Case Study: FeI₂","title":"Get Julia and Sunny","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Sunny is implemented in Julia. This is a relatively new programming language that allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users may wish to take a look at our Getting Started with Julia guide. Sunny requires Julia 1.9 or later.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"From the Julia prompt, load Sunny. For plotting, one can choose either GLMakie (a pop-up window) or WGLMakie (inline plots for a Jupyter notebook or VSCode).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"If these packages are not yet installed, Julia should offer to install them using its built-in package management system. If old versions are installed, you may need to update them to run this tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html#Crystals","page":"Case Study: FeI₂","title":"Crystals","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A Crystal describes the crystallographic unit cell and will usually be loaded from a .cif file. Here, we instead build a crystal by listing all atoms and their types.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"a = b = 4.05012 # Lattice constants for triangular lattice\nc = 6.75214 # Spacing in the z-direction\n\nlatvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that\n # define the conventional unit cell\npositions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions\n # of lattice vectors\ntypes = [\"Fe\", \"I\", \"I\"]\nFeI2 = Crystal(latvecs, positions; types)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Observe that Sunny inferred the space group, 'P -3 m 1' (164) and labeled the atoms according to their point group symmetries.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Only the Fe atoms are magnetic, so we discard the I ions using subcrystal.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"cryst = subcrystal(FeI2, \"Fe\")","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Observe that cryst retains the spacegroup symmetry of the full FeI₂ crystal. This information will be used, for example, to propagate exchange interactions between symmetry-equivalent bonds.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In a running Julia environment, the crystal can be viewed interactively using view_crystal.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"view_crystal(cryst, 8.0)","category":"page"},{"location":"examples/fei2_tutorial.html#Symmetry-analysis","page":"Case Study: FeI₂","title":"Symmetry analysis","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The command print_symmetry_table provides a list of all the symmetry-allowed interactions up to a cutoff distance.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"print_symmetry_table(cryst, 8.0)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The allowed g-tensor is expressed as a 3×3 matrix in the free coefficients A, B, ... The allowed single-ion anisotropy is expressed as a linear combination of Stevens operators. The latter correspond to polynomials of the spin operators, as we will describe below.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The allowed exchange interactions are given as a 3×3 matrix for representative bonds. The notation Bond(i, j, n) indicates a bond between atom indices i and j, with cell offset n. In the general case, it will be necessary to associate atom indices with their positions in the unit cell; these can be viewed with display(cryst). Note that the order of the pair (i j) is significant if the exchange tensor contains antisymmetric Dzyaloshinskii–Moriya (DM) interactions.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In the case of FeI₂, Bond(1, 1, [1,0,0]) is one of the 6 nearest-neighbor Fe-Fe bonds on a triangular lattice layer, and Bond(1, 1, [0,0,1]) is an Fe-Fe bond between layers.","category":"page"},{"location":"examples/fei2_tutorial.html#Building-a-spin-System","page":"Case Study: FeI₂","title":"Building a spin System","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In constructing a spin System, we must provide several additional details about the spins.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"sys = System(cryst, (4, 4, 4), [SpinInfo(1, S=1, g=2)], :SUN, seed=2)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"This system includes 444 unit cells, i.e. 64 Fe atoms, each with spin S=1 and a g-factor of 2. Quantum mechanically, spin S=1 involves a superposition of 2S+1=3 distinct angular momentum states. In :SUN mode, this superposition will be modeled explicitly using the formalism of SU(3) coherent states, which captures both dipolar and quadrupolar fluctuations. For the more traditional dipole dynamics, use :dipole mode instead.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Next we will use set_exchange! to assign interaction to bonds. Sunny will automatically propagate each interaction to all symmetry-equivalent bonds in the unit cell. The FeI₂ interactions below follow Bai et al.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"J1pm = -0.236\nJ1pmpm = -0.161\nJ1zpm = -0.261\nJ2pm = 0.026\nJ3pm = 0.166\nJ′0pm = 0.037\nJ′1pm = 0.013\nJ′2apm = 0.068\n\nJ1zz = -0.236\nJ2zz = 0.113\nJ3zz = 0.211\nJ′0zz = -0.036\nJ′1zz = 0.051\nJ′2azz = 0.073\n\nJ1xx = J1pm + J1pmpm\nJ1yy = J1pm - J1pmpm\nJ1yz = J1zpm\n\nset_exchange!(sys, [J1xx 0.0 0.0;\n 0.0 J1yy J1yz;\n 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))\nset_exchange!(sys, [J2pm 0.0 0.0;\n 0.0 J2pm 0.0;\n 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))\nset_exchange!(sys, [J3pm 0.0 0.0;\n 0.0 J3pm 0.0;\n 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))\nset_exchange!(sys, [J′0pm 0.0 0.0;\n 0.0 J′0pm 0.0;\n 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))\nset_exchange!(sys, [J′1pm 0.0 0.0;\n 0.0 J′1pm 0.0;\n 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))\nset_exchange!(sys, [J′2apm 0.0 0.0;\n 0.0 J′2apm 0.0;\n 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function set_onsite_coupling! assigns a single-ion anisotropy. The argument can be constructed using spin_matrices or stevens_matrices. Here we use Julia's anonymous function syntax to assign an easy-axis anisotropy along the direction hatz.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"D = 2.165\nset_onsite_coupling!(sys, S -> -D*S[3]^2, 1)","category":"page"},{"location":"examples/fei2_tutorial.html#Calculating-structure-factor-intensities","page":"Case Study: FeI₂","title":"Calculating structure factor intensities","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In the remainder of this tutorial, we will examine Sunny's tools for calculating the dynamical structure factor using a multi-boson generalization of linear spin wave theory (LSWT). This theory describes non-interacting quasi-particle excitations that hybridize dipolar and quadrupolar modes.","category":"page"},{"location":"examples/fei2_tutorial.html#Finding-the-ground-state","page":"Case Study: FeI₂","title":"Finding the ground state","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Begin with a random configuration and use minimize_energy! to find a configuration of the SU(3) coherent states (i.e. spin dipoles and quadrupoles) that locally minimizes energy.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"randomize_spins!(sys)\nminimize_energy!(sys)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A positive number above indicates that the procedure has converged to a local energy minimum. The configuration, however, may still have defects. This can be checked by visualizing the spins, colored according to their z-components.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"plot_spins(sys; color=[s[3] for s in sys.dipoles])","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A different understanding of the magnetic ordering can be obtained by moving to Fourier space. The 'instant' structure factor 𝒮(𝐪) is an experimental observable. To investigate 𝒮(𝐪) as true 3D data, Sunny provides instant_correlations and related functions. Here, however, we will use print_wrapped_intensities, which gives average intensities for the individual Bravais sublattices (in effect, all wavevectors are wrapped to the first Brillouin zone).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"print_wrapped_intensities(sys)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The result will likely be approximately consistent with the known zero-field energy-minimizing magnetic structure of FeI₂, which is single-Q (two-up, two-down antiferromagnetic order). Mathematically, spontaneous symmetry breaking should select one of Q = 0 -14 14, 14 0 14, or -141414, associated with the three-fold rotational symmetry of the crystal spacegroup. In nature, however, one will frequently encounter competing \"domains\" associated with the three possible orientations of the ground state.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"If the desired ground state is already known, as with FeI₂, it could be entered by hand using set_dipole!. Alternatively, in the case of FeI₂, we could repeatedly employ the above randomization and minimization procedure until a defect-free configuration is found. Some systems will have more complicated ground states, which can be much more challenging to find. For this, Sunny provides experimental support for powerful simulated annealing via parallel tempering, but that is outside the scope of this tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Here, let's break the three-fold symmetry of FeI₂ by hand. Given one or more desired Q modes, Sunny can suggest a magnetic supercell with appropriate periodicity. Let's arbitrarily select one of the three possible ordering wavevectors, Q = 0 -14 14. Sunny suggests a corresponding magnetic supercell in units of the crystal lattice vectors.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"suggest_magnetic_supercell([[0, -1/4, 1/4]])","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The system returned by reshape_supercell is smaller, and is sheared relative to the original system. This makes it much easier to find the global energy minimum.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"sys_min = reshape_supercell(sys, [1 0 0; 0 2 1; 0 -2 1])\nrandomize_spins!(sys_min)\nminimize_energy!(sys_min);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Plot the system again, now including \"ghost\" spins out to 12Å","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"plot_spins(sys_min; color=[s[3] for s in sys_min.dipoles], ghost_radius=12)","category":"page"},{"location":"examples/fei2_tutorial.html#Linear-spin-wave-theory","page":"Case Study: FeI₂","title":"Linear spin wave theory","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Now that we have found the ground state for a magnetic supercell, we can immediately proceed to perform zero-temperature calculations using linear spin wave theory. We begin by instantiating a SpinWaveTheory type using the supercell.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"swt = SpinWaveTheory(sys_min)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Select a sequence of wavevectors that will define a piecewise linear interpolation in reciprocal lattice units (RLU).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"q_points = [[0,0,0], [1,0,0], [0,1,0], [1/2,0,0], [0,1,0], [0,0,0]];\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function reciprocal_space_path will linearly sample a path between the provided q-points with a given density. The xticks return value provides labels for use in plotting.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"density = 50\npath, xticks = reciprocal_space_path(cryst, q_points, density);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The dispersion function defines the quasiparticle excitation energies ω_i(𝐪) for each point 𝐪 along the reciprocal space path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp = dispersion(swt, path);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In addition to the band energies ω_i(𝐪), Sunny can calculate the inelastic neutron scattering intensity I_i(𝐪) for each band i according to an intensity_formula. We choose to apply a polarization correction (1 - 𝐪𝐪) by setting the mode argument to :perp. Selecting delta_function_kernel specifies that we want the energy and intensity of each band individually.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function intensities_bands uses linear spin wave theory to calculate both the dispersion and intensity data for the provided path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp, intensity = intensities_bands(swt, path, formula);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"These can be plotted in GLMakie.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"fig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nylims!(ax, 0.0, 7.5)\nxlims!(ax, 1, size(disp, 1))\ncolorrange = extrema(intensity)\nfor i in axes(disp, 2)\n lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange)\nend\nfig","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"To make comparisons with inelastic neutron scattering (INS) data, it is helpful to employ an empirical broadening kernel, e.g., a lorentzian.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"γ = 0.15 # width in meV\nbroadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The intensities_broadened function requires an energy range in addition to the 𝐪-space path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"energies = collect(0:0.01:10) # 0 < ω < 10 (meV).\nis1 = intensities_broadened(swt, path, energies, broadened_formula);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A real FeI₂ sample will exhibit competing magnetic domains associated with spontaneous symmetry breaking of the 6-fold rotational symmetry of the triangular lattice. Note that the wavevectors 𝐪 and -𝐪 are equivalent in the structure factor, which leaves three distinct domain orientations, which are related by 120° rotations about the z-axis. Rather than rotating the spin configuration directly, on can rotate the 𝐪-space path. Below, we use rotation_in_rlu to average the intensities over all three possible orientations.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"R = rotation_in_rlu(cryst, [0, 0, 1], 2π/3)\nis2 = intensities_broadened(swt, [R*q for q in path], energies, broadened_formula)\nis3 = intensities_broadened(swt, [R*R*q for q in path], energies, broadened_formula)\nis_averaged = (is1 + is2 + is3) / 3\n\nfig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nheatmap!(ax, 1:size(is_averaged, 1), energies, is_averaged)\nfig","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"This result can be directly compared to experimental neutron scattering data from Bai et al.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"(The publication figure accidentally used a non-standard coordinate system to label the wave vectors.)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"To get this agreement, the use of SU(3) coherent states is essential. In other words, we needed a theory of multi-flavored bosons. The lower band has large quadrupolar character, and arises from the strong easy-axis anisotropy of FeI₂. By setting mode = :SUN, the calculation captures this coupled dipole-quadrupole dynamics.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"An interesting exercise is to repeat the same study, but using mode = :dipole instead of :SUN. That alternative choice would constrain the coherent state dynamics to the space of dipoles only.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The full dynamical spin structure factor (DSSF) can be retrieved as a 33 matrix with the dssf function, for a given path of 𝐪-vectors.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp, is = dssf(swt, path);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The first output disp is identical to that obtained from dispersion. The second output is contains a list of 33 matrix of intensities. For example, is[q,n][2,3] yields the (yz) component of the structure factor intensity for nth mode at the qth wavevector in the path.","category":"page"},{"location":"examples/fei2_tutorial.html#What's-next?","page":"Case Study: FeI₂","title":"What's next?","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The multi-boson linear spin wave theory, applied above, can be understood as the quantization of a certain generalization of the Landau-Lifshitz spin dynamics. Rather than dipoles, this dynamics takes places on the space of SU(N) coherent states.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The full SU(N) coherent state dynamics, with appropriate quantum correction factors, can be useful to model finite temperature scattering data. In particular, it captures certain anharmonic effects due to thermal fluctuations. This is the subject of our FeI₂ at Finite Temperature tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The classical dynamics is also a good starting point to study non-equilibrium phenomena. Empirical noise and damping terms can be used to model coupling to a thermal bath. This yields a Langevin dynamics of SU(N) coherent states. Our CP² Skyrmion Quench tutorial shows how this dynamics gives rise to the formation of novel topological defects in a temperature quench.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Relative to LSWT calculations, it can take much more time to estimate mathcalS(𝐪ω) intensities using classical dynamics simulation. See the SunnyTutorials notebooks for examples of \"production-scale\" simulations.","category":"page"},{"location":"structure-factor.html#Structure-Factor-Calculations","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"","category":"section"},{"location":"structure-factor.html#Overview","page":"Structure Factor Calculations","title":"Overview","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The dynamical structure factor is of fundamental importance for characterizing a magnetic system, and facilitates quantitative comparison between theory and experimental scattering data.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Consider, for example, a two-point dynamical spin correlation function, s^α(𝐱+Δ𝐱 t+Δt) s^β(𝐱 t). Here s^α(𝐱 t) represents the time dynamics of a spin dipole component α at position 𝐱, and brackets represent an average over equilibrium initial conditions and over (𝐱 t). The dynamical structure factor is defined as the Fourier transform of this two-point correlation in both space and time, up to an overall scaling factor. Using the convolution theorem, the result is,","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ(𝐪 ω) = frac1V s^α(𝐪 ω)^ast s^β(𝐪 ω) ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"with V the system volume. We will restrict attention to lattice systems with periodic boundaries.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Consider a crystal unit cell defined by three lattice vectors 𝐚_1 𝐚_2 𝐚_3, and linear system sizes L_1 L_2 L_3 measured in unit cells. The allowed momentum vectors take on discrete values 𝐪 = sum_α=1^3 m_α 𝐛_α L_α, where m_α are an integers and the reciprocal lattice vectors 𝐛_α are defined to satisfy 𝐚_α 𝐛_β = 2π δ_αβ. For a Bravais lattice, 𝐪 will be periodic in the first Brillouin zone, i.e., under any shift 𝐪 𝐪 𝐛_α. More generally, consider a non-Bravais lattice such that each unit cell may contain multiple spins. By partitioning spins s_j(𝐱t) according to their sublattice index j, the relevant momenta 𝐪 remain discretized as above, but now periodicity in the first Brillouin zone is lost. The structure factor may be written as a phase-average over the displacements between sublattices 𝐫_jk,","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ(𝐪 ω) = _jk e^i 𝐫_jk 𝐪 𝒮^αβ_jk(𝐪 ω) ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"From a theoretical perspective, the quantity","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ_jk(𝐪 ω) = frac1V s_j^α(𝐪 ω)^ast s_k^β(𝐪 ω)","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"is fundamental. For each sublattice j, the data s_j^α(𝐪 ω) can be efficiently obtained by fast Fourier tranformation of a real space configuration s_j^α(𝐱 t). Internally, Sunny will calculate and store the discrete 𝒮^αβ_jk(𝐪 ω) correlation data, and use this to construct 𝒮^αβ(𝐪ω) intensities that can be compared with experiment.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Calculating this structure factor involves several steps, with various possible settings. Sunny provides a number of tools to facilitate this calculation and to extract information from the results. These tools are briefly outlined below. Please see the Examples for a \"real life\" use case. Detailed function information is available in the Library API.","category":"page"},{"location":"structure-factor.html#Estimating-stucture-factors-with-classical-dynamics","page":"Structure Factor Calculations","title":"Estimating stucture factors with classical dynamics","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Classical dynamics may be used to estimate structure factor data by analyzing the spin-spin correlations of dynamical trajectories. This is fundamentally a Monte Carlo approach, as the trajectories must be started from an initial spin configuration that is sampled at thermal equilibrium. (Note that it is not possible to estimate a true T=0 dynamical structure factor using this method, but the temperature may be very low.) Samples are accumulated into a SampledCorrelations, from which intensity information may be extracted. The user does not typically build their own SampledCorrelations but instead initializes one by calling either dynamical_correlations or instant_correlations, as described below.","category":"page"},{"location":"structure-factor.html#Estimating-a-dynamical-structure-factor:-𝒮(𝐪,ω)","page":"Structure Factor Calculations","title":"Estimating a dynamical structure factor: 𝒮(𝐪ω)","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A SampledCorrelations for estimating the dynamical structure factor, 𝒮^αβ(𝐪ω), may be created by calling dynamical_correlations. This requires three keyword arguments. These will determine the dynamics used to calculate samples and, consequently, the ω information that will be available. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Δt: Determines the step size used for simulating the dynamics. A smaller number will require proportionally more calculation time. While a smaller Δt will enable the resolution of higher energies, Δt is typically selected to ensure numerical stability rather than to maximize the largest ω value. A safe choice is to use the smaller value of Δt = 0.1/(J* S^2) or Δt = 0.1/(D * S), where S is magnetic moment of the largest local spin (as specified in SpinInfo), J is the parameter governing the largest bilinear interaction (e.g. exchange), and D is the parameter governing the largest single-site term of the Hamiltonian (e.g., anisotropy or Zeeman term).\nωmax: Sets the maximum resolved energy. Note that this is not independent of Δt. If ωmax too large, Sunny will throw an error and ask you to choose a smaller Δt. \nnω: Determines the number of energy bins to resolve. A larger number will require more calculation time.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A sample may be added by calling add_sample!(sc, sys). The input sys must be a spin configuration in good thermal equilibrium, e.g., using the continuous Langevin dynamics or using single spin flip trials with LocalSampler. The statistical quality of the 𝒮^αβ(𝐪ω) can be improved by repeatedly generating decorrelated spin configurations in sys and calling add_sample! on each configuration.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The outline of typical use case might look like this:","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"# Make a `SampledCorrelations`\nsc = dynamical_correlations(sys; Δt=0.05, ωmax=10.0, nω=100) \n\n# Add samples\nfor _ in 1:nsamples\n decorrelate_system(sys) # Perform some type of Monte Carlo simulation\n add_sample!(sc, sys) # Use spins to calculate trajectory and accumulate new sample of 𝒮(𝐪,ω)\nend","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The calculation may be configured in a number of ways; see the dynamical_correlations documentation for a list of all keywords.","category":"page"},{"location":"structure-factor.html#Estimating-an-instantaneous-(\"static\")-structure-factor:-𝒮(𝐪)","page":"Structure Factor Calculations","title":"Estimating an instantaneous (\"static\") structure factor: 𝒮(𝐪)","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Sunny provides two methods for calculating instantaneous, or static, structure factors: 𝒮^αβ(𝐪). The first involves calculating spatial spin-spin correlations at single time slices. The second involves calculating a dynamic structure factor first and integrating out the ω information. The advantage of the latter approach is that it enables application of an ω-dependent classical-to-quantum rescaling of structure factor intensities, a method that should be preferred whenever comparing results to experimental data or spin wave calculations. A disadvantage of this approach is that it is computationally more expensive. There are also many cases when it is not straightforward to calculate a meaningful dynamics, as when working with Ising spins. In this section we will discuss how to calculate instantaneous structure factors from static spin configurations. Information about calculating instantaneous data from a dynamical correlations can be found in the following section.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The basic usage for the instantaneous case is very similar to the dynamic case, except one calls instant_correlations instead of dynamical_correlations to configure a SampledCorrelations. Note that there are no required keywords as there is no need to specify any dynamics. instant_correlations will return a SampledCorrelations containing no data. Samples may be added by calling add_sample!(sc, sys), where sc is the SampledCorrelations. When performing a finite-temperature calculation, it is important to ensure that the spin configuration in the sys represents a good equilibrium sample, as in the dynamical case. Note, however, that we recommend calculating instantaneous correlations at finite temperature calculations by using full dynamics (i.e., using dynamical_correlations) and then integrating out the energy axis. An approach to doing this is described in the next section.","category":"page"},{"location":"structure-factor.html#Extracting-information-from-sampled-correlation-data","page":"Structure Factor Calculations","title":"Extracting information from sampled correlation data","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The basic function for extracting information from a SampledCorrelations at a particular wave vector, 𝐪, is intensities_interpolated. It takes a SampledCorrelations, a list of wave vectors, and an intensity_formula. The intensity_formula specifies how to contract and correct correlation data to arrive at a physical intensity. A simple example is formula = intensity_formula(sc, :perp), which will instruct Sunny apply polarization corrections: sum_αβ(I-q_α q_β) 𝒮^αβ(𝐪ω). An intensity at the wave vector 𝐪 = (𝐛_2 + 𝐛_3)2 may then be retrieved with intensities_interpolated(sf, [[0.0, 0.5, 0.5]], formula) . intensities_interpolated returns a list of nω elements at each wavevector. The corresponding ω values can be retrieved by calling available_energies on sf.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Since Sunny only calculates the structure factor on a finite lattice when performing classical simulations, it is important to realize that exact information is only available at a discrete set of wave vectors. Specifically, for each axis index i, we will get information at q_i = fracnL_i, where n runs from (frac-L_i2+1) to fracL_i2 and L_i is the linear dimension of the lattice used for the calculation. If you request a wave vector that does not fall into this set, Sunny will automatically round to the nearest 𝐪 that is available. If intensities_interpolated is given the keyword argument interpolation=:linear, Sunny will use trilinear interpolation to determine a result at the requested wave vector. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"To retrieve the intensities at all wave vectors for which there is exact data, first call the function available_wave_vectors to generate a list of qs. This takes an optional keyword argument bzsize, which must be given a tuple of three integers specifying the number of Brillouin zones to calculate, e.g., bzsize=(2,2,2). The resulting list of wave vectors may then be passed to intensities_interpolated.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Alternatively, intensities_binned can be used to place the exact data into histogram bins for comparison with experiment.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The convenience function reciprocal_space_path returns a list of wavevectors sampled along a path that connects specified 𝐪 points. This list can be used as an input to intensities. Another convenience method, reciprocal_space_shell will generate points on a sphere of a given radius. This is useful for powder averaging. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A number of arguments for intensity_formula are available which modify the calculation of structure factor intensity. It is generally recommended to provide a value of kT corresponding to the temperature of sampled configurations. Given kT, Sunny will include an energy- and temperature-dependent classical-to-quantum rescaling of intensities in the formula.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"To retrieve intensity data from a instantaneous structure factor, use instant_intensities_interpolated, which accepts similar arguments to intensities_interpolated. This function may also be used to calculate instantaneous information from a dynamical correlation data, i.e. from a SampledCorrelations created with dynamical_correlations. Note that it is important to supply a value to kT to reap the benefits of this approach over simply calculating a static structure factor at the outset. ","category":"page"},{"location":"writevtk.html#ParaView-Rendering","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The 4D correlation data produced by Sunny is too high-dimensional to visualize directly. This page describes how to export 3D slices of correlation data from Sunny to the Visual ToolKit (VTK) format, which is compatible with the ParaView visualization software. ParaView supports volumetric rendering:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html#Simulation-data","page":"ParaView Rendering","title":"Simulation data","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"First, generate some correlation data in Sunny. We will use a 2D lattice, since the correlation data S(Q_xQ_yomega) is 3D and can be exported in its entirety. The following code sets up the system, thermalizes it, and records the correlation data in a SampledCorrelations called dsf.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"using Sunny\n\n# Single layer 12x12 periodic square lattice\nlatsize = (12,12,1)\n\nlatvecs = lattice_vectors(8.,8.,12.,90,100,90)\npositions = [[0,0,0]]\ntypes = [\"Cu\"]\nformfactors = [FormFactor(\"Cu2\")]\nxtal = Crystal(latvecs, positions; types)\n\nsys = System(xtal, latsize, [SpinInfo(1, S=1/2, g=2)], :SUN; seed=1)\n\nJ = 10.\nset_exchange!(sys, J, Bond(1,1,[1,0,0]))\nset_exchange!(sys, J, Bond(1,1,[0,1,0]))\n\nΔt = 0.01\nkT = 0.5\nlangevin = Langevin(Δt; λ=0.5, kT)\nrandomize_spins!(sys)\nfor i in 1:10_000 # Long enough to reach equilibrium\n step!(sys, langevin)\nend \n\nωmax=10.\n\ndsf = dynamical_correlations(sys\n ;Δt=Δt\n ,nω=48\n ,ωmax=ωmax\n ,process_trajectory=:symmetrize)\n\nnsamples = 10\nfor _ in 1:nsamples\n for _ in 1:1000 \n step!(sys, langevin)\n end\n add_sample!(dsf, sys)\nend","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The default histogram BinningParameters are already integrated over the z direction because the system is 2D:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"unit_resolution_binning_parameters(dsf)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 12 bins from -0.042 to +0.958 along [+1.27 dx] (Δ = 0.065)\n⊡ 12 bins from -0.042 to +0.958 along [+1.27 dy] (Δ = 0.065)\n∫ Integrated from +0.000 to +0.000 along [-0.33 dx +1.88 dz] (Δ = 0.524)\n⊡ 48 bins from -0.107 to +10.134 along [+1.00 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The histogram is very oblong; it's approximately 1x1x10. To make it a nicer shape, we will rescale the energy axis to be be fractions of ωmax:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"params = unit_resolution_binning_parameters(dsf)\nscale_factor = ωmax\nparams.binend[4] /= scale_factor\nparams.binstart[4] /= scale_factor\nparams.binwidth[4] /= scale_factor\nparams.covectors[4,:] ./= scale_factor","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Doing this changes the last axis of the histogram to fit in [0,1]:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 49 bins from -0.011 to +1.013 along [+0.10 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that our histogram is a cube, we compute the intensity in the histogram bins using the usual intensities_binned:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"formula = intensity_formula(dsf,:trace)\nsignal, counts = intensities_binned(dsf, params; formula)\nintensity = signal ./ counts","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that we have our intensity data and the binning parameters, we can export to VTK format using export_vtk and move to ParaView for the visualization.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"# Importing WriteVTK enables Sunny's export-to-VTK functions\nimport WriteVTK\n\n# [1,2,4] specifies that the (x,y,z) axes in ParaView are (Qx,Qy,ω)\nexport_vtk(\"square_lattice\", params, intensity; dims_kept = [1,2,4])\n# Writes a file square_lattice.vti in the current directory","category":"page"},{"location":"writevtk.html#Loading-in-ParaView","page":"ParaView Rendering","title":"Loading in ParaView","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In ParaView, use File > Open to open square_lattice.vti. This will add the file to the Pipeline Browser with a closed eye icon, indicating that the data is ready to be loaded.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In the Properties panel, both bin_centers and data will be selected for import by default. Uncheck bin_centers because we don't need that information for the visualization. Click the green Apply button to load the data.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"By default, only the outline of the data is shown in the 3D viewport. Since we adjusted the energy axis, the outline is a 1x1x1 cube. Optionally enable the axes grid under \"View\", and customize using the adjacent edit button.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"To enable the volumetric render:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Select \"Volume\" from the \"Representation\" drop-down menu under \"Display\".\nThe \"Coloring\" drop-down should automatically select data because it's the only data loaded.\nOpen the Color Map Editor to adjust the opacity of the fog, which may be too faint to see by default.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Depending on your computer and your dataset size, the volumetric rendering may be slow, but our dataset is relatively small, so the render should be fast.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"If nothing shows up at first, don't despair. Often, there are Bragg-like peaks in the correlation data which outshine everything else. To see this, enable Display Data Histogram in the Color Map Editor panel. To zoom in on the lower-intensity data, click and drag the right side handle of the opacity transfer function box to the middle a few times.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"After suitable color mapping, the dispersion curve should become visible:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html#Experiment-data","page":"ParaView Rendering","title":"Experiment data","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Note that since only the data and binning parameters are required for exporting to VTK, experiment data can be exported in the same way. For example, to visualize S(Q_xQ_yQ_z), do this:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"# Load 4D histogram data from Mantid\nparams, signal = load_nxs(\"experiment_data.nxs\")\n\n# Integrate out the energy axis so we are 3D\nintegrate_axes!(params; axes = 4)\nsignal = sum(signal; dims = 4)\n\n# Export to ParaView\nexport_vtk(\"experiment_data_as_vtk\", params, signal)","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"EditURL = \"../../../examples/ising2d.jl\"","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/ising2d.html#Classical-Ising-model","page":"Classical Ising model","title":"Classical Ising model","text":"","category":"section"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"This tutorial illustrates simulation of the classical 2D Ising model.","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"Sunny expects a 3D Crystal unit cell. To model a square lattice, we create an orthogonal unit cell where the z-spacing is distinct from the x and y spacing.","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"a = 1\nlatvecs = lattice_vectors(a,a,10a,90,90,90)\ncrystal = Crystal(latvecs, [[0,0,0]])","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"Create a System of spins with linear size L in the x and y directions, and only one layer in the z direction. The option :dipole means that the system will store Heisenberg spins, as opposed to SU(N) coherent states. Polarize the initial spin configuration using polarize_spins!. Following the Ising convention, we will restrict these spins to the z-axis and give them magnitude S=1.","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"By default, Sunny uses physical units, e.g. magnetic field in tesla. Here we specify an alternative Units system, so that the Zeeman coupling between the spin dipole 𝐬 and an external field 𝐁 has the dimensionless form -𝐁𝐬.","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"L = 128\nsys = System(crystal, (L,L,1), [SpinInfo(1, S=1, g=1)], :dipole, units=Units.theory, seed=0)\npolarize_spins!(sys, (0,0,1))","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"Use set_exchange! to include a ferromagnetic Heisenberg interaction along nearest-neighbor bonds. The Bond below connects two spins displaced by one lattice constant in the x-direction. This interaction will be propagated to all nearest-neighbors bonds in the system, consistent with the symmetries of the square lattice.","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"set_exchange!(sys, -1.0, Bond(1,1,(1,0,0)))","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"If an external field is desired, it can be set using set_external_field!.","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"B = 0\nset_external_field!(sys, (0,0,B))","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"The critical temperature for the Ising model is known analytically.","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"Tc = 2/log(1+√2)","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"Use a LocalSampler to perform nsweeps Monte Carlo sweeps. A sweep consists of, on average, one trial update per spin in the system. Each proposed update is accepted or rejected according to the Metropolis acceptance probability. As its name suggests, the propose_flip function will only propose pure spin flips, 𝐬 rightarrow -𝐬.","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"nsweeps = 4000\nsampler = LocalSampler(kT=Tc, propose=propose_flip)\nfor i in 1:nsweeps\n step!(sys, sampler)\nend","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"Plot the Ising spins by extracting the z-component of the dipoles","category":"page"},{"location":"examples/ising2d.html","page":"Classical Ising model","title":"Classical Ising model","text":"heatmap(reshape([s.z for s in sys.dipoles], (L,L)))","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"EditURL = \"../../../examples/powder_averaging.jl\"","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/powder_averaging.html#Powder-Averaged-CoRhO","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"","category":"section"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"This tutorial illustrates the calculation of the powder-averaged structure factor by performing an orientational average. We consider a simple model of the diamond-cubic crystal CoRh₂O₄, with parameters extracted from Ge et al., Phys. Rev. B 96, 064413.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"Construct a diamond Crystal in the conventional (non-primitive) cubic unit cell. Sunny will populate all eight symmetry-equivalent sites when given the international spacegroup number 227 (\"Fd-3m\") and the appropriate setting. For this spacegroup, there are two conventional translations of the unit cell, and it is necessary to disambiguate through the setting keyword argument. (On your own: what happens if setting is omitted?)","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"a = 8.5031 # (Å)\nlatvecs = lattice_vectors(a, a, a, 90, 90, 90)\ncryst = Crystal(latvecs, [[0,0,0]], 227, setting=\"1\")","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"In a running Julia environment, the crystal can be viewed interactively using view_crystal.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"view_crystal(cryst, 8.0)","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"Construct a System with an antiferromagnetic nearest neighbor interaction J. Because the diamond crystal is bipartite, the ground state will have unfrustrated Néel order. Selecting latsize=(1,1,1) is sufficient because the ground state is periodic over each cubic unit cell. By passing an explicit seed, the system's random number generator will give repeatable results.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"latsize = (2, 2, 2)\nseed = 0\nS = 3/2\nJ = 7.5413*meV_per_K # (~ 0.65 meV)\nsys = System(cryst, latsize, [SpinInfo(1; S, g=2)], :dipole; seed=0)\nset_exchange!(sys, J, Bond(1, 3, [0,0,0]))","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"In the ground state, each spin is exactly anti-aligned with its 4 nearest-neighbors. Because every bond contributes an energy of -JS^2, the energy per site is -2JS^2. In this calculation, a factor of 1/2 avoids double-counting the bonds. Due to lack of frustration, direct energy minimization is successful in finding the ground state.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"randomize_spins!(sys)\nminimize_energy!(sys)\n\n@assert energy_per_site(sys) ≈ -2J*S^2","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"Plotting the spins confirms the expected Néel order. Note that the overall, global rotation of dipoles is arbitrary.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"s0 = sys.dipoles[1,1,1,1]\nplot_spins(sys; color=[s'*s0 for s in sys.dipoles])","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"For numerical efficiency, it will be helpful to work with the smallest possible magnetic supercell. Here, it is the primitive unit cell, which contains just two sites. The variable shape below defines the primitive lattice vectors cryst.prim_latvecs in units of the conventional lattice vectors. This result is used as input to reshape_supercell. The energy per site remains the same, which verifies that the magnetic supercell is valid.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"shape = cryst.latvecs \\ cryst.prim_latvecs\nsys_prim = reshape_supercell(sys, shape)\n@assert energy_per_site(sys_prim) ≈ -2J*S^2","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"Now estimate 𝒮(𝐪ω) with SpinWaveTheory and an intensity_formula. The mode :perp contracts with a dipole factor to return the unpolarized intensity. The formula also employs lorentzian broadening. The isotropic FormFactor for Cobalt(2+) dampens intensities at large 𝐪.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"swt = SpinWaveTheory(sys_prim)\nη = 0.4 # (meV)\nkernel = lorentzian(η)\nformfactors = [FormFactor(\"Co2\")]\nformula = intensity_formula(swt, :perp; kernel, formfactors)","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"For the \"single crystal\" result, we may use reciprocal_space_path to construct a path that connects high-symmetry points in reciprocal space. The intensities_broadened function collects intensities along this path for the given set of energy values.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"qpoints = [[0.0, 0.0, 0.0], [0.5, 0.0, 0.0], [0.5, 0.5, 0.0], [0.0, 0.0, 0.0]]\npath, xticks = reciprocal_space_path(cryst, qpoints, 50)\nenergies = collect(0:0.01:6)\nis = intensities_broadened(swt, path, energies, formula)\n\nfig = Figure()\nax = Axis(fig[1,1]; aspect=1.4, ylabel=\"ω (meV)\", xlabel=\"𝐪 (r.l.u.)\",\n xticks, xticklabelrotation=π/10)\nheatmap!(ax, 1:size(is, 1), energies, is, colormap=:gnuplot2)\nfig","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"A powder measurement effectively involves an average over all possible crystal orientations. We use the function reciprocal_space_shell to sample n wavevectors on a sphere of a given radius (inverse angstroms), and then calculate the spherically-averaged intensity.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"radii = 0.01:0.02:3 # (1/Å)\noutput = zeros(Float64, length(radii), length(energies))\nfor (i, radius) in enumerate(radii)\n n = 300\n qs = reciprocal_space_shell(cryst, radius, n)\n is = intensities_broadened(swt, qs, energies, formula)\n output[i, :] = sum(is, dims=1) / size(is, 1)\nend\n\nfig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Q (Å⁻¹)\", ylabel=\"ω (meV)\")\nheatmap!(ax, radii, energies, output, colormap=:gnuplot2)\nfig","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"This result can be compared to experimental neutron scattering data from Fig. 5 of Ge et al.","category":"page"},{"location":"examples/powder_averaging.html","page":"Powder Averaged CoRh₂O₄","title":"Powder Averaged CoRh₂O₄","text":"","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"EditURL = \"../../../examples/out_of_equilibrium.jl\"","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/out_of_equilibrium.html#CP-Skyrmion-Quench","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"","category":"section"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"This example demonstrates Sunny's ability to simulate the out-of-equilibrium dynamics of generalized spin systems. We will implement the model Hamiltonian of Zhang et al., Nature Communications 14, 3626 (2023), which supports a novel type of topological defect, a CP² skyrmion, that involves both the dipolar and quadrupolar parts of a quantum spin.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Beginning from an initial high-temperature state, a disordered gas of CP² skyrmions can be formed by rapidly quenching to low temperature. To model the coupled dynamics of dipoles and quadrupoles, Sunny uses a recently developed generalization of the Landau-Lifshitz spin dynamics, Dahlbom et al., Phys. Rev. B 106, 235154 (2022).","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The Hamiltonian we will implement,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"mathcalH = sum_langle ij rangle J_ij( hatS_i^x hatS_j^x + hatS_i^y hatS_j^y + DeltahatS_i^z hatS_j^z) - hsum_ihatS_i^z + Dsum_i(hatS_i^z)^2","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"contains competing ferromagnetic nearest-neightbor and antiferromagnetic next-nearest-neighbor exchange terms on a triangular lattice. Both exchanges exhibit anisotropy on the z-term. Additionally, there is an external magnetic field, h, and easy-plane single-ion anisotropy, D 0. We begin by implementing the Crystal.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"lat_vecs = lattice_vectors(1.0, 1.0, 2.0, 90, 90, 120)\nbasis_vecs = [[0,0,0]]\ncryst = Crystal(lat_vecs, basis_vecs)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The crystal is then used to create a spin System. All parameters in this model system are dimensionless, so we select \"theory\" units and set the g-factor to one.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"L = 40\ndims = (L, L, 1)\nsys = System(cryst, dims, [SpinInfo(1, S=1, g=1)], :SUN; seed=101, units=Units.theory)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"We proceed to implement each term of the Hamiltonian, selecting our parameters so that the system occupies a region of the phase diagram that supports skyrmions. The exchange interactions are set as follows.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"J1 = -1 # Nearest-neighbor ferromagnetic\nJ2 = (2.0/(1+√5)) # Tune competing exchange to set skyrmion scale length\nΔ = 2.6 # Exchange anisotropy\n\nex1 = J1 * [1.0 0.0 0.0;\n 0.0 1.0 0.0;\n 0.0 0.0 Δ]\nex2 = J2 * [1.0 0.0 0.0;\n 0.0 1.0 0.0;\n 0.0 0.0 Δ]\nset_exchange!(sys, ex1, Bond(1, 1, [1, 0, 0]))\nset_exchange!(sys, ex2, Bond(1, 1, [1, 2, 0]))","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Next we add the external field,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"h = 15.5\nfield = set_external_field!(sys, [0.0 0.0 h])","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"and finally an easy-plane single-ion anisotropy,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"D = 19.0\nset_onsite_coupling!(sys, S -> D*S[3]^2, 1)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Initialize system to an infinite temperature (fully randomized) initial condition.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"randomize_spins!(sys)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"We are now ready to simulate the quenching process using a generalized Langevin spin dynamics. If we were working with spin dipoles only, then Langevin dynamics would be the usual Landau-Lifshitz spin dynamics, augmented with damping and noise terms. In the present study, we are instead working with quantum spin-1 (an (N=3)-level system that includes both dipoles and quadrupoles). Here, Langevin captures the coupled dipole-quadrupole dynamics using the formalism of SU(N) coherent states.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Selecting kT = 0 in the Langevin dynamics will effective disable the noise term. Then the parameter λ effectively determines the damping time-scale.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Δt = 0.2/D # Integration time step (inverse meV). Typically this will be\n # inversely proportional to the largest energy scale in the\n # system. We can use a fairly large time-step here because\n # accuracy isn't critical.\nkT = 0 # Target equilibrium temperature (meV)\nλ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)\nintegrator = Langevin(Δt; kT, λ)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Finally we run the dynamics. We will record the state of the system at three different times during the quenching process by copying the coherents field of the System.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"τs = [4., 16, 256] # Times to record snapshots\nframes = [] # Empty array to store snapshots\nfor i in eachindex(τs)\n dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate\n numsteps = round(Int, dur/Δt)\n for _ in 1:numsteps # Perform the integration\n step!(sys, integrator)\n end\n push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration\nend","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"To visualize the state of the system contained in each snapshot, we will calculate and plot the skyrmion density on each plaquette of our lattice. The function plot_triangular_plaquettes is not part of the core Sunny package, but rather something you could define yourself. We are using the definition in plotting2d.jl from the Sunny examples/extra directory.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"include(pkgdir(Sunny, \"examples\", \"extra\", \"plotting2d.jl\"))\n\nfunction sun_berry_curvature(z₁, z₂, z₃)\n z₁, z₂, z₃ = normalize.((z₁, z₂, z₃))\n n₁ = z₁ ⋅ z₂\n n₂ = z₂ ⋅ z₃\n n₃ = z₃ ⋅ z₁\n return angle(n₁ * n₂ * n₃)\nend\n\nplot_triangular_plaquettes(sun_berry_curvature, frames; resolution=(1800,600),\n offset_spacing=10, texts = [\"\\tt = \"*string(τ) for τ in τs], text_offset = (0.0, 6.0)\n)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The times are given in hbarJ_1. The white background corresponds to a quantum paramagnetic state, where the local spin exhibits a strong quadrupole moment and little or no dipole moment. Observe that the process has generated a number of well-formed skyrmions of both positive (red) and negative (blue) charge in addition to a number of other metastable spin configurations. A full-sized version of this figure is available in Dahlbom et al..","category":"page"},{"location":"library.html#Library-API","page":"Library API","title":"Library API","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"This page describes the public types and functions exported by Sunny. This documentation can be also be accessed using the Julia help system (enter ? at the Julia command prompt).","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"Sunny.plot_spins\nSunny.view_crystal\nSunny.export_vtk","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"Modules = [Sunny]\nPrivate = false","category":"page"},{"location":"library.html#Sunny.Site","page":"Library API","title":"Sunny.Site","text":"(cell1, cell2, cell3, i) :: Site\n\nFour indices identifying a single site in a System. The first three indices select the lattice cell and the last selects the sublattice (i.e., the atom within the unit cell).\n\nThis object can be used to index dipoles and coherents fields of a System. A Site is also required to specify inhomogeneous interactions via functions such as set_external_field_at! or set_exchange_at!.\n\nNote that the definition of a cell may change when a system is reshaped. In this case, it is convenient to construct the Site using position_to_site, which always takes a position in fractional coordinates of the original lattice vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Units","page":"Library API","title":"Sunny.Units","text":"Units.meV\nUnits.theory\n\nThe unit system is implicitly determined by the definition of two physical constants: the vacuum permeability μ₀ and the Bohr magneton μ_B. Temperatures are effectively measured in units of energy (k_B = 1) and time is effectively measured in units of inverse energy (ħ = 1). The default unit system, Units.meV, employs (meV, Å, tesla). Select alternatively Units.theory for a units system defined so that μ₀ = μ_B = 1.\n\nSee also meV_per_K\n\n\n\n\n\n","category":"constant"},{"location":"library.html#Sunny.meV_per_K","page":"Library API","title":"Sunny.meV_per_K","text":"meV_per_K = 0.086173332621451774\n\nA physical constant. Useful for converting kelvin into the default energy units, meV.\n\n\n\n\n\n","category":"constant"},{"location":"library.html#Sunny.BinningParameters","page":"Library API","title":"Sunny.BinningParameters","text":"BinningParameters(binstart,binend,binwidth;covectors = I(4))\nBinningParameters(binstart,binend;numbins,covectors = I(4))\n\nDescribes a 4D parallelepided histogram in a format compatible with experimental Inelasitic Neutron Scattering data. See generate_mantid_script_from_binning_parameters to convert BinningParameters to a format understandable by the Mantid software, or load_nxs to load BinningParameters from a Mantid .nxs file.\n\nThe coordinates of the histogram axes are specified by multiplication of (q,ω) with each row of the covectors matrix, with q given in [R.L.U.]. Since the default covectors matrix is the identity matrix, the default axes are (qx,qy,qz,ω) in absolute units.\n\nThe convention for the binning scheme is that:\n\nThe left edge of the first bin starts at binstart\nThe bin width is binwidth\nThe last bin contains binend\nThere are no \"partial bins;\" the last bin may contain values greater than binend. C.f. count_bins.\n\nA value can be binned by computing its bin index:\n\ncoords = covectors * value\nbin_ix = 1 .+ floor.(Int64,(coords .- binstart) ./ binwidth)\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Bond","page":"Library API","title":"Sunny.Bond","text":"Bond(i, j, n)\n\nRepresents a bond between atom indices i and j. n is a vector of three integers specifying unit cell displacement in terms of lattice vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Crystal","page":"Library API","title":"Sunny.Crystal","text":"An object describing a crystallographic unit cell and its space group symmetry. Constructors are as follows:\n\nCrystal(filename; symprec=1e-5)\n\nReads the crystal from a .cif file located at the path filename. The optional parameter symprec controls the precision tolerance for spacegroup symmetries.\n\nCrystal(latvecs, positions; types=nothing, symprec=1e-5)\n\nConstructs a crystal from the complete list of atom positions positions, with coordinates (between 0 and 1) in units of lattice vectors latvecs. Spacegroup symmetry information is automatically inferred. The optional parameter types is a list of strings, one for each atom, and can be used to break symmetry-equivalence between atoms.\n\nCrystal(latvecs, positions, spacegroup_number; types=nothing, setting=nothing, symprec=1e-5)\n\nBuilds a crystal by applying symmetry operators for a given international spacegroup number. For certain spacegroups, there are multiple possible unit cell settings; in this case, a warning message will be printed, and a list of crystals will be returned, one for every possible setting. Alternatively, the optional setting string will disambiguate between unit cell conventions.\n\nCurrently, crystals built using only the spacegroup number will be missing some symmetry information. It is generally preferred to build a crystal from a .cif file or from the full specification of the unit cell.\n\nExamples\n\n# Read a Crystal from a .cif file\nCrystal(\"filename.cif\")\n\n# Build an FCC crystal using the primitive unit cell. The spacegroup number\n# 225 is inferred.\nlatvecs = [1 1 0;\n 1 0 1;\n 0 1 1] / 2\npositions = [[0, 0, 0]]\nCrystal(latvecs, positions)\n\n# Build a CsCl crystal (two cubic sublattices). By providing distinct type\n# strings, the spacegroup number 221 is inferred.\nlatvecs = lattice_vectors(1, 1, 1, 90, 90, 90)\npositions = [[0,0,0], [0.5,0.5,0.5]]\ntypes = [\"Na\", \"Cl\"]\ncryst = Crystal(latvecs, positions; types)\n\n# Build a diamond cubic crystal from its spacegroup number 227. This\n# spacegroup has two possible settings (\"1\" or \"2\"), which determine an\n# overall unit cell translation.\nlatvecs = lattice_vectors(1, 1, 1, 90, 90, 90)\npositions = [[1, 1, 1] / 4]\ncryst = Crystal(latvecs, positions, 227; setting=\"1\")\n\nSee also lattice_vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.FormFactor-Tuple{String}","page":"Library API","title":"Sunny.FormFactor","text":"FormFactor(ion::String; g_lande=2)\n\nThe magnetic form factor for a given magnetic ion and charge state. When passed to an intensity_formula, determines a 𝐪-dependent scaling of the structure factor.\n\nThe parameter ion must be one of the following strings:\n\nAm2, Am3, Am4, Am5, Am6, Am7, Au1, Au2, Au3, Au4, Au5, Ce2, Co0, Co1, Co2, Co3,\nCo4, Cr0, Cr1, Cr2, Cr3, Cr4, Cu0, Cu1, Cu2, Cu3, Cu4, Dy2, Dy3, Er2, Er3, Eu2,\nEu3, Fe0, Fe1, Fe2, Fe3, Fe4, Gd2, Gd3, Hf2, Hf3, Ho2, Ho3, Ir0a, Ir0b, Ir0c,\nIr1a, Ir1b, Ir2, Ir3, Ir4, Ir5, Ir6, Mn0, Mn1, Mn2, Mn3, Mn4, Mo0, Mo1, Nb0,\nNb1, Nd2, Nd3, Ni0, Ni1, Ni2, Ni3, Ni4, Np3, Np4, Np5, Np6, Os0a, Os0b, Os0c,\nOs1a, Os1b, Os2, Os3, Os4, Os5, Os6, Os7, Pd0, Pd1, Pr3, Pt1, Pt2, Pt3, Pt4,\nPt5, Pt6, Pu3, Pu4, Pu5, Pu6, Re0a, Re0b, Re0c, Re1a, Re1b, Re2, Re3, Re4, Re5,\nRe6, Rh0, Rh1, Ru0, Ru1, Sc0, Sc1, Sc2, Sm2, Sm3, Ta2, Ta3, Ta4, Tb2, Tb3, Tc0,\nTc1, Ti0, Ti1, Ti2, Ti3, Tm2, Tm3, U3, U4, U5, V0, V1, V2, V3, V4, W0a, W0b,\nW0c, W1a, W1b, W2c, W3, W4, W5, Y0, Yb2, Yb3, Zr0, Zr1\n\nThe trailing number denotes ionization state. For example, \"Fe0\" denotes a neutral iron atom, while \"Fe2\" denotes Fe²⁺. If multiple electronic configurations are possible, they will be distinguished by a trailing letter (a, b, ...). Omitting this letter will print an informative error,\n\nFormFactor(\"Ir0\")\n\nERROR: Disambiguate form factor according to electronic configuration:\n \"Ir0a\" -- 6s⁰5d⁹\n \"Ir0b\" -- 6s¹5d⁸\n \"Ir0c\" -- 6s²5d⁷\n\nThe form factor is approximated as\n\nF(s) = j_0(s) + frac2-gg j_2(s) s^2,\n\ninvolving the Landé g-factor. The j_l(s) are radial integrals associated with the lth Bessel function of the magnetic dipole, where s = k4π, and k is the magnitude of momentum transfer. \n\nThe radial integrals have been calculated using Hartree-Fock for transition metals, or Dirac-Fock for the rare earths and actinide series [1–3]. Sunny uses approximate fits as a sum of Gaussians,\n\nj_0(s) = A e^-as^2 + B e^-bs^2 + C e^-cs^2 + D e^-ds^2 + E \nj_l(s) = (A e^-as^2 + B e^-bs^2 + C e^-cs^2 + D e^-ds^2 + E) s^2\n\nReferences:\n\nhttps://www.ill.eu/sites/ccsl/ffacts/ffachtml.html\nJ. Brown, The Neutron Data Booklet, 2nd ed., Sec. 2.5 Magnetic Form Factors (2003)\nK. Kobayashi, T. Nagao, M. Ito, Acta Cryst. A, 67 pp 473–480 (2011)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.ImplicitMidpoint","page":"Library API","title":"Sunny.ImplicitMidpoint","text":"ImplicitMidpoint(Δt::Float64; atol=1e-12) where N\n\nEnergy-conserving spin dynamics. One call to the step! function will advance a System by Δt units of time.\n\nUses the spherical midpoint integration scheme for dipole systems and the Schrödinger midpoint integration scheme for SU(N) spin systems. Both integration schemes are symplectic, and therefore avoid energy drift over long periods of simulation time.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Langevin","page":"Library API","title":"Sunny.Langevin","text":"Langevin(Δt::Float64; λ::Float64, kT::Float64)\n\nSpin dynamics with coupling to a Langevin thermostat, which includes damping and noise terms. One call to the step! function will advance a System by Δt units of time.\n\nAssuming ergodicity, the Langevin dynamics will sample from thermal equilibrium for the target temperature kT. The empirical parameter λ determines the strength of the coupling to the thermal bath. In other words, 1/λ is the decorrelation time-scale. If λ = 0, then the spin dynamics coincides with ImplicitMidpoint.\n\nAn alternative approach to sampling is LocalSampler, which may be preferred when the allowed spin values become effective discrete (e.g. Ising spins).\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.LocalSampler","page":"Library API","title":"Sunny.LocalSampler","text":"LocalSampler(; kT, nsweeps=1.0, propose=propose_uniform)\n\nMonte Carlo simulation involving Metropolis updates to individual spins. One call to the step! function will perform nsweeps of MCMC sampling for a provided System. The default value of 1.0 means that step! performs, on average, one trial update per spin.\n\nAssuming ergodicity, the LocalSampler will sample from thermal equilibrium for the target temperature kT. \n\nThe trial spin updates are sampled using the propose function. Built-in options include propose_uniform, propose_flip, and propose_delta. Multiple proposals can be mixed with the macro @mix_proposals.\n\nThe returned object stores fields ΔE and Δs, which represent the cumulative change to the net energy and dipole, respectively.\n\nAn alternative approach to sampling is Langevin, which may be preferred for simulating continuous spins, especially in the presence of long-range dipole-dipole interactions (cf. enable_dipole_dipole!).\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SampledCorrelations","page":"Library API","title":"Sunny.SampledCorrelations","text":"SampledCorrelations\n\nBasic data type for storing sampled correlation data. A SampleCorrelations is initialized by calling either dynamical_correlations or instant_correlations.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SpinInfo","page":"Library API","title":"Sunny.SpinInfo","text":"SpinInfo(atom::Int; S, g=2)\n\nCharacterizes the spin at a given atom index within the crystal unit cell. S is an integer multiple of 1/2 and gives the spin angular momentum in units of ħ. g is the g-factor or tensor, such that an angular momentum dipole s produces a magnetic moment g s in units of the Bohr magneton.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SpinWaveTheory","page":"Library API","title":"Sunny.SpinWaveTheory","text":"SpinWaveTheory(sys, energy_ϵ::Float64=1e-8)\n\nConstructs an object to perform linear spin wave theory. Use it with dispersion and dssf functions.\n\nThe optional parameter energy_ϵ adds a small positive shift to the diagonal of the dynamical matrix D to avoid numerical issues with zero-energy quasi-particle modes.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.System-Tuple{Crystal, Tuple{Int64, Int64, Int64}, Vector{SpinInfo}, Symbol}","page":"Library API","title":"Sunny.System","text":"System(crystal::Crystal, latsize, infos, mode; units=Units.meV, seed::Int)\n\nConstruct a System of spins for a given Crystal symmetry. The latsize parameter determines the number of unit cells in each lattice vector direction. The infos parameter is a list of SpinInfo objects, which determine the magnitude S and g-tensor of each spin.\n\nThe two primary options for mode are :SUN and :dipole. In the former, each spin-S degree of freedom is described as an SU(N) coherent state, i.e. a quantum superposition of N = 2S + 1 levels. This formalism can be useful to capture multipolar spin fluctuations or local entanglement effects. \n\nMode :dipole projects the SU(N) dynamics onto the restricted space of pure dipoles. In practice this means that Sunny will simulate Landau-Lifshitz dynamics, but single-ion anisotropy and biquadratic exchange interactions will be renormalized to improve accuracy. To disable this renormalization, use the mode :dipole_large_S which applies the S classical limit. For details, see the documentation page: Interaction Strength Renormalization.\n\nThe default units system of (meV, Å, tesla) can be overridden by with the units parameter; see Units. \n\nAn optional seed may be provided to achieve reproducible random number generation.\n\nAll spins are initially polarized in the z-direction.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.add_sample!-Tuple{SampledCorrelations, System}","page":"Library API","title":"Sunny.add_sample!","text":"add_sample!(sc::SampledCorrelations, sys::System)\n\nadd_trajectory uses the spin configuration contained in the System to generate a correlation data and accumulate it into sc. For static structure factors, this involves analyzing the spin-spin correlations of the spin configuration provided. For a dynamic structure factor, a trajectory is calculated using the given spin configuration as an initial condition. The spin-spin correlations are then calculating in time and accumulated into sc. \n\nThis function will change the state of sys when calculating dynamical structure factor data. To preserve the initial state of sys, it must be saved separately prior to calling add_sample!. Alternatively, the initial spin configuration may be copied into a new System and this new System can be passed to add_sample!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.available_energies-Tuple{SampledCorrelations}","page":"Library API","title":"Sunny.available_energies","text":"available_energies(sc::SampledCorrelations; negative_energies=false)\n\nReturn the ω values for the energy index of a SampledCorrelations. By default, only returns values for non-negative energies, which corresponds to the default output of intensities. Set negative_energies to true to retrieve all ω values.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.available_wave_vectors-Tuple{SampledCorrelations}","page":"Library API","title":"Sunny.available_wave_vectors","text":"available_wave_vectors(sc::SampledCorrelations; bzsize=(1,1,1))\n\nReturns all wave vectors for which sc contains exact values. bsize specifies the number of Brillouin zones to be included.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.axes_bincenters-Tuple{Any, Any, Any}","page":"Library API","title":"Sunny.axes_bincenters","text":"axes_bincenters(params::BinningParameters)\n\nReturns tick marks which label the bins of the histogram described by BinningParameters by their bin centers.\n\nThe following alternative syntax can be used to compute bin centers for a single axis:\n\naxes_bincenters(binstart,binend,binwidth)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.broaden_energy-Tuple{SampledCorrelations, Any, Function}","page":"Library API","title":"Sunny.broaden_energy","text":"broaden_energy(sc::SampledCorrelations, vals, kernel::Function; negative_energies=false)\n\nPerforms a real-space convolution along the energy axis of an array of intensities. Assumes the format of the intensities array corresponds to what would be returned by intensities_interpolated. kernel must be a function that takes two numbers: kernel(ω, ω₀), where ω is a frequency, and ω₀ is the center frequency of the kernel. Sunny provides lorentzian for the most common use case:\n\nnewvals = broaden_energy(sc, vals, (ω, ω₀) -> lorentzian(ω-ω₀, 0.2))\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.count_bins-Tuple{Any, Any, Any}","page":"Library API","title":"Sunny.count_bins","text":"count_bins(binstart,binend,binwidth)\n\nReturns the number of bins in the binning scheme implied by binstart, binend, and binwidth. To count the bins in a BinningParameters, use params.numbins.\n\nThis function defines how partial bins are handled, so it should be used preferentially over computing the number of bins manually.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dispersion-Tuple{SpinWaveTheory, Any}","page":"Library API","title":"Sunny.dispersion","text":"dispersion(swt::SpinWaveTheory, qs)\n\nComputes the spin excitation energy dispersion relations given a SpinWaveTheory and an array of wave vectors qs. Each element q of qs must be a 3-vector in units of reciprocal lattice units. I.e., qᵢ is given in 2πaᵢ with aᵢ the lattice constant of the original chemical lattice.\n\nThe first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of the array is an energy.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dmvec-Tuple{Any}","page":"Library API","title":"Sunny.dmvec","text":"dmvec(D)\n\nAntisymmetric matrix representation of the Dzyaloshinskii-Moriya pseudo-vector,\n\n [ 0 D[3] -D[2]\n -D[3] 0 D[1]\n D[2] -D[1] 0 ]\n\nUseful in the context of set_exchange!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dssf-Tuple{SpinWaveTheory, Any}","page":"Library API","title":"Sunny.dssf","text":"dssf(swt::SpinWaveTheory, qs)\n\nGiven a SpinWaveTheory object, computes the dynamical spin structure factor,\n\n 𝒮^αβ(𝐤 ω) = 1(2πN)dt _𝐫 expi(ωt - 𝐤𝐫) S^α(𝐫 t)S^β(0 0)\n\nusing the result from linear spin-wave theory,\n\n 𝒮^αβ(𝐤 ω) = _n A_n^αβ(𝐤)^2 δω-ω_n(𝐤)\n\nqs is an array of wave vectors of arbitrary dimension. Each element q of qs must be a 3-vector in reciprocal lattice units (RLU), i.e., in the basis of reciprocal lattice vectors.\n\nThe first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of this array is a tensor (3×3 matrix) corresponding to the indices α and β.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dynamical_correlations-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.dynamical_correlations","text":"dynamical_correlations(sys::System; Δt, nω, ωmax, \n process_trajectory=:none, observables=nothing, correlations=nothing)\n\nCreates an empty SampledCorrelations object for calculating and storing dynamical structure factor intensities 𝒮(𝐪ω). Call add_sample! to accumulate data for the given configuration of a spin system. Internally, this will run a dynamical trajectory and measure time correlations. The 𝒮(𝐪ω) data can be retrieved by calling intensities_interpolated. Alternatively, instant_intensities_interpolated will integrate out ω to obtain 𝒮(𝐪), optionally applying classical-to-quantum correction factors.\n\nThree keywords are required to specify the dynamics used for the trajectory calculation.\n\nΔt: The time step used for calculating the trajectory from which dynamic spin-spin correlations are calculated. The trajectories are calculated with an ImplicitMidpoint integrator.\nωmax: The maximum energy, ω, that will be resolved.\nnω: The number of energy bins to calculated between 0 and ωmax.\n\nAdditional keyword options are the following:\n\nobservables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.\ncorrelations: Specify which correlation functions are calculated, i.e. which matrix elements αβ of 𝒮^αβ(qω) are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.eachsite-Tuple{System}","page":"Library API","title":"Sunny.eachsite","text":"eachsite(sys::System)\n\nAn iterator over all Sites in the system. \n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.enable_dipole_dipole!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.enable_dipole_dipole!","text":"enable_dipole_dipole!(sys::System)\n\nEnables long-range dipole-dipole interactions,\n\n -(μ_04π) _ij (3 (𝐌_j𝐫_ij)(𝐌_i𝐫_ij) - 𝐌_i𝐌_j) 𝐫_ij^3\n\nwhere the sum is over all pairs of spins (singly counted), including periodic images, regularized using the Ewald summation convention. The magnetic moments are 𝐌_i = μ_B g 𝐒_i where g is the g-factor or g-tensor, and 𝐒_i is the spin angular momentum dipole in units of ħ. The Bohr magneton μ_B and vacuum permeability μ_0 are physical constants, with numerical values determined by the unit system.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.energy-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.energy","text":"energy(sys::System)\n\nThe total system energy. See also energy_per_site.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.energy_per_site-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.energy_per_site","text":"energy_per_site(sys::System)\n\nThe total system energy divided by the number of sites.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.generate_mantid_script_from_binning_parameters-Tuple{Any}","page":"Library API","title":"Sunny.generate_mantid_script_from_binning_parameters","text":"generate_mantid_script_from_binning_parameters(params::BinningParameters)\n\nGenerate a Mantid script which bins data according to the given BinningParameters.\n\nwarning: Units\nTake care to ensure the units are correct (R.L.U. or absolute). You may want to call Sunny.bin_rlu_as_absolute_units! or Sunny.bin_absolute_units_as_rlu! first.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.global_position-Tuple{System, Any}","page":"Library API","title":"Sunny.global_position","text":"global_position(sys::System, site::Site)\n\nPosition of a Site in global coordinates.\n\nTo precompute a full list of positions, one can use eachsite as below:\n\npos = [global_position(sys, site) for site in eachsite(sys)]\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.instant_correlations-Tuple{System}","page":"Library API","title":"Sunny.instant_correlations","text":"instant_correlations(sys::System; process_trajectory=:none, observables=nothing, correlations=nothing)\n\nCreates an empty SampledCorrelations object for calculating and storing instantaneous structure factor intensities 𝒮(𝐪). Call add_sample! to accumulate data for the given configuration of a spin system. Call instant_intensities_interpolated to retrieve averaged 𝒮(𝐪) data.\n\nImportant note: When dealing with continuous (non-Ising) spins, consider creating using dynamical_correlations instead of instant_correlations. The former will provide full 𝒮(𝐪ω) data, from which 𝒮(𝐪) can be obtained by integrating out ω. During this integration step, Sunny can incorporate temperature- and ω-dependent classical-to-quantum correction factors to produce more accurate 𝒮(𝐪) estimates. See instant_intensities_interpolated for more information.\n\nThe following optional keywords are available:\n\nobservables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.\ncorrelations: Specify which correlation functions are calculated, i.e. which matrix elements αβ of 𝒮^αβ(qω) are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.instant_intensities_interpolated-Tuple{SampledCorrelations, Any, Any}","page":"Library API","title":"Sunny.instant_intensities_interpolated","text":"instant_intensities_interpolated(sc::SampledCorrelations, qs; kwargs...)\n\nReturn 𝒮(𝐪) intensities at wave vectors qs. The functionality is very similar to intensities_interpolated, except the returned array has dimensions identical to qs. If called on a SampledCorrelations with dynamical information, i.e., 𝒮(𝐪ω), the ω information is integrated out.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.integrate_axes!-Tuple{BinningParameters}","page":"Library API","title":"Sunny.integrate_axes!","text":"integrate_axes!(params::BinningParameters; axes)\n\nIntegrate over one or more axes of the histogram by setting the number of bins in that axis to 1. Examples:\n\nintegrate_axes!(params; axes = [2,3])\nintegrate_axes!(params; axes = 2)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.integrated_lorentzian-Tuple{Float64}","page":"Library API","title":"Sunny.integrated_lorentzian","text":"integrated_lorentzian(η)\n\nReturns x mapsto atan(xη)π for use with intensities_binned.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_bands-Tuple{SpinWaveTheory, Any, Sunny.SpinWaveIntensityFormula}","page":"Library API","title":"Sunny.intensities_bands","text":"dispersion, intensities = intensities_bands(swt::SpinWaveTheory, ks, formula::SpinWaveIntensityFormula)\n\nComputes the scattering intensities at each energy band for each momentum transfer k in ks, according to Linear Spin Wave Theory and the given intensity formula. The formula must have a delta-function kernel, e.g.:\n\nformula = intensity_formula(swt, :perp, formula; kernel = delta_function_kernel)\n\nor else the bands will be broadened, and their intensity can not be computed.\n\nThe outputs will be arrays with indices identical to ks, with the last index giving the band index. dispersions reports the energy of each band, while intensities reports the scattering intensity.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_binned-Tuple{SampledCorrelations, BinningParameters, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.intensities_binned","text":"intensity, counts = intensities_binned(sc::SampledCorrelations, params::BinningParameters, formula; integrated_kernel)\n\nGiven correlation data contained in a SampledCorrelations and BinningParameters describing the shape of a histogram, compute the intensity and normalization for each histogram bin using a given intensity_formula.\n\nThe BinningParameters are expected to accept (q,ω) in R.L.U. for the (possibly reshaped) crystal associated with sc.\n\nThis is an alternative to intensities_interpolated which bins the scattering intensities into a histogram instead of interpolating between them at specified qs values. See unit_resolution_binning_parameters for a reasonable default choice of BinningParameters which roughly emulates intensities_interpolated with interpolation = :round.\n\nIf a function integrated_kernel(Δω) is passed, it will be used as the CDF of a kernel function for energy broadening. For example, integrated_kernel = Δω -> atan(Δω/η)/pi (c.f. integrated_lorentzian implements Lorentzian broadening with parameter η. Energy-dependent energy broadening can be achieved by providing an integrated_kernel(ω,Δω) whose first argument is the energy transfer ω.\n\nCurrently, energy broadening is only supported if the BinningParameters are such that the first three axes are purely spatial and the last (energy) axis is [0,0,0,1].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_broadened-Tuple{SpinWaveTheory, Any, Any, Any}","page":"Library API","title":"Sunny.intensities_broadened","text":"intensities_broadened(swt::SpinWaveTheory, ks, ωvals, formula)\n\nComputes the scattering intensities at each (Q,ω) according to Linear Spin Wave Theory and the given intensity formula. The required formula must have a non-delta-function kernel, e.g.:\n\nformula = intensity_formula(swt, :perp; kernel = lorentzian(0.05))\n\nor else the intensity at ωvals which are not exactly on the dispersion curve can not be calculated.\n\nThe intensity is computed at each wave vector in ks and each energy in ωvals. The output will be an array with indices identical to ks, with the last index matching ωvals.\n\nNote that ks is an array of wave vectors of arbitrary dimension. Each element k of ks must be a 3-wavevector in absolute units.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_interpolated-Tuple{SampledCorrelations, Any, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.intensities_interpolated","text":"intensities_interpolated(sc::SampledCorrelations, qs, formula:ClassicalIntensityFormula; interpolation=nothing, negative_energies=false)\n\nThe basic function for retrieving 𝒮(𝐪ω) information from a SampledCorrelations. Maps an array of wave vectors qs to an array of structure factor intensities, including an additional energy index. The values of ω associated with the energy index can be retrieved by calling available_energies. The three coordinates of each wave vector are measured in reciprocal lattice units, i.e., multiples of the reciprocal lattice vectors.\n\ninterpolation: Since 𝒮(𝐪 ω) is calculated on a finite lattice, data is only available at discrete wave vectors. By default, Sunny will round a requested q to the nearest available wave vector. Linear interpolation can be applied by setting interpolation=:linear.\nnegative_energies: If set to true, Sunny will return the periodic extension of the energy axis. Most users will not want this.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SampledCorrelations, AbstractVector{Int64}}","page":"Library API","title":"Sunny.intensity_formula","text":"formula = intensity_formula(sc::SampledCorrelations)\n\nEstablish a formula for computing the intensity of the discrete scattering modes (q,ω) using the correlation data 𝒮^αβ(qω) stored in the SampledCorrelations. The formula returned from intensity_formula can be passed to intensities_interpolated or intensities_binned.\n\nintensity_formula(sc,...; kT = Inf, formfactors = ...)\n\nThere are keyword arguments providing temperature and form factor corrections:\n\nkT: If a temperature is provided, the intensities will be rescaled by a temperature- and ω-dependent classical-to-quantum factor. kT should be specified when making comparisons with spin wave calculations or experimental data. If kT is not specified, infinite temperature (no correction) is assumed.\nformfactors: To apply form factor corrections, provide this keyword with a list of FormFactors, one for each symmetry-distinct site in the crystal. The order of FormFactors must correspond to the order of site symmetry classes, e.g., as they appear when printed in display(crystal).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SampledCorrelations, Any}","page":"Library API","title":"Sunny.intensity_formula","text":"A custom intensity formula can be specifed by providing a function intensity = f(q,ω,correlations) and specifying which correlations it requires:\n\nintensity_formula(f,sc::SampledCorrelations, required_correlations; kwargs...)\n\nThe function is intended to be specified using do notation. For example, this custom formula sums the off-diagonal correlations:\n\nrequired = [(:Sx,:Sy),(:Sy,:Sz),(:Sx,:Sz)]\nintensity_formula(sc,required,return_type = ComplexF64) do k, ω, off_diagonal_correlations\n sum(off_diagonal_correlations)\nend\n\nIf your custom formula returns a type other than Float64, use the return_type keyword argument to flag this.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SpinWaveTheory, AbstractVector{Int64}}","page":"Library API","title":"Sunny.intensity_formula","text":"formula = intensity_formula(swt::SpinWaveTheory; kernel = ...)\n\nEstablish a formula for computing the scattering intensity by diagonalizing the hamiltonian H(q) using Linear Spin Wave Theory.\n\nIf kernel = delta_function_kernel, then the resulting formula can be used with intensities_bands.\n\nIf kernel is an energy broadening kernel function, then the resulting formula can be used with intensities_broadened. Energy broadening kernel functions can either be a function of Δω only, e.g.:\n\nkernel = Δω -> ...\n\nor a function of both the energy transfer ω and of Δω, e.g.:\n\nkernel = (ω,Δω) -> ...\n\nThe integral of a properly normalized kernel function over all Δω is one.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{SpinWaveTheory, Symbol}","page":"Library API","title":"Sunny.intensity_formula","text":"intensity_formula([swt or sc], contraction_mode::Symbol)\n\nSunny has several built-in formulas that can be selected by setting contraction_mode to one of these values:\n\n:trace (default), which yields operatornametr 𝒮(qω) = _α 𝒮^αα(qω)\n:perp, which contracts 𝒮^αβ(qω) with the dipole factor δ_αβ - q_αq_β, returning the unpolarized intensity.\n:full, which will return all elements 𝒮^αβ(𝐪ω) without contraction.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lattice_params-Tuple{StaticArraysCore.SMatrix{3, 3, Float64, 9}}","page":"Library API","title":"Sunny.lattice_params","text":"lattice_params(latvecs::Mat3)\n\nCompute the lattice parameters (a b c α β γ) for the three lattice vectors provided as columns of latvecs. The inverse mapping is lattice_vectors.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lattice_vectors-NTuple{6, Any}","page":"Library API","title":"Sunny.lattice_vectors","text":"lattice_vectors(a, b, c, α, β, γ)\n\nReturn the lattice vectors, as columns of the 33 output matrix, that correspond to the conventional unit cell defined by the lattice constants (a b c) and the angles (α β γ) in degrees. The inverse mapping is lattice_params.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.load_nxs-Tuple{Any}","page":"Library API","title":"Sunny.load_nxs","text":"params, signal = load_nxs(filename)\n\nGiven the name of a Mantid-exported MDHistoWorkspace file, load the BinningParameters and the signal from that file.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lorentzian-Tuple{Any, Any}","page":"Library API","title":"Sunny.lorentzian","text":"lorentzian(x, η)\n\nReturns η(π(x^2 + η^2)).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.magnetic_moment-Tuple{System, Any}","page":"Library API","title":"Sunny.magnetic_moment","text":"magnetic_moment(sys::System, site::Site)\n\nGet the magnetic moment for a Site. This is the spin dipole multiplied by the Bohr magneton and the local g-tensor.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.merge_correlations-Union{Tuple{Array{SampledCorrelations{N}, 1}}, Tuple{N}} where N","page":"Library API","title":"Sunny.merge_correlations","text":"merge_correlations(scs::Vector{SampledCorrelations)\n\nAccumulate a list of SampledCorrelations into a single, summary SampledCorrelations. Useful for reducing the results of parallel computations.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.minimize_energy!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.minimize_energy!","text":"minimize_energy!(sys::System{N}; maxiters=100, subiters=20,\n method=Optim.ConjugateGradient(), kwargs...) where N\n\nOptimizes the spin configuration in sys to minimize energy. A total of maxiters iterations will be attempted, with restarts after every subiters iterations. The remaining kwargs will be forwarded to the optimize method of the Optim.jl package.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.polarize_spins!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.polarize_spins!","text":"polarize_spins!(sys::System, dir)\n\nPolarize all spins in the system along the direction dir.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.position_to_site-Tuple{System, Any}","page":"Library API","title":"Sunny.position_to_site","text":"position_to_site(sys::System, r)\n\nConverts a position r to four indices of a Site. The coordinates of r are given in units of the lattice vectors for the original crystal. This function can be useful for working with systems that have been reshaped using reshape_supercell.\n\nExample\n\n# Find the `site` at the center of a unit cell which is displaced by four\n# multiples of the first lattice vector\nsite = position_to_site(sys, [4.5, 0.5, 0.5])\n\n# Print the dipole at this site\nprintln(sys.dipoles[site])\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.powder_average_binned-Tuple{SampledCorrelations, Any, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.powder_average_binned","text":"powder_average_binned(sc::SampledCorrelations, radial_binning_parameters; formula\n ω_binning_parameters, integrated_kernel = nothing, bzsize = nothing)\n\nThis function emulates the experimental situation of \"powder averaging,\" where only the magnitude (and not the direction) of the momentum transfer is resolvable. The intensities are binned similarly to intensities_binned, but the histogram x-axis is |k| in absolute units, which is a nonlinear function of kx,ky,kz. The y-axis is energy.\n\nRadial binning parameters are specified as tuples (start,end,bin_width), e.g. radial_binning_parameters = (0,6π,6π/55).\n\nEnergy broadening is supported in the same way as intensities_binned, and this function accepts the same kind of intensity_formula.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.primitive_cell_shape-Tuple{Crystal}","page":"Library API","title":"Sunny.primitive_cell_shape","text":"primitive_cell_shape(cryst::Crystal)\n\nReturns the shape of the primitive cell as a 3×3 matrix, in fractional coordinates of the conventional lattice vectors. May be useful for constructing inputs to reshape_supercell.\n\nExamples\n\n# Valid if `cryst` has not been reshaped\n@assert cryst.prim_latvecs ≈ cryst.latvecs * primitive_cell_shape(cryst)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_bond-Tuple{Crystal, Bond}","page":"Library API","title":"Sunny.print_bond","text":"print_bond(cryst::Crystal, bond::Bond; b_ref::Bond)\n\nPrints symmetry information for bond bond. A symmetry-equivalent reference bond b_ref can optionally be provided to fix the meaning of the coefficients A, B, ...\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_site-Tuple{Any, Any}","page":"Library API","title":"Sunny.print_site","text":"print_site(cryst, i; R=I)\n\nPrint symmetry information for the site i, including allowed g-tensor and allowed anisotropy operator. An optional rotation matrix R can be provided to define the reference frame for expression of the anisotropy.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_stevens_expansion-Tuple{AbstractMatrix}","page":"Library API","title":"Sunny.print_stevens_expansion","text":"function print_stevens_expansion(op)\n\nPrints a local Hermitian operator as a linear combination of Stevens operators. The operator op may be a finite-dimensional matrix or an abstract spin polynomial in the large-S limit.\n\nExamples\n\nS = spin_matrices(2)\nprint_stevens_expansion(S[1]^4 + S[2]^4 + S[3]^4)\n# Prints: (1/20)𝒪₄₀ + (1/4)𝒪₄₄ + 102/5\n\nS = spin_matrices(Inf)\nprint_stevens_expansion(S[1]^4 + S[2]^4 + S[3]^4)\n# Prints: (1/20)𝒪₄₀ + (1/4)𝒪₄₄ + (3/5)𝒮⁴\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_suggested_frame-Tuple{Crystal, Int64}","page":"Library API","title":"Sunny.print_suggested_frame","text":"print_suggested_frame(cryst, i; digits=4)\n\nPrint a suggested reference frame, as a rotation matrix R, that can be used as input to print_site(). The purpose is to simplify the description of allowed anisotropies.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_symmetry_table-Tuple{Crystal, Any}","page":"Library API","title":"Sunny.print_symmetry_table","text":"print_symmetry_table(cryst::Crystal, max_dist)\n\nPrint symmetry information for all equivalence classes of sites and bonds, up to a maximum bond distance of max_dist. Equivalent to calling print_bond(cryst, b) for every bond b in reference_bonds(cryst, max_dist), where Bond(i, i, [0,0,0]) refers to a single site i.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_wrapped_intensities-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.print_wrapped_intensities","text":"print_wrapped_intensities(sys::System; nmax=10)\n\nFor Bravais lattices: Prints up to nmax wavevectors according to their instantaneous (static) structure factor intensities, listed in descending order. For non-Bravais lattices: Performs the same analysis for each spin sublattice independently; the output weights are naïvely averaged over sublattices, without incorporating phase shift information. This procedure therefore wraps all wavevectors into the first Brillouin zone. Each wavevector coordinate is given between -12 and 12 in reciprocal lattice units (RLU). The output from this function will typically be used as input to suggest_magnetic_supercell.\n\nBecause this function does not incorporate phase information in its averaging over sublattices, the printed weights are not directly comparable with experiment. For that purpose, use instant_correlations instead.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_delta-Tuple{Any}","page":"Library API","title":"Sunny.propose_delta","text":"propose_delta(magnitude)\n\nGenerate a proposal function that adds a Gaussian perturbation to the existing spin state. In :dipole mode, the procedure is to first introduce a random three-vector perturbation 𝐬 = 𝐬 + 𝐬 ξ and then return the properly normalized spin 𝐬 (𝐬𝐬). Each component of the random vector ξ is Gaussian distributed with a standard deviation of magnitude; the latter is dimensionless and typically smaller than one. \n\nIn :SUN mode, the procedure is analogous, but now involving Gaussian perturbations to each of the N complex components of an SU(N) coherent state.\n\nIn the limit of very large magnitude, this function coincides with propose_uniform.\n\nFor use with LocalSampler.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_flip-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.propose_flip","text":"propose_flip\n\nFunction to propose pure spin flip updates in the context of a LocalSampler. Dipoles are flipped as 𝐬 -𝐬. SU(N) coherent states are flipped using the time-reversal operator.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_uniform","page":"Library API","title":"Sunny.propose_uniform","text":"propose_uniform\n\nFunction to propose a uniformly random spin update in the context of a LocalSampler. In :dipole mode, the result is a random three-vector with appropriate normalization. In :SUN mode, the result is a random SU(N) coherent state with appropriate normalization.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.randomize_spins!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.randomize_spins!","text":"randomize_spins!(sys::System)\n\nRandomizes all spins under appropriate the uniform distribution.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_path-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.reciprocal_space_path","text":"reciprocal_space_path(cryst::Crystal, qs, density)\n\nReturns a pair (path, xticks). The path return value is a list of wavevectors that samples linearly between the provided wavevectors qs. The xticks return value can be used to label the special 𝐪 values on the x-axis of a plot.\n\nSpecial note about units: the wavevectors qs must be provided in reciprocal lattice units (RLU) for the given crystal, but the sampling density must be specified in the global frame. Specifically, the density is given as number of sample points per unit of radian inverse length, where the unit of length is the same as that used to specify the lattice vectors of the Crystal. The path will therefore include more samples between q-points that are further apart in absolute Fourier distance.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_path_bins-Tuple{Any, Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.reciprocal_space_path_bins","text":"reciprocal_space_path_bins(sc,qs,density,args...;kwargs...)\n\nTakes a list of wave vectors, qs in R.L.U., and builds a series of histogram BinningParameters whose first axis traces a path through the provided points. The second and third axes are integrated over according to the args and kwargs, which are passed through to slice_2D_binning_parameters.\n\nAlso returned is a list of marker indices corresponding to the input points, and a list of ranges giving the indices of each histogram x-axis within a concatenated histogram. The density parameter is given in samples per reciprocal lattice unit (R.L.U.).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_shell-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.reciprocal_space_shell","text":"reciprocal_space_shell(cryst::Crystal, radius, n)\n\nSample n points on the reciprocal space sphere with a given radius (units of inverse length).\n\nExamples\n\n# Sample wavevectors on the sphere at fixed density\nreciprocal_space_shell(cryst, r, 4π*r^2*density)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reference_bonds-Tuple{Crystal, Float64}","page":"Library API","title":"Sunny.reference_bonds","text":"reference_bonds(cryst::Crystal, max_dist)\n\nReturns a full list of bonds, one for each symmetry equivalence class, up to distance max_dist. The reference bond b for each equivalence class is selected according to a scoring system that prioritizes simplification of the elements in basis_for_symmetry_allowed_couplings(cryst, b).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.remove_periodicity!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.remove_periodicity!","text":"remove_periodicity!(sys::System, dims)\n\nRemove periodic interactions along the dimensions where dims is true. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nExample\n\n# Remove periodic boundaries along the 1st and 3rd dimensions\nremove_periodicity!(sys::System, (true, false, true))\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.repeat_periodically-Union{Tuple{N}, Tuple{System{N}, Tuple{Int64, Int64, Int64}}} where N","page":"Library API","title":"Sunny.repeat_periodically","text":"repeat_periodically(sys::System{N}, counts::NTuple{3,Int}) where N\n\nCreates a System identical to sys but repeated a given number of times in each dimension, specified by the tuple counts.\n\nSee also reshape_supercell.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reshape_supercell-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.reshape_supercell","text":"reshape_supercell(sys::System, shape)\n\nMaps an existing System to a new one that has the shape and periodicity of a requested supercell. The columns of the 33 integer matrix shape represent the supercell lattice vectors measured in units of the original crystal lattice vectors.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.resize_supercell-Union{Tuple{N}, Tuple{System{N}, Tuple{Int64, Int64, Int64}}} where N","page":"Library API","title":"Sunny.resize_supercell","text":"resize_supercell(sys::System{N}, latsize::NTuple{3,Int}) where N\n\nCreates a System with a given number of conventional unit cells in each lattice vector direction. Interactions and other settings will be inherited from sys.\n\nConvenience function for:\n\nreshape_supercell(sys, [latsize[1] 0 0; 0 latsize[2] 0; 0 0 latsize[3]])\n\nSee also reshape_supercell.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.rotate_operator-Tuple{LinearAlgebra.Hermitian{ComplexF64, Matrix{ComplexF64}}, Any}","page":"Library API","title":"Sunny.rotate_operator","text":"rotate_operator(A, R)\n\nRotates the local quantum operator A according to the 33 rotation matrix R.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.rotation_in_rlu-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.rotation_in_rlu","text":"rotation_in_rlu(cryst::Crystal, axis, angle)\n\nReturns a 33 matrix that rotates wavevectors in reciprocal lattice units (RLU). The axis vector is a real-space direction in absolute units (but arbitrary magnitude), and the angle is in radians.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_coherent!-Union{Tuple{N}, Tuple{System{N}, Any, Any}} where N","page":"Library API","title":"Sunny.set_coherent!","text":"set_coherent!(sys::System, Z, site::Site)\n\nSet a coherent spin state at a Site using the N complex amplitudes in Z.\n\nFor a standard SpinInfo, these amplitudes will be interpreted in the eigenbasis of 𝒮ᶻ. That is, Z[1] represents the amplitude for the basis state fully polarized along the z-direction, and subsequent components represent states with decreasing angular momentum along this axis (m = S S-1 -S).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_dipole!-Union{Tuple{N}, Tuple{System{N}, Any, Any}} where N","page":"Library API","title":"Sunny.set_dipole!","text":"set_dipole!(sys::System, dir, site::Site)\n\nPolarize the spin at a Site along the direction dir.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_exchange!-Union{Tuple{N}, Tuple{System{N}, Any, Bond}} where N","page":"Library API","title":"Sunny.set_exchange!","text":"set_exchange!(sys::System, J, bond::Bond)\n\nSets a 3×3 spin-exchange matrix J along bond, yielding a pairwise interaction energy 𝐒_iJ 𝐒_j. This interaction will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells.\n\nThe parameter J may be scalar or matrix-valued. As a convenience, dmvec(D) can be used to construct the antisymmetric part of the exchange, where D is the Dzyaloshinskii-Moriya pseudo-vector. The resulting interaction will be 𝐃(𝐒_i𝐒_j).\n\nFor more general interactions, such as biquadratic, use set_pair_coupling! instead.\n\nExamples\n\n# An explicit exchange matrix\nJ1 = [2 3 0;\n -3 2 0;\n 0 0 2]\nset_exchange!(sys, J1, bond)\n\n# An equivalent Heisenberg + DM exchange \nJ2 = 2*I + dmvec([0,0,3])\nset_exchange!(sys, J2, bond)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_exchange_at!-Union{Tuple{N}, Tuple{System{N}, Any, Union{NTuple{4, Int64}, CartesianIndex{4}}, Union{NTuple{4, Int64}, CartesianIndex{4}}}} where N","page":"Library API","title":"Sunny.set_exchange_at!","text":"set_exchange_at!(sys::System, J, site1::Site, site2::Site; offset=nothing)\n\nSets an exchange interaction `𝐒_i⋅J 𝐒_j along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nIf the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.\n\nFor more general interactions, such as biquadratic, use set_pair_coupling_at! instead.\n\nSee also set_exchange!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_external_field!-Tuple{System, Any}","page":"Library API","title":"Sunny.set_external_field!","text":"set_external_field!(sys::System, B::Vec3)\n\nSets the external field B that couples to all spins.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_external_field_at!-Tuple{System, Any, Any}","page":"Library API","title":"Sunny.set_external_field_at!","text":"set_external_field_at!(sys::System, B::Vec3, site::Site)\n\nSets a Zeeman coupling between a field B and a single spin. Site includes a unit cell and a sublattice index.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_onsite_coupling!-Tuple{System, Any, Int64}","page":"Library API","title":"Sunny.set_onsite_coupling!","text":"set_onsite_coupling!(sys::System, op, i::Int)\n\nSet the single-ion anisotropy for the ith atom of every unit cell, as well as all symmetry-equivalent atoms. The operator op may be provided as an abstract function of the local spin operators, as a polynomial of spin_matrices, or as a linear combination of stevens_matrices.\n\nExamples\n\n# An easy axis anisotropy in the z-direction\nset_onsite_coupling!(sys, S -> -D*S[3]^3, i)\n\n# The unique quartic single-ion anisotropy for a site with cubic point group\n# symmetry\nset_onsite_coupling!(sys, S -> 20*(S[1]^4 + S[2]^4 + S[3]^4), i)\n\n# An equivalent expression of this quartic anisotropy, up to a constant shift\nO = stevens_matrices(spin_label(sys, i))\nset_onsite_coupling!(sys, O[4,0] + 5*O[4,4], i)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_onsite_coupling_at!-Tuple{System, Any, Union{NTuple{4, Int64}, CartesianIndex{4}}}","page":"Library API","title":"Sunny.set_onsite_coupling_at!","text":"set_onsite_coupling_at!(sys::System, op, site::Site)\n\nSets the single-ion anisotropy operator op for a single Site, ignoring crystal symmetry. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nSee also set_onsite_coupling!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_pair_coupling!-Union{Tuple{N}, Tuple{System{N}, AbstractMatrix, Any}} where N","page":"Library API","title":"Sunny.set_pair_coupling!","text":"set_pair_coupling!(sys::System, op, bond)\n\nSets an arbitrary coupling op along bond. This coupling will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites.\n\nExamples\n\n# Bilinear+biquadratic exchange involving 3×3 matrices J1 and J2\nset_pair_coupling!(sys, (Si, Sj) -> Si'*J1*Sj + (Si'*J2*Sj)^2, bond)\n\n# Equivalent expression using an appropriate fixed matrix representation\nS = spin_matrices(1/2)\nSi, Sj = to_product_space(S, S)\nset_pair_coupling!(sys, Si'*J1*Sj + (Si'*J2*Sj)^2, bond)\n\nSee also spin_matrices, to_product_space.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_pair_coupling_at!-Union{Tuple{N}, Tuple{System{N}, AbstractMatrix, Union{NTuple{4, Int64}, CartesianIndex{4}}, Union{NTuple{4, Int64}, CartesianIndex{4}}}} where N","page":"Library API","title":"Sunny.set_pair_coupling_at!","text":"set_pair_coupling_at!(sys::System, op, bond)\n\nSets an arbitrary coupling along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites. The documentation for set_pair_coupling! provides examples constructing op.\n\nThe system must support inhomogeneous interactions via to_inhomogeneous.\n\nIf the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_spiral_order!-Tuple{Any}","page":"Library API","title":"Sunny.set_spiral_order!","text":"set_spiral_order!(sys; q, axis, S0)\n\nInitializes the system with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0 at the real-space origin. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.\n\nExample\n\n# Spiral order for a wavevector propagating in the direction of the first\n# reciprocal lattice vector (i.e., orthogonal to the lattice vectors ``𝐚_2``\n# and ``𝐚_3``), repeating with a period of 10 lattice constants, and spiraling\n# about the ``ẑ``-axis. The spin at the origin will point in the direction\n# ``𝐒_0 = ŷ + ẑ``. Here, ``(x̂, ŷ, ẑ)`` are the axes of Cartesian coordinate\n# system in the global frame.\nset_spiral_order!(sys; q=[1/10, 0, 0], axis=[0, 0, 1], S0=[0, 1, 1])\n\nSee also set_spiral_order_on_sublattice!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_spiral_order_on_sublattice!-Tuple{Any, Any}","page":"Library API","title":"Sunny.set_spiral_order_on_sublattice!","text":"set_spiral_order_on_sublattice!(sys, i; q, axis, S0)\n\nInitializes sublattice i with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0. The phase is selected such that the spin at sys.dipole[1,1,1,i] will point in the direction of S0. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.\n\nThis function is not available for systems with reshaped unit cells.\n\nSee also set_spiral_order!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_vacancy_at!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.set_vacancy_at!","text":"set_vacancy_at!(sys::System, site::Site)\n\nMake a single site nonmagnetic. Site includes a unit cell and a sublattice index.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.slice_2D_binning_parameters-Tuple{Vector{Float64}, Any, Any, Int64, Any}","page":"Library API","title":"Sunny.slice_2D_binning_parameters","text":"slice_2D_binning_parameter(sc::SampledCorrelations, cut_from_q, cut_to_q, cut_bins::Int64, cut_width::Float64; plane_normal = [0,0,1],cut_height = cutwidth)\n\nCreates BinningParameters which make a cut along one dimension of Q-space.\n\nThe x-axis of the resulting histogram consists of cut_bins-many bins ranging from cut_from_q to cut_to_q. The width of the bins in the transverse direciton is controlled by cut_width and cut_height.\n\nThe binning in the transverse directions is defined in the following way, which sets their normalization and orthogonality properties:\n\ncut_covector = normalize(cut_to_q - cut_from_q)\ntransverse_covector = normalize(plane_normal × cut_covector)\ncotransverse_covector = normalize(transverse_covector × cut_covector)\n\nIn other words, the axes are orthonormal with respect to the Euclidean metric.\n\nIf the cut is too narrow, there will be very few scattering vectors per bin, or the number per bin will vary substantially along the cut. If the output appears under-resolved, try increasing cut_width.\n\nThe four axes of the resulting histogram are:\n\nAlong the cut\nFist transverse Q direction\nSecond transverse Q direction\nEnergy\n\nThis function can be used without reference to a SampledCorrelations using this alternate syntax to manually specify the bin centers for the energy axis:\n\nslice_2D_binning_parameter(ω_bincenters, cut_from, cut_to,...)\n\nwhere ω_bincenters specifies the energy axis, and both cut_from and cut_to are arbitrary covectors, in any units.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.spin_label-Tuple{System, Int64}","page":"Library API","title":"Sunny.spin_label","text":"spin_label(sys::System, i::Int)\n\nIf atom i carries a single spin-S moment, then returns the half-integer label S. Otherwise, throws an error.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.spin_matrices-Tuple{Any}","page":"Library API","title":"Sunny.spin_matrices","text":"spin_matrices(S)\n\nReturns a triple of NN spin matrices, where N = 2S+1. These are the generators of SU(2) in the spin-S representation.\n\nIf S == Inf, then the return values are abstract symbols denoting infinite-dimensional matrices that commute. These can be useful for repeating historical studies, or modeling micromagnetic systems. A technical discussion appears in the Sunny documentation page: Interaction Strength Renormalization.\n\nExample\n\nS = spin_matrices(3/2)\n@assert S'*S ≈ (3/2)*(3/2+1)*I\n@assert S[1]*S[2] - S[2]*S[1] ≈ im*S[3]\n\nS = spin_matrices(Inf)\n@assert S[1]*S[2] - S[2]*S[1] == 0\n\nSee also print_stevens_expansion.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.step!","page":"Library API","title":"Sunny.step!","text":"step!(sys::System, dynamics)\n\nAdvance the spin configuration one dynamical time-step. The dynamics object may be a continuous spin dynamics, such as Langevin or ImplicitMidpoint, or it may be a discrete Monte Carlo sampling scheme such as LocalSampler.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.stevens_matrices-Tuple{Any}","page":"Library API","title":"Sunny.stevens_matrices","text":"stevens_matrices(S)\n\nReturns a generator of Stevens operators in the spin-S representation. The return value O can be indexed as O[k,q], where 0 k 6 labels an irrep of SO(3) and -k q k. This will produce an NN matrix where ``N = 2S\n\n1``. Linear combinations of Stevens operators can be used as a \"physical\n\nbasis\" for decomposing local observables. To see this decomposition, use print_stevens_expansion.\n\nIf S == Inf, then symbolic operators will be returned. In this infinite dimensional limit, the Stevens operators become homogeneous polynomials of commuting spin operators.\n\nExample\n\nO = stevens_matrices(2)\nS = spin_matrices(2)\n\nA = (1/20)O[4,0] + (1/4)O[4,4] + (102/5)I\nB = S[1]^4 + S[2]^4 + S[3]^4\n@assert A ≈ B\n\nSee also spin_matrices and Interaction Strength Renormalization.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.subcrystal-Union{Tuple{N}, Tuple{Crystal, Vararg{String, N}}} where N","page":"Library API","title":"Sunny.subcrystal","text":"subcrystal(cryst, types) :: Crystal\n\nFilters sublattices of a Crystal by atom types, keeping the space group unchanged.\n\nsubcrystal(cryst, classes) :: Crystal\n\nFilters sublattices of Crystal by equivalence classes, keeping the space group unchanged.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.suggest_magnetic_supercell-Tuple{Any}","page":"Library API","title":"Sunny.suggest_magnetic_supercell","text":"suggest_magnetic_supercell(qs; tol=1e-12, maxsize=100)\n\nSuggests a magnetic supercell, in units of the crystal lattice vectors, that is consistent with periodicity of the wavevectors qs in RLU. If the wavevectors are incommensurate (with respect to the maximum supercell size maxsize), one can select a larger error tolerance tol to find a supercell that is almost commensurate.\n\nPrints a 33 matrix of integers that is suitable for use in reshape_supercell.\n\nExamples\n\n# A magnetic supercell for a single-Q structure. Will print\nq1 = [0, -1/4, 1/4]\nsuggest_magnetic_supercell([q1]) # [1 0 0; 0 2 1; 0 -2 1]\n\n# A larger magnetic supercell for a double-Q structure\nq2 = [1/4, 0, 1/4]\nsuggest_magnetic_supercell([q1, q2]) # [1 2 2; -1 2 -2; -1 2 2]\n\n# If given incommensurate wavevectors, find an approximate supercell that\n# is exactly commensurate for nearby wavevectors.\nsuggest_magnetic_supercell([[0, 0, 1/√5], [0, 0, 1/√7]]; tol=1e-2)\n\n# This prints [1 0 0; 0 1 0; 0 0 16], which becomes commensurate under the\n# approximations `1/√5 ≈ 7/16` and `1/√7 ≈ 3/8`.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.symmetry_equivalent_bonds-Tuple{System, Bond}","page":"Library API","title":"Sunny.symmetry_equivalent_bonds","text":"symmetry_equivalent_bonds(sys::System, bond::Bond)\n\nGiven a Bond for the original (unreshaped) crystal, return all symmetry equivalent bonds in the System. Each returned bond is represented as a pair of Sites, which may be used as input to set_exchange_at!. Reverse bonds are not included (no double counting).\n\nExample\n\nfor (site1, site2, offset) in symmetry_equivalent_bonds(sys, bond)\n @assert site1 < site2\n set_exchange_at!(sys, J, site1, site2; offset)\nend\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.to_inhomogeneous-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.to_inhomogeneous","text":"to_inhomogeneous(sys::System)\n\nReturns a copy of the system that allows for inhomogeneous interactions, which can be set using set_onsite_coupling_at!, set_exchange_at!, and set_vacancy_at!.\n\nInhomogeneous systems do not support symmetry-propagation of interactions or system reshaping.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.to_product_space-Tuple{Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.to_product_space","text":"to_product_space(A, B, Cs...)\n\nGiven lists of operators acting on local Hilbert spaces individually, return the corresponding operators that act on the tensor product space. In typical usage, the inputs will represent local physical observables and the outputs will be used to define quantum couplings.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.unit_resolution_binning_parameters-Tuple{Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.unit_resolution_binning_parameters","text":"unit_resolution_binning_parameters(sc::SampledCorrelations)\n\nCreate BinningParameters which place one histogram bin centered at each possible (q,ω) scattering vector of the crystal. This is the finest possible binning without creating bins with zero scattering vectors in them.\n\nThis function can be used without reference to a SampledCorrelations using an alternate syntax to manually specify the bin centers for the energy axis and the lattice size:\n\nunit_resolution_binning_parameters(ω_bincenters,latsize,[reciprocal lattice vectors])\n\nThe last argument may be a 3x3 matrix specifying the reciprocal lattice vectors, or a Crystal.\n\nLastly, binning parameters for a single axis may be specifed by their bin centers:\n\n(binstart,binend,binwidth) = unit_resolution_binning_parameters(bincenters::Vector{Float64})\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.@mix_proposals-Tuple","page":"Library API","title":"Sunny.@mix_proposals","text":"@mix_proposals weight1 propose1 weight2 propose2 ...\n\nMacro to generate a proposal function that randomly selects among the provided functions according to the provided probability weights. For use with LocalSampler.\n\nExample\n\n# A proposal function that proposes a spin flip 40% of the time, and a\n# Gaussian perturbation 60% of the time.\n@mix_proposals 0.4 propose_flip 0.6 propose_delta(0.2)\n\n\n\n\n\n","category":"macro"},{"location":"library.html#Optional-Makie-extensions","page":"Library API","title":"Optional Makie extensions","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"The following will be enabled through a package extension if either GLMakie or WGLMakie is loaded.","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"plot_spins\nview_crystal","category":"page"},{"location":"library.html#Sunny.plot_spins","page":"Library API","title":"Sunny.plot_spins","text":"plot_spins(sys::System; arrowscale=1.0, color=:red, colorfn=nothing,\n colormap=:viridis, colorrange=nothing, show_cell=true, orthographic=false,\n ghost_radius=0, dims=3\n\nPlot the spin configuration defined by sys. Optional parameters are:\n\narrowscale: Scale all arrows by dimensionless factor.\ncolor: Arrow colors. May be symbolic or numeric. If scalar, will be shared among all sites.\ncolorfn: Function that dynamically maps from a site index to a numeric color value. Useful for animations.\ncolormap, colorrange: Used to populate colors from numbers following Makie conventions.\nshow_cell: Show original crystallographic unit cell.\northographic: Use camera with orthographic projection.\nghost_radius: Show translucent periodic images up to a given distance (length units).\ndims: Spatial dimensions of system (1, 2, or 3).\n\nCalling notify on the return value will animate the figure.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.view_crystal","page":"Library API","title":"Sunny.view_crystal","text":"view_crystal(crystal::Crystal, max_dist::Real; show_axis=true, orthographic=false)\n\nAn interactive crystal viewer, with bonds up to max_dist.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Optional-WriteVTK-extensions","page":"Library API","title":"Optional WriteVTK extensions","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"The following will be enabled through a package extension if WriteVTK is loaded.","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"export_vtk","category":"page"},{"location":"library.html#Sunny.export_vtk","page":"Library API","title":"Sunny.export_vtk","text":"export_vtk(filename,params::BinningParameters,data)\n\nExport a VTK-compatible file to filename (do not include file extension when specifying the file name) which contains the data as VTK Cell Data on a grid parameterized by params.\n\nAt least one axis of the BinningParameters must be integrated over, since VTK does not support 4D data. See integrate_axes!.\n\n\n\n\n\n","category":"function"},{"location":"examples/contributed/MgCr2O4-tutorial.html#MgCr2O4-at-Finite-Temperature","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Author: Martin Mourigal Date: September 9, 2022 (Updated by October 28, 2023 using Sunny 0.5.5)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"In this tutorial, we will walk through an example in Sunny and calculate the spin dynamical properties of the Heisenberg pyrochlore antiferromagnet and apply this knowledge to MgCr2O4 and ZnCr2O4, which are known to approximate this model. Relevant publications include:","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"[1] P. H. Conlon and J. T. Chalker, Phys. Rev. Lett. 102, 237206 (2009)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"[2] P. H. Conlon and J. T. Chalker, Phys. Rev. B 81, 224413 (2010)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"[3] X. Bai, J. A. M. Paddison, et al. Phys. Rev. Lett. 122, 097201 (2019)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Setting-up-Julia","page":"MgCr2O4 at Finite Temperature","title":"Setting up Julia","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To run the examples in the tutorial, you will need a working installation of the Julia programming language and the Sunny package. Some useful references for getting started are:","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Getting started with Julia for Sunny\nSunny Docs","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We will begin by loading the relevant packages.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"using Sunny # The main package\nusing GLMakie # Plotting package","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Setting-up-the-crystal-structure","page":"MgCr2O4 at Finite Temperature","title":"Setting up the crystal structure","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Before specifying the interactions of our system, we first must set up the crystal. We will begin by specifying the pyrochlore lattice (illustrated below) in the manner that is typical of theorists.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Picture Credits: Theory of Quantum Matter Unit, OIST","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#\"Theorist\"-Method","page":"MgCr2O4 at Finite Temperature","title":"\"Theorist\" Method","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"In this approach, we directly define the lattice vectors and specify the position of each atom in fractional coordinates.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"latvecs = lattice_vectors(8.3342, 8.3342, 8.3342, 90, 90, 90)\npositions = [[0.875, 0.625, 0.375],\n [0.625, 0.125, 0.625],\n [0.875, 0.875, 0.125],\n [0.625, 0.875, 0.375],\n [0.875, 0.125, 0.875],\n [0.625, 0.625, 0.125],\n [0.875, 0.375, 0.625],\n [0.625, 0.375, 0.875],\n [0.375, 0.625, 0.875],\n [0.125, 0.125, 0.125],\n [0.375, 0.875, 0.625],\n [0.125, 0.875, 0.875],\n [0.375, 0.125, 0.375],\n [0.125, 0.625, 0.625],\n [0.375, 0.375, 0.125],\n [0.125, 0.375, 0.375]];\ntypes = [\"B\" for _ in positions]\nxtal_pyro = Crystal(latvecs, positions; types) # We will call this crystal the Theoretical Pyrochlore","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'F d -3 m' (227)\nLattice params a=8.334, b=8.334, c=8.334, α=90°, β=90°, γ=90°\nCell volume 578.9\nType 'B', Wyckoff 16c (point group '.-3m'):\n 1. [7/8, 5/8, 3/8]\n 2. [5/8, 1/8, 5/8]\n 3. [7/8, 7/8, 1/8]\n 4. [5/8, 7/8, 3/8]\n 5. [7/8, 1/8, 7/8]\n 6. [5/8, 5/8, 1/8]\n 7. [7/8, 3/8, 5/8]\n 8. [5/8, 3/8, 7/8]\n 9. [3/8, 5/8, 7/8]\n 10. [1/8, 1/8, 1/8]\n 11. [3/8, 7/8, 5/8]\n 12. [1/8, 7/8, 7/8]\n 13. [3/8, 1/8, 3/8]\n 14. [1/8, 5/8, 5/8]\n 15. [3/8, 3/8, 1/8]\n 16. [1/8, 3/8, 3/8]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To examine the result interactively, we can call view_crystal.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"view_crystal(xtal_pyro, 3.2)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#\"Experimentalist\"-Method-#1-(Incorrect)","page":"MgCr2O4 at Finite Temperature","title":"\"Experimentalist\" Method #1 (Incorrect)","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"A real crystal is more complicated than this, however, and we will now construct the system using the actual CIF file of MgCr2O4 from ICSD. This can be done by copying over the data from a CIF file by hand, but this can be dangerous, as shown below.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"latvecs = lattice_vectors(8.3342, 8.3342, 8.3342, 90, 90, 90)\npositions = [[0.1250, 0.1250, 0.1250],\n [0.5000, 0.5000, 0.5000],\n [0.2607, 0.2607, 0.2607]]\ntypes = [\"Mg\",\"Cr\",\"O\"]\nxtal_mgcro_1 = Crystal(latvecs, positions; types)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'R 3 m' (160)\nLattice params a=8.334, b=8.334, c=8.334, α=90°, β=90°, γ=90°\nCell volume 578.9\nType 'Mg', Wyckoff 3a (point group '3m'):\n 1. [1/8, 1/8, 1/8]\nType 'Cr', Wyckoff 3a (point group '3m'):\n 2. [1/2, 1/2, 1/2]\nType 'O', Wyckoff 3a (point group '3m'):\n 3. [0.2607, 0.2607, 0.2607]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Sunny returned a valid crystal, but it did get right space group for MgCr2O4. This can be fixed by modifying the input to include the space group and the setting.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#\"Experimentalist\"-Method-#2-(Correct)","page":"MgCr2O4 at Finite Temperature","title":"\"Experimentalist\" Method #2 (Correct)","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"As above, we will define the crystal structure of MgCr2O4 by copying the info from a CIF file, but we will also specify the space group and setting explicitly.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"latvecs = lattice_vectors(8.3342, 8.3342, 8.3342, 90, 90, 90)\npositions = [[0.1250, 0.1250, 0.1250],\n [0.5000, 0.5000, 0.5000],\n [0.2607, 0.2607, 0.2607]]\ntypes = [\"Mg\",\"Cr\",\"O\"]\nspacegroup = 227 # Space Group Number\nsetting = \"2\" # Space Group setting\nxtal_mgcro_2 = Crystal(latvecs, positions, spacegroup; types, setting)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'F d -3 m' (227)\nLattice params a=8.334, b=8.334, c=8.334, α=90°, β=90°, γ=90°\nCell volume 578.9\nType 'Mg', Wyckoff 8b (point group '-43m'):\n 1. [1/8, 1/8, 1/8]\n 2. [5/8, 5/8, 1/8]\n 3. [7/8, 3/8, 3/8]\n 4. [3/8, 7/8, 3/8]\n 5. [5/8, 1/8, 5/8]\n 6. [1/8, 5/8, 5/8]\n 7. [3/8, 3/8, 7/8]\n 8. [7/8, 7/8, 7/8]\nType 'Cr', Wyckoff 16c (point group '.-3m'):\n 9. [1/2, 0, 0]\n 10. [3/4, 1/4, 0]\n 11. [0, 1/2, 0]\n 12. [1/4, 3/4, 0]\n 13. [3/4, 0, 1/4]\n 14. [1/2, 1/4, 1/4]\n 15. [1/4, 1/2, 1/4]\n 16. [0, 3/4, 1/4]\n 17. [0, 0, 1/2]\n 18. [1/4, 1/4, 1/2]\n 19. [1/2, 1/2, 1/2]\n 20. [3/4, 3/4, 1/2]\n 21. [1/4, 0, 3/4]\n 22. [0, 1/4, 3/4]\n 23. [3/4, 1/2, 3/4]\n 24. [1/2, 3/4, 3/4]\nType 'O', Wyckoff 32e (point group '.3m'):\n 25. [0.7393, 0.0107, 0.0107]\n 26. [0.5107, 0.2393, 0.0107]\n 27. [0.2393, 0.5107, 0.0107]\n 28. [0.0107, 0.7393, 0.0107]\n 29. [0.5107, 0.0107, 0.2393]\n 30. [0.7393, 0.2393, 0.2393]\n 31. [0.0107, 0.5107, 0.2393]\n 32. [0.2393, 0.7393, 0.2393]\n 33. [0.2607, 0.2607, 0.2607]\n 34. [0.4893, 0.4893, 0.2607]\n 35. [0.7607, 0.7607, 0.2607]\n 36. [0.9893, 0.9893, 0.2607]\n 37. [0.4893, 0.2607, 0.4893]\n 38. [0.2607, 0.4893, 0.4893]\n 39. [0.9893, 0.7607, 0.4893]\n 40. [0.7607, 0.9893, 0.4893]\n 41. [0.2393, 0.0107, 0.5107]\n 42. [0.0107, 0.2393, 0.5107]\n 43. [0.7393, 0.5107, 0.5107]\n 44. [0.5107, 0.7393, 0.5107]\n 45. [0.0107, 0.0107, 0.7393]\n 46. [0.2393, 0.2393, 0.7393]\n 47. [0.5107, 0.5107, 0.7393]\n 48. [0.7393, 0.7393, 0.7393]\n 49. [0.7607, 0.2607, 0.7607]\n 50. [0.9893, 0.4893, 0.7607]\n 51. [0.2607, 0.7607, 0.7607]\n 52. [0.4893, 0.9893, 0.7607]\n 53. [0.9893, 0.2607, 0.9893]\n 54. [0.7607, 0.4893, 0.9893]\n 55. [0.4893, 0.7607, 0.9893]\n 56. [0.2607, 0.9893, 0.9893]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"This result is correct, but at this point we might as well import the CIF file directly, which we now proceed to do.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#\"Experimentalist\"-Method-#3-(Correct-–-if-your-CIF-file-is)","page":"MgCr2O4 at Finite Temperature","title":"\"Experimentalist\" Method #3 (Correct – if your CIF file is)","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To import a CIF file, simply give the path to Crystal. One may optionally specify a precision parameter to apply to the symmetry analysis.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"cif = pkgdir(Sunny, \"examples\", \"longer_examples\", \"MgCr2O4_160953_2009.cif\")\nxtal_mgcro_3 = Crystal(cif; symprec=0.001)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'F d -3 m' (227)\nLattice params a=8.333, b=8.333, c=8.333, α=90°, β=90°, γ=90°\nCell volume 578.6\nType 'Mg1', Wyckoff 8b (point group '-43m'):\n 1. [1/8, 1/8, 1/8]\n 2. [5/8, 5/8, 1/8]\n 3. [7/8, 3/8, 3/8]\n 4. [3/8, 7/8, 3/8]\n 5. [5/8, 1/8, 5/8]\n 6. [1/8, 5/8, 5/8]\n 7. [3/8, 3/8, 7/8]\n 8. [7/8, 7/8, 7/8]\nType 'Cr1', Wyckoff 16c (point group '.-3m'):\n 9. [1/2, 0, 0]\n 10. [3/4, 1/4, 0]\n 11. [0, 1/2, 0]\n 12. [1/4, 3/4, 0]\n 13. [3/4, 0, 1/4]\n 14. [1/2, 1/4, 1/4]\n 15. [1/4, 1/2, 1/4]\n 16. [0, 3/4, 1/4]\n 17. [0, 0, 1/2]\n 18. [1/4, 1/4, 1/2]\n 19. [1/2, 1/2, 1/2]\n 20. [3/4, 3/4, 1/2]\n 21. [1/4, 0, 3/4]\n 22. [0, 1/4, 3/4]\n 23. [3/4, 1/2, 3/4]\n 24. [1/2, 3/4, 3/4]\nType 'O1', Wyckoff 32e (point group '.3m'):\n 25. [0.7388, 0.0112, 0.0112]\n 26. [0.5112, 0.2388, 0.0112]\n 27. [0.2388, 0.5112, 0.0112]\n 28. [0.0112, 0.7388, 0.0112]\n 29. [0.5112, 0.0112, 0.2388]\n 30. [0.7388, 0.2388, 0.2388]\n 31. [0.0112, 0.5112, 0.2388]\n 32. [0.2388, 0.7388, 0.2388]\n 33. [0.2612, 0.2612, 0.2612]\n 34. [0.4888, 0.4888, 0.2612]\n 35. [0.7612, 0.7612, 0.2612]\n 36. [0.9888, 0.9888, 0.2612]\n 37. [0.4888, 0.2612, 0.4888]\n 38. [0.2612, 0.4888, 0.4888]\n 39. [0.9888, 0.7612, 0.4888]\n 40. [0.7612, 0.9888, 0.4888]\n 41. [0.2388, 0.0112, 0.5112]\n 42. [0.0112, 0.2388, 0.5112]\n 43. [0.7388, 0.5112, 0.5112]\n 44. [0.5112, 0.7388, 0.5112]\n 45. [0.0112, 0.0112, 0.7388]\n 46. [0.2388, 0.2388, 0.7388]\n 47. [0.5112, 0.5112, 0.7388]\n 48. [0.7388, 0.7388, 0.7388]\n 49. [0.7612, 0.2612, 0.7612]\n 50. [0.9888, 0.4888, 0.7612]\n 51. [0.2612, 0.7612, 0.7612]\n 52. [0.4888, 0.9888, 0.7612]\n 53. [0.9888, 0.2612, 0.9888]\n 54. [0.7612, 0.4888, 0.9888]\n 55. [0.4888, 0.7612, 0.9888]\n 56. [0.2612, 0.9888, 0.9888]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Finally, we wish to restrict attention to the magnetic atoms in the unit cell while maintaining symmetry information for the full crystal, which is required to determine the correct exchange and g-factor anisotropies. This can be achieved with the subcrystal function.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"xtal_mgcro = subcrystal(xtal_mgcro_2,\"Cr\")","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'F d -3 m' (227)\nLattice params a=8.334, b=8.334, c=8.334, α=90°, β=90°, γ=90°\nCell volume 578.9\nType 'Cr', Wyckoff 16c (point group '.-3m'):\n 1. [1/2, 0, 0]\n 2. [3/4, 1/4, 0]\n 3. [0, 1/2, 0]\n 4. [1/4, 3/4, 0]\n 5. [3/4, 0, 1/4]\n 6. [1/2, 1/4, 1/4]\n 7. [1/4, 1/2, 1/4]\n 8. [0, 3/4, 1/4]\n 9. [0, 0, 1/2]\n 10. [1/4, 1/4, 1/2]\n 11. [1/2, 1/2, 1/2]\n 12. [3/4, 3/4, 1/2]\n 13. [1/4, 0, 3/4]\n 14. [0, 1/4, 3/4]\n 15. [3/4, 1/2, 3/4]\n 16. [1/2, 3/4, 3/4]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Making-a-System-and-assigning-interactions","page":"MgCr2O4 at Finite Temperature","title":"Making a System and assigning interactions","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Making-a-System","page":"MgCr2O4 at Finite Temperature","title":"Making a System","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Before assigning any interactions, we first have to set up a System using our Crystal.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"dims = (6, 6, 6) # Supercell dimensions\nspininfos = [SpinInfo(1, S=3/2, g=2)] # Specify spin information, note that all sites are symmetry equivalent\nsys_pyro = System(xtal_pyro, dims, spininfos, :dipole) # Make a system in dipole (Landau-Lifshitz) mode on pyrochlore lattice\nsys_mgcro = System(xtal_mgcro, dims, spininfos, :dipole); # Same on MgCr2O4 crystal","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To understand what interactions we may assign to this system, we have to understand the the symmetry properties of the crystal, which we turn to next.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Symmetry-analysis-for-exchange-and-single-ion-anisotropies","page":"MgCr2O4 at Finite Temperature","title":"Symmetry analysis for exchange and single-ion anisotropies","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"print_symmetry_table reports all the exchange interactions, single-site anisotropies, and g-factors allowed on our crystal. It takes a Cyrstal and a distance. We'll look at both the \"theorist's\" pyrochlore lattice,","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"print_symmetry_table(xtal_pyro, 5.9)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Atom 1\nType 'B', position [7/8, 5/8, 3/8], multiplicity 16\nAllowed g-tensor: [ A B -B\n B A B\n -B B A]\nAllowed anisotropy in Stevens operators:\n c₁*(𝒪[2,-2]+2𝒪[2,-1]-2𝒪[2,1]) +\n c₂*(7𝒪[4,-3]+2𝒪[4,-2]-𝒪[4,-1]+𝒪[4,1]+7𝒪[4,3]) + c₃*(𝒪[4,0]+5𝒪[4,4]) +\n c₄*(-11𝒪[6,-6]-8𝒪[6,-3]+𝒪[6,-2]-8𝒪[6,-1]+8𝒪[6,1]-8𝒪[6,3]) + c₅*(𝒪[6,0]-21𝒪[6,4]) + c₆*((9/5)𝒪[6,-6]+(24/5)𝒪[6,-5]+𝒪[6,-2]+(8/5)𝒪[6,-1]-(8/5)𝒪[6,1]-(24/5)𝒪[6,5])\n\nSunny.Bond(1, 3, [0, 0, 0])\nDistance 2.94658466788246, coordination 6\nConnects 'B' at [7/8, 5/8, 3/8] to 'B' at [7/8, 7/8, 1/8]\nAllowed exchange matrix:[ A -D D\n D B C\n -D C B]\nAllowed DM vector: [0 -D -D]\n\nSunny.Bond(1, 2, [0, 0, 0])\nDistance 5.10363435357589, coordination 12\nConnects 'B' at [7/8, 5/8, 3/8] to 'B' at [5/8, 1/8, 5/8]\nAllowed exchange matrix:[ A C-E D-F\n C+E B -C+E\n D+F -C-E A]\nAllowed DM vector: [E F -E]\n\nSunny.Bond(2, 6, [0, 0, 0])\nDistance 5.89316933576492, coordination 6\nConnects 'B' at [5/8, 1/8, 5/8] to 'B' at [5/8, 5/8, 1/8]\nAllowed exchange matrix:[A D D\n D B C\n D C B]\n\nSunny.Bond(1, 5, [0, 0, 0])\nDistance 5.89316933576492, coordination 6\nConnects 'B' at [7/8, 5/8, 3/8] to 'B' at [7/8, 1/8, 7/8]\nAllowed exchange matrix:[ A D -D\n D B C\n -D C B]\n\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"and for the the MgCrO4 crystal,","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"print_symmetry_table(xtal_mgcro, 6.0)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Atom 1\nType 'Cr', position [1/2, 0, 0], multiplicity 16\nAllowed g-tensor: [A B B\n B A B\n B B A]\nAllowed anisotropy in Stevens operators:\n c₁*(𝒪[2,-2]+2𝒪[2,-1]+2𝒪[2,1]) +\n c₂*(-7𝒪[4,-3]-2𝒪[4,-2]+𝒪[4,-1]+𝒪[4,1]+7𝒪[4,3]) + c₃*(𝒪[4,0]+5𝒪[4,4]) +\n c₄*(-11𝒪[6,-6]-8𝒪[6,-3]+𝒪[6,-2]-8𝒪[6,-1]-8𝒪[6,1]+8𝒪[6,3]) + c₅*(𝒪[6,0]-21𝒪[6,4]) + c₆*((9/5)𝒪[6,-6]+(24/5)𝒪[6,-5]+𝒪[6,-2]+(8/5)𝒪[6,-1]+(8/5)𝒪[6,1]+(24/5)𝒪[6,5])\n\nSunny.Bond(1, 2, [0, 0, 0])\nDistance 2.94658466788246, coordination 6\nConnects 'Cr' at [1/2, 0, 0] to 'Cr' at [3/4, 1/4, 0]\nAllowed exchange matrix:[A C -D\n C A -D\n D D B]\nAllowed DM vector: [-D D 0]\n\nSunny.Bond(1, 7, [0, 0, 0])\nDistance 5.10363435357589, coordination 12\nConnects 'Cr' at [1/2, 0, 0] to 'Cr' at [1/4, 1/2, 1/4]\nAllowed exchange matrix:[ A C-E D-F\n C+E B -C+E\n D+F -C-E A]\nAllowed DM vector: [E F -E]\n\nSunny.Bond(1, 3, [0, 0, 0])\nDistance 5.89316933576492, coordination 6\nConnects 'Cr' at [1/2, 0, 0] to 'Cr' at [0, 1/2, 0]\nAllowed exchange matrix:[A D C\n D A C\n C C B]\n\nSunny.Bond(1, 3, [1, 0, 0])\nDistance 5.89316933576492, coordination 6\nConnects 'Cr' at [1/2, 0, 0] to 'Cr' at [1, 1/2, 0]\nAllowed exchange matrix:[A D C\n D A C\n C C B]\n\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Note that the exchange anisotropies allowed on the the pyrochlore lattice are slightly different from those on the MgCr2O4 cyrstal due to the role of Mg and O in the bonds. Also note that Sunny has correctly identified the two inequivalent bonds 3a and 3b having the same length. A question may arises to know which bond is J3a and which is J3b, let's plot the structure.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"view_crystal(xtal_mgcro, 5.9)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"The crystal viewer shows that the second interaction – cyan color with distance of 5.89Å – is in fact the one hopping through a chromium site, meaning it is J3a! We will need to be careful with that later.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Building-the-exchange-interactions-for-our-system","page":"MgCr2O4 at Finite Temperature","title":"Building the exchange interactions for our system","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We begin by setting the scale of our exchange interactions on each bond.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"J1 = 3.27 # value of J1 in meV from Bai's PRL paper\nJ_pyro = [1.00,0.000,0.000,0.000]*J1 # pure nearest neighbor pyrochlore\nJ_mgcro = [1.00,0.0815,0.1050,0.085]*J1; # further neighbor pyrochlore relevant for MgCr2O4\n# val_J_mgcro = [1.00,0.000,0.025,0.025]*val_J1; # this is a funny setting from Conlon-Chalker","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"These values are then assigned to their corresponding bonds with set_exchange!.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"# === Assign exchange interactions to pyrochlore system ===\nset_exchange!(sys_pyro, J_pyro[1], Bond(1, 3, [0,0,0])) # J1\nset_exchange!(sys_pyro, J_pyro[2], Bond(1, 2, [0,0,0])) # J2\nset_exchange!(sys_pyro, J_pyro[3], Bond(2, 6, [0,0,0])) # J3a\nset_exchange!(sys_pyro, J_pyro[4], Bond(1, 5, [0,0,0])) # J3b\n\n# === Assign exchange interactions to MgCr2O4 system ===\nset_exchange!(sys_mgcro, J_mgcro[1], Bond(1, 2, [0,0,0])) # J1\nset_exchange!(sys_mgcro, J_mgcro[2], Bond(1, 7, [0,0,0])) # J2\nset_exchange!(sys_mgcro, J_mgcro[3], Bond(1, 3, [0,0,0])) # J3a -- Careful here!\nset_exchange!(sys_mgcro, J_mgcro[4], Bond(1, 3, [1,0,0])); # J3b -- And here!","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We will not be assigning any single-ion anisotropies, so we have finished specifying our models. For good measure, we will set both systems to a random (infinite temperature) initial condition.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"randomize_spins!(sys_pyro)\nrandomize_spins!(sys_mgcro);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Cooling-our-System-amd-calculating-the-instantaneous-and-dynamic-structure-factors-at-the-final-temperature","page":"MgCr2O4 at Finite Temperature","title":"Cooling our System amd calculating the instantaneous and dynamic structure factors at the final temperature","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We begin by thermalizing our system at a particular temperature. We will accomplish this by running Langevin dynamics. To do this, we must set up a Langevin integrator.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Δt = 0.05 # Integration time step in meV^-1\nλ = 0.1 # Phenomenological damping parameter\nkT = 1.8 # Desired temperature in meV\nlangevin = Langevin(Δt; λ, kT); # Construct integrator","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We can now thermalize our systems by running the integrator.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"for _ in 1:2000\n step!(sys_pyro, langevin)\n step!(sys_mgcro, langevin)\nend","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"As a sanity check, we'll plot the real-space spin configurations of both systems after themalization. First the pyrochlore,","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"plot_spins(sys_pyro)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"and then the MgCr2O4,","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"plot_spins(sys_mgcro)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Instantaneous-Structure-Factor","page":"MgCr2O4 at Finite Temperature","title":"Instantaneous Structure Factor","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Next we can examine the instantaneous structure factor.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"isf_pyro = instant_correlations(sys_pyro)\nisf_mgcro = instant_correlations(sys_mgcro);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"These are currently empty. Let's add correlation data from 10 trajectories.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"for _ in 1:10\n # Run dynamics to decorrelate\n for _ in 1:500\n step!(sys_pyro, langevin)\n step!(sys_mgcro, langevin)\n end\n # Add samples\n add_sample!(isf_pyro, sys_pyro)\n add_sample!(isf_mgcro, sys_mgcro)\nend","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To retrieve the intensities, we set up a formula and call intensities_interpolated on an array of wave vectors.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"qvals = -4.0:0.025:4.0\nqs = [(qa, qb, 0) for qa in qvals, qb in qvals] # Wave vectors to query\n\nformula_pyro = intensity_formula(isf_pyro, :perp)\nformula_mgcro = intensity_formula(isf_mgcro, :perp)\nSq_pyro = instant_intensities_interpolated(isf_pyro, qs, formula_pyro)\nSq_mgcro = instant_intensities_interpolated(isf_mgcro, qs, formula_mgcro);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Finally, we can plot the results.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"fig = Figure(; resolution=(1200,500))\naxparams = (aspect = true, xticks=-4:4, yticks=-4:4, titlesize=20,\n xlabel = \"𝐪a (2π a⁻¹)\", ylabel = \"𝐪b (2π b⁻¹)\", xlabelsize = 18, ylabelsize=18,)\nax_pyro = Axis(fig[1,1]; title=\"Pyrochlore\", axparams...)\nax_mgcro = Axis(fig[1,3]; title=\"MgCr2O4\", axparams...)\nhm = heatmap!(ax_pyro, qvals, qvals, Sq_pyro)\nColorbar(fig[1,2], hm)\nhm = heatmap!(ax_mgcro, qvals, qvals, Sq_mgcro)\nColorbar(fig[1,4], hm)\nfig","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Dynamical-Structure-Factor","page":"MgCr2O4 at Finite Temperature","title":"Dynamical Structure Factor","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We can also estimate the dynamical structure factor.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"sc_pyro = dynamical_correlations(sys_pyro; Δt, ωmax = 10.0, nω = 100)\nsc_mgcro = dynamical_correlations(sys_mgcro; Δt, ωmax = 10.0, nω = 100);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Next we add some sample trajectories.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"for _ in 1:3\n # Run dynamics to decorrelate\n for _ in 1:500\n step!(sys_pyro, langevin)\n step!(sys_mgcro, langevin)\n end\n # Add samples\n add_sample!(sc_pyro, sys_pyro)\n add_sample!(sc_mgcro, sys_mgcro)\nend","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We can now examine the structure factor intensities along a path in momentum space.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"fig = Figure(; resolution=(1200,900))\naxsqw = (xticks=-4:4, yticks=0:2:10, ylabel=\"E (meV)\", ylabelsize=18, xlabelsize=18, )\n\nqbs = 0.0:0.5:1.5 # Determine q_b for each slice\nfor (i, qb) in enumerate(qbs)\n path, _ = reciprocal_space_path(xtal_pyro, [[-4.0, qb, 0.0],[4.0, qb, 0.0]], 40) # Generate a path of wave\n # vectors through the BZ\n formula = intensity_formula(sc_pyro, :perp; kT) # Temperature keyword enables intensity rescaling\n Sqω_pyro = intensities_interpolated(sc_pyro, path, formula)\n\n ax = Axis(fig[fldmod1(i,2)...]; xlabel = \"q = (x, $qb, 0)\", axsqw...)\n ωs = available_energies(sc_pyro)\n heatmap!(ax, [p[1] for p in path], ωs, Sqω_pyro; colorrange=(0.0, 4.0))\nend\nfig","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"And let's take a look at the same slices for MgCr2O4.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"fig = Figure(; resolution=(1200,900))\n\nqbs = 0.0:0.5:1.5\nfor (i, qb) in enumerate(qbs)\n path, _ = reciprocal_space_path(xtal_mgcro, [[-4.0, qb, 0.0],[4.0, qb, 0.0]], 40) # Generate a path of wave\n # vectors through the BZ\n formula = intensity_formula(sc_mgcro, :perp; kT) # Temperature keyword enables intensity rescaling\n Sqω_mgcro = intensities_interpolated(sc_mgcro, path, formula)\n\n ax = Axis(fig[fldmod1(i,2)...]; xlabel = \"q = (x, $qb, 0)\", axsqw...)\n ωs = available_energies(sc_mgcro)\n heatmap!(ax, [p[1] for p in path], ωs, Sqω_mgcro; colorrange=(0.0, 4.0))\nend\nfig","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Instantaneous-structure-factor-from-a-dynamical-structure-factor","page":"MgCr2O4 at Finite Temperature","title":"Instantaneous structure factor from a dynamical structure factor","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Finally, we note that the instant structure factor can be calculated from the dynamical structure factor. We simply call instant_intensities rather than intensities. This will calculate the instantaneous structure factor from from 𝒮(𝐪ω) by integrating out ω . An advantage of doing this (as opposed to using instant_correlations) is that Sunny is able to apply a temperature- and energy-dependent intensity rescaling before integrating out the dynamical information. The results of this approach are more suitable for comparison with experimental data.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"qvals = -4.0:0.05:4.0\nqs = [(qa, qb, 0) for qa in qvals, qb in qvals] # Wave vectors to query\n\nformula_pyro = intensity_formula(sc_pyro, :perp; kT) # Temperature keyword enables intensity rescaling\nformula_mgcro = intensity_formula(sc_mgcro, :perp; kT) # Temperature keyword enables intensity rescaling\n\nSq_pyro = instant_intensities_interpolated(sc_pyro, qs, formula_pyro)\nSq_mgcro = instant_intensities_interpolated(sc_mgcro, qs, formula_mgcro);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We can plot the results below. It is useful to compare these to the plot above generated with an instant_correlations.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"fig = Figure(; resolution=(1200,500))\nax_pyro = Axis(fig[1,1]; title=\"Pyrochlore\", axparams...)\nax_mgcro = Axis(fig[1,3]; title=\"MgCr2O4\", axparams...)\nhm = heatmap!(ax_pyro, qvals, qvals, Sq_pyro)\nColorbar(fig[1,2], hm)\nhm = heatmap!(ax_mgcro, qvals, qvals, Sq_mgcro)\nColorbar(fig[1,4], hm)\nfig","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Powder-Averaged-CoRhO-at-Finite-Temperature","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Inspired by: Ge et al., Phys. Rev. B 96, 064413 (2017)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Authors: Martin Mourigal, David Dahlbom","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Date: October 28, 2023 (Sunny 0.5.5)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Goal: This script is to calculate the temperature dependence of the magnon excitations in the spin-3/2 Heisenberg Diamond Antiferromagnet and compare to powder-averaged results obtained for the compound CoRh₂O₄","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Load-pacakges.","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Load pacakges.","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"using Sunny, GLMakie, ProgressMeter, Statistics, Random, Brillouin\ncif_path = pkgdir(Sunny, \"examples\", \"longer_examples\", \"CoRh2O4_#109301.cif\");","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Define-custom-functions.","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Define custom functions.","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"The function quench! randomizes the spins of a given System, fixes a target temperature, and lets the system relax at this temperature for nrelax integration steps.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function quench!(sys, integrator; kTtarget, nrelax)\n randomize_spins!(sys);\n integrator.kT = kTtarget;\n prog = Progress(nrelax; dt=10.0, desc=\"Quenched and now relaxing: \", color=:green);\n for _ in 1:nrelax\n step!(sys, integrator)\n next!(prog)\n end\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"quench! (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"dwell! takes a System, sets a target temperature, and has the system dwell at this temperature for ndwell integration steps.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function dwell!(sys, integrator; kTtarget, ndwell)\n integrator.kT = kTtarget;\n prog = Progress(ndwell; dt=10.0, desc=\"Dwelling: \", color=:green);\n for _ in 1:ndwell\n step!(sys, integrator)\n next!(prog)\n end\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"dwell! (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"anneal! takes a temperature schedule and cools the System through it, with ndwell steps of the integrator at each temperature in the schedule. Returns the energy at the end of the dwell for each scheduled temperature.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function anneal!(sys, integrator; kTschedule, ndwell)\n nspins = prod(size(sys.dipoles));\n ensys = zeros(length(kTschedule))\n prog = Progress(ndwell*length(kTschedule); dt=10.0, desc=\"Annealing: \", color=:red);\n for (i, kT) in enumerate(kTschedule)\n integrator.kT = kT\n for _ in 1:ndwell\n step!(sys, integrator)\n next!(prog)\n end\n ensys[i] = energy(sys)\n end\n return ensys/nspins\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"anneal! (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"sample_sf! samples a structure factor, which may be either an instant or dynamical structure factor. The integrator is run ndecorr times before each one of the samples is taken.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function sample_sf!(sf, sys, integrator; nsamples, ndecorr)\n prog = Progress(nsamples*ndecorr; dt=10.0, desc=\"Sampling SF: \", color=:red);\n for _ in 1:nsamples\n for _ in 1:ndecorr\n step!(sys, integrator)\n next!(prog)\n end\n add_sample!(sf, sys) # Accumulate the newly sampled structure factor into `sf`\n end\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"sample_sf! (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"powder_average powder averages a structure factor. Works for both instant and dynamical structure factors. To prevent smearing, removes Bragg peaks before introducing energy broadening. Bragg peaks are added back at ω=0 after broadening.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function powder_average(sc, rs, npts, formula; η=0.1)\n prog = Progress(length(rs); dt=10., desc=\"Powder Averaging: \", color=:blue)\n ωs = available_energies(sc)\n output = zeros(Float64, length(rs), length(ωs))\n for (i, r) in enumerate(rs)\n qs = reciprocal_space_shell(sc.crystal, r, npts)\n vals = intensities_interpolated(sc, qs, formula)\n bragg_idxs = findall(x -> x > maximum(vals)*0.9, vals)\n bragg_vals = vals[bragg_idxs]\n vals[bragg_idxs] .= 0\n vals = broaden_energy(sc, vals, (ω,ω₀)->lorentzian(ω-ω₀, η))\n vals[bragg_idxs] .= bragg_vals\n output[i,:] .= mean(vals, dims=1)[1,:]\n next!(prog)\n end\n return output\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"powder_average (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#System-Definition-for-CoRhO","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"System Definition for CoRh₂O₄","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Define the crystal structure of CoRh₂O₄ in the conventional cell.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"xtal = Crystal(cif_path; symprec=1e-4)\nmagxtal = subcrystal(xtal,\"Co1\")\nview_crystal(magxtal, 6.0)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Print the symmetry-allowed interactions.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"print_symmetry_table(magxtal, 4.0)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Atom 1\nType 'Co1', position [0, 0, 0], multiplicity 8\nAllowed g-tensor: [A 0 0\n 0 A 0\n 0 0 A]\nAllowed anisotropy in Stevens operators:\n c₁*(𝒪[4,0]+5𝒪[4,4]) +\n c₂*(𝒪[6,0]-21𝒪[6,4])\n\nSunny.Bond(1, 3, [0, 0, 0])\nDistance 3.6784429025744, coordination 4\nConnects 'Co1' at [0, 0, 0] to 'Co1' at [1/4, 1/4, 1/4]\nAllowed exchange matrix:[A B B\n B A B\n B B A]\n\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Assign local Hilbert space","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"S = 3/2\nlhs = [SpinInfo(1; S, g=2)]\nformfactors = [FormFactor(\"Co2\")];","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Create System and randomize it","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"sunmode = :dipole\nlatsize = (10,10,10)\nsys = System(magxtal, latsize, lhs, sunmode; seed=1)\nrandomize_spins!(sys)\nplot_spins(sys)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Define exchange interactions.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"scaleJ = 0.63\nvalJ1 = 1.00*scaleJ\nset_exchange!(sys, valJ1, Bond(1, 3, [0, 0, 0]));","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Thermalize-system-to-an-ordered,-yet-finite-temperature,-state","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Thermalize system to an ordered, yet finite temperature, state","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Define Langevin Integrator and Initialize it","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Δt0 = 0.05/abs(scaleJ*S); ## Time steps in Langevin\nλ0 = 0.1; ## Langevin damping, usually 0.05 or 0.1 is good.\nkT0 = 0.01*abs(scaleJ*S); ## Initialize at some temperature\nintegrator = Langevin(Δt0; λ=λ0, kT=kT0);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Thermalization Option 1: Quench the system from infinite temperature to a target temperature. Note: this may lead to a poorly thermalized sample","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"quench!(sys, integrator; kTtarget=kT0, nrelax=10000);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Option 2: Anneal (according to a temperature schedule) then dwell once we've reach base temperature. (Uncomment to execute.)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"# kTs = [abs(scaleJ)*valS*100 * 0.9^k for k in 0:100]\n# anneal!(sys,integrator;kTschedule=kTs,ndwell=500)\n# dwell!(sys,integrator;kTtarget=kTs[end],ndwell=2000)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot the resulting spin system to check ordering in real space","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"plot_spins(sys)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Calculation-of-Neutron-Scattering-Responses","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Calculation of Neutron Scattering Responses","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Fourier-transformed-instantaneous-two-point-correlation-functions","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Fourier transformed instantaneous two-point correlation functions","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Calculate the instantaneous/equal-time structure factor.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"eqsf = instant_correlations(sys)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"SampledCorrelations (6.252 MiB)\n[S(q) | 0 sample]\nLattice: (10, 10, 10)×8\n6 correlations in Dipole mode:\n╔ ⬤ ⬤ ⬤ Sx\n║ ⋅ ⬤ ⬤ Sy\n╚ ⋅ ⋅ ⬤ Sz\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"If desired, add additional samples by decorrelating and then re-calculating the eqsf.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"nsamples = 1\nndecorr = 1000\n@time sample_sf!(eqsf, sys, integrator; nsamples=nsamples, ndecorr=ndecorr);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":" 0.966485 seconds (1.89 M allocations: 118.815 MiB, 6.25% gc time, 50.12% compilation time)\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Project onto a constant Q-Slice in momentum space.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"nQpts = 200\nQxpts = range(-10.0, 10.0, length=nQpts)\nQypts = range(-10.0, 10.0, length=nQpts)\nqz = 1.0\nQpts = [[qx, qy, qz] for qx in Qxpts, qy in Qypts]\ninstant_formula = intensity_formula(eqsf, :perp; formfactors)\niq = instant_intensities_interpolated(eqsf, Qpts, instant_formula);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot the resulting I(Q)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"heatmap(Qxpts, Qypts, iq;\n colorrange = (0, maximum(iq)/20),\n axis = (\n xlabel=\"Momentum Transfer Qx (r.l.u)\", xlabelsize=16,\n ylabel=\"Momentum Transfer Qy (r.l.u)\", ylabelsize=16,\n aspect=true,\n )\n)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Dynamical-and-energy-integrated-two-point-correlation-functions","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Dynamical and energy-integrated two-point correlation functions","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Calculate the time traces and Fourier transform: Dynamical Structure Factor (first sample).","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"ωmax = 6.0 # Maximum energy to resolve\nnω = 100 # Number of energies to resolve\nsc = dynamical_correlations(sys; Δt=Δt0, nω=nω, ωmax=ωmax, process_trajectory=:symmetrize)\n@time add_sample!(sc, sys) # Add a sample trajectory","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":" 5.471269 seconds (14.33 M allocations: 291.526 MiB, 0.75% gc time, 0.02% compilation time)\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"If desired, add additional decorrelated samples.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"nsamples = 10\nndecorr = 1000\n@time sample_sf!(sc, sys, integrator; nsamples=nsamples, ndecorr=ndecorr);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Sampling SF: 27%|██████████▎ | ETA: 0:00:33\u001b[KSampling SF: 49%|██████████████████▊ | ETA: 0:00:25\u001b[KSampling SF: 68%|█████████████████████████▉ | ETA: 0:00:17\u001b[KSampling SF: 84%|████████████████████████████████ | ETA: 0:00:09\u001b[KSampling SF: 100%|██████████████████████████████████████| Time: 0:00:53\u001b[K\n 59.070784 seconds (143.58 M allocations: 2.866 GiB, 0.56% gc time, 0.24% compilation time)\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Can use the Brillouin package for help on determining high symmetry points","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"kp = irrfbz_path(227,[[1,0,0], [0,1,0], [0,0,1]])\nkpc = cartesianize(kp)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"KPath{3} (6 points, 2 paths, 8 points in paths):\n points: :U => [1.570796, 6.283185, 1.570796]\n :W => [3.141593, 6.283185, 0.0]\n :K => [4.712389, 4.712389, 0.0]\n :Γ => [0.0, 0.0, 0.0]\n :L => [3.141593, 3.141593, 3.141593]\n :X => [0.0, 6.283185, 0.0]\n paths: [:Γ, :X, :U]\n [:K, :Γ, :L, :W, :X]\n basis: [-6.283185, 6.283185, 6.283185]\n [6.283185, -6.283185, 6.283185]\n [6.283185, 6.283185, -6.283185]","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Project onto a constant QE-Slice in momentum-energy space.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"densQpts = 50\nsymQpts = [[0.75, 0.75, 0.00], # List of wave vectors that define a path\n [0.00, 0.00, 0.00],\n [0.50, 0.50, 0.50],\n [0.50, 1.00, 0.00],\n [0.00, 1.00, 0.00],\n [0.25, 1.00, 0.25],\n [0.00, 1.00, 0.00],\n [0.00,-4.00, 0.00]]\n(Qpts, xticks) = reciprocal_space_path(magxtal, symQpts, densQpts)\nformula = intensity_formula(sc, :perp; formfactors, kT=integrator.kT)\niqw = intensities_interpolated(sc, Qpts, formula);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"If desired, broaden the sc in energy.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"η = 0.1 ## Lorentzian energy broadening parameter\niqwc = broaden_energy(sc, iqw, (ω, ω₀) -> lorentzian(ω-ω₀, η));","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"If desired, calculated the energy-integrated structure factor","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"iqt = instant_intensities_interpolated(sc, Qpts, formula);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot the resulting I(Q,W).","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"ωs = available_energies(sc)\nheatmap(1:size(iqwc, 1), ωs, iqwc;\n colorrange = (0, maximum(iqwc)/20000.0),\n axis = (;\n xlabel=\"Momentum Transfer (r.l.u)\",\n ylabel=\"Energy Transfer (meV)\",\n xticks,\n xticklabelrotation=π/5,\n aspect = 1.4,\n )\n)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Projection into a powder-averaged neutron scattering intensity .","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Qmax = 3.5\nnQpts = 100\nQpow = range(0, Qmax, nQpts)\nnpoints = 100\npqw = powder_average(sc, Qpow, npoints, formula; η);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Powder Averaging: 78%|█████████████████████████▊ | ETA: 0:00:03\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot resulting Ipow(|Q|,W).","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"heatmap(Qpow, ωs, pqw;\n axis = (\n xlabel=\"|Q| (Å⁻¹)\",\n ylabel=\"Energy Transfer (meV)\",\n aspect = 1.4,\n ),\n colorrange = (0, 40.0)\n)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Calculation-of-temperature-dependent-powder-average-spectrum","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Calculation of temperature-dependent powder average spectrum","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Define a temperature schedule","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"kTs = [60 40 25 20 15 12 10 4] * Sunny.meV_per_K\npqw_res = []\nfor kT in kTs\n dwell!(sys, integrator; kTtarget=kT, ndwell=1000);\n sc_loc = dynamical_correlations(sys; Δt=Δt0, nω, ωmax, process_trajectory=:symmetrize);\n add_sample!(sc_loc, sys)\n formula = intensity_formula(sc, :perp; formfactors, kT)\n push!(pqw_res, powder_average(sc_loc, Qpow, npoints, formula; η))\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"┌ Warning: Assignment to `formula` in soft scope is ambiguous because a global variable by the same name exists: `formula` will be treated as a new local. Disambiguate by using `local formula` to suppress this warning or `global formula` to assign to the existing global variable.\n└ @ ~/Research/SunnyContributed/contributed-docs/build/CoRh2O4-tutorial.md:7\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 81%|██████████████████████████▊ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 81%|██████████████████████████▊ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 87%|████████████████████████████▊ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:11\u001b[K\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot the resulting Ipow(|Q|,W) as a function of temperature, to compare with Fig.6 of https://arxiv.org/abs/1706.05881","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"fig = Figure(; resolution=(1200,600))\nfor i in 1:8\n r, c = fldmod1(i, 4)\n ax = Axis(fig[r, c];\n title = \"kT = \"*string(round(kTs[9-i], digits=3))*\" (meV)\",\n xlabel = r == 2 ? \"|Q| (Å⁻¹)\" : \"\",\n ylabel = c == 1 ? \"Energy Transfer (meV)\" : \"\",\n aspect = 1.4,\n )\n heatmap!(ax, Qpow, ωs, pqw_res[9-i]; colorrange = (0, 20.0))\nend\nfig","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"EditURL = \"../../../../examples/spinw_ports/08_Kagome_AFM.jl\"","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html#Tutorial-8-Kagome-Antiferromagnet","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"","category":"section"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"This is a Sunny port of SpinW Tutorial 8, originally authored by Bjorn Fak and Sandor Toth. The goal is to calculate the linear spin wave theory spectrum for the sqrt3 times sqrt3 order of a Kagome antiferromagnet.","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Load Packages","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Build a Crystal with Poverline3 space group,","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"a = 1\nlatvecs = lattice_vectors(a, a, 10a, 90, 90, 120)\ncrystal = Crystal(latvecs, [[1/2,0,0]], 147)","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Build a System with antiferrogmanetic nearest neighbor exchange J=1.","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"S = 1\nsys = System(crystal, (3,3,1), [SpinInfo(1; S, g=2)], :dipole)\nJ = 1.0\nset_exchange!(sys, J, Bond(2,3,[0,0,0]))","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Initialize to an energy minimizing magnetic structure, for which nearest-neighbor spins are at 120° angles.","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"q = -[1/3, 1/3, 0]\naxis = [0,0,1]\nset_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[cos(0),sin(0),0])\nset_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[cos(0),sin(0),0])\nset_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[cos(2π/3),sin(2π/3),0])\nplot_spins(sys; dims=2)","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Check energy. Each site participates in 4 bonds with energy JS^2cos(2π3). Factor of 1/2 avoids double counting.","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"@assert energy_per_site(sys) ≈ (4/2)*J*S^2*cos(2π/3)","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Define a path in reciprocal space.","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"points_rlu = [[-1/2, 0, 0], [0, 0, 0], [1/2, 1/2, 0]]\ndensity = 100\npath, xticks = reciprocal_space_path(crystal, points_rlu, density);\nnothing #hide","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Calculate discrete intensities","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"swt = SpinWaveTheory(sys)\nformula = intensity_formula(swt, :perp; kernel=delta_function_kernel)\ndisp, intensity = intensities_bands(swt, path, formula);\nnothing #hide","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"Plot over a restricted color range from [0,1e-2]. Note that the intensities of the flat band at zero-energy are off-scale.","category":"page"},{"location":"examples/spinw/08_Kagome_AFM.html","page":"Tutorial 8 - Kagome Antiferromagnet","title":"Tutorial 8 - Kagome Antiferromagnet","text":"fig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nylims!(ax, -1e-1, 2.3)\nfor i in axes(disp, 2)\n lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange=(0,1e-2))\nend\nfig","category":"page"},{"location":"renormalization.html#Interaction-Strength-Renormalization","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"A unique feature of Sunny is its support for building classical models where quantum spin is represented as an N-level system, rather than just an expected dipole. This generalization can be important when modeling quantum spin Hamiltonians that include, e.g., a single-ion anisotropy, or a biquadratic coupling between sites. Sunny also supports constraining quantum spin to the space of pure dipoles; in this case, Sunny will automatically perform an interaction strength renormalization that maximizes accuracy.","category":"page"},{"location":"renormalization.html#Local-operators","page":"Interaction Strength Renormalization","title":"Local operators","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"A quantum spin-S state has N = 2S + 1 levels. Each local spin operator hatS^xyz is faithfully represented as an NN matrix. These matrices can be accessed using spin_matrices for a given label S. For example, the Pauli matrices are associated with S = 12.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"When S 12, it is possible to construct multipole moments beyond the spin-dipole. For example,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"S = spin_matrices(3/2)\n@assert S[3] ≈ diagm([3/2, 1/2, -1/2, -3/2])\n@assert S[3]^2 ≈ diagm([9/4, 1/4, 1/4, 9/4])","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"If the operator -S[3]^2 is passed to set_onsite_coupling!, it would set an easy-axis anisotropy in the hatz direction.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Any Hermitian operator can be expanded in the basis of Stevens operators hatmathcalO_kq up to a constant shift. To see this expansion, use print_stevens_expansion:","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"print_stevens_expansion((S[1]^2 + S[2]^2)) # Prints -(1/3)𝒪₂₀ + 5/2","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Alternatively, the same operator could have been constructed directly from stevens_matrices:","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"O = stevens_matrices(3/2)\n@assert S[1]^2 + S[2]^2 ≈ -O[2, 0]/3 + (5/2)*I","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"See below for an explicit definition of Stevens operators as polynomials of the spin operators.","category":"page"},{"location":"renormalization.html#Renormalization-procedure-for-:dipole-mode","page":"Interaction Strength Renormalization","title":"Renormalization procedure for :dipole mode","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Sunny will typically operate in one of two modes: :SUN or :dipole. The former faithfully represents quantum spin as an SU(N) coherent-state which, for our purposes, is an N-component complex vector. In contrast, :dipole mode constrains the coherent-state to the space of pure dipoles. Here, Sunny will automatically renormalize the magnitude of each Stevens operator to achieve maximal consistency with :SUN mode. This procedure was derived in D. Dahlbom et al., [arXiv:2304.03874].","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"By way of illustration, consider a quantum operator hatmathcalH_mathrmlocal giving a single-ion anisotropy for one site. In Stevens operators,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"hatmathcal H_mathrmlocal = sum_k q A_kq hatmathcalO_kq","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"for some coefficients A_kq.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"In :SUN mode, Sunny will faithfully represent hatmathcal H_mathrmlocal as an NN matrix. In :dipole mode, the expected energy langle hatmathcal H_mathrmlocal rangle must somehow be approximated as a function of the expected dipole mathbfs.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"One approach is to formally take S to infty, and this yields the traditional classical limit of a spin system. In this, limit spin operators commute, and expectation values of polynomials become polynomials of expectation values. For example, langle hatS^alpha hatS^betarangle to langle hatS^alpha rangle langle hatS^betarangle, because any corrections are damped by the factor S^-1 to 0. The expectation of a Stevens operator langle hatmathcalO_kq rangle becomes a Stevens function mathcalO_kq(mathbfs), i.e., a polynomial of expected dipole mathbfs = langle hatmathbfS rangle.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"In a real magnetic compound, however, the spin magnitude S is not necessarily large. To obtain a better approximation, one should avoid the assumption S to infty. Our approach is to start with the full dynamics of SU(N) coherent states, and then constrain it to the space of pure dipole states mathbfsrangle. The latter are defined such that expectation values,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"langle mathbfs hatmathbfS mathbfsrangle = mathbfs","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"have magnitude mathbfs = S, which is maximal.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"For pure dipole states, expectations can be computed exactly,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"langle mathbfs hatmathcalO mathbfsrangle = c_k mathcalO_kq(mathbfs)","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"The right-hand side involves a renormalization of the Stevens functions, where","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"beginalign*\nc_1 = 1 \nc_2 = 1-frac12S^-1 \nc_3 = 1-frac32S^-1+frac12S^-2 \nc_4 = 1-3S^-1+frac114S^-2-frac34S^-3 \nc_5 = 1-5S^-1+frac354S^-2-frac254S^-3+frac32S^-4 \nc_6 = 1-frac152S^-1+frac854S^-2-frac2258S^-3+frac1378S^-4-frac154S^-5 \nvdots\nendalign*","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Constrained to the space of dipoles, the expected local energy becomes","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"E_mathrmlocal(mathbfs) = langle mathbfs hatmathcal H_mathrmlocal mathbfsrangle = sum_k q c_k A_kq mathcalO_kq(mathbfs)","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"It can be shown that SU(N) dynamics reduces to the usual Landau-Lifshitz dynamics of dipoles, but involving E_mathrmlocal(mathbfs) as the classical Hamiltonian. Through the renormalization factors c_k, Sunny avoids the large-S assumption, and gives a more accurate result.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Renormalization also applies to the coupling between different sites. In Sunny, couplings will often be expressed as a polynomial of spin operators using set_pair_coupling!, but any such coupling can be decomposed as sum of tensor products of Stevens operators. Without loss of generality, consider a single coupling between two Stevens operators hatmathcalH_mathrmcoupling = hatmathcalO_kq otimes hatmathcalO_kq along a bond connecting sites i and j. Upon constraining to pure dipole states mathbfs_irangle and mathbfs_jrangle, the expected energy takes the form E_mathrmcoupling = c_k c_k mathcalO_kq(mathbfs_i) mathcalO_kq(mathbfs_j), which now involves a product of renormalized Stevens functions. ","category":"page"},{"location":"renormalization.html#Use-:dipole_large_S-mode-to-disable-renormalization","page":"Interaction Strength Renormalization","title":"Use :dipole_large_S mode to disable renormalization","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Although we generally recommend the above renormalization procedure, there are circumstances where it is not desirable. Examples include reproducing a model-system study, or describing a micromagnetic system for which the Stoinfty limit is a good approximation. To simulate dipoles without interaction strength renormalization, construct a System using the mode :dipole_large_S. Symbolic operators in the large-S limit can be constructed by passing Inf to either spin_matrices or stevens_matrices.","category":"page"},{"location":"renormalization.html#Definition-of-Stevens-operators","page":"Interaction Strength Renormalization","title":"Definition of Stevens operators","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"The Stevens operators hatmathcalO_kq are defined as polynomials of angular momentum operators hatS_xyz in some spin-S representation.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Using","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"beginalign*\nX = mathbfhatS cdot mathbfhatS = S (S+1) \nhatS_pm = hatS_x pm i hatS_y \nphi_+ = frac14quad phi_- = frac14 i\nendalign*","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"the relevant Stevens operators are defined as,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"beginalign*\nhatmathcalO_00 =1\n\nhatmathcalO_1pm1 =phi_pm(hatS_+pmhatS_-)+mathrmhc\nhatmathcalO_10 =hatS_z\n\nhatmathcalO_2pm2 =phi_pm(hatS_+^2pmhatS_-^2)+mathrmhc\nhatmathcalO_2pm1 =phi_pm(hatS_+pmhatS_-)hatS_z+mathrmhc\nhatmathcalO_20 =3hatS_z^2-X\n\nhatmathcalO_3pm3 =phi_pm(hatS_+^3pmhatS_-^3)+mathrmhc\nhatmathcalO_3pm2 =phi_pm(hatS_+^2pmhatS_-^2)hatS_z+mathrmhc\nhatmathcalO_3pm1 =phi_pm(hatS_+pmhatS_-)(5hatS_z^2-X-12)+mathrmhc\nhatmathcalO_30 =5hatS_z^3-(3X-1)hatS_z\n\nhatmathcalO_4pm4 =phi_pm(hatS_+^4pmhatS_-^4)+mathrmhc\nhatmathcalO_4pm3 =phi_pm(hatS_+^3pmhatS_-^3)hatS_z+mathrmhc\nhatmathcalO_4pm2 =phi_pm(hatS_+^2pmhatS_-^2)(7hatS_z^2-(X+5))+mathrmhc\nhatmathcalO_4pm1 =phi_pm(hatS_+pmhatS_-)(7hatS_z^3-(3X+1)hatS_z)+mathrmhc\nhatmathcalO_40 =35hatS_z^4-(30X-25)hatS_z^2+(3X^2-6X)\n\nhatmathcalO_5pm5 =phi_pm(hatS_+^5pmhatS_-^5)+mathrmhc\nhatmathcalO_5pm4 =phi_pm(hatS_+^4pmhatS_-^4)hatS_z+mathrmhc\nhatmathcalO_5pm3 =phi_pm(hatS_+^3pmhatS_-^3)(9hatS_z^2-(X+332))+mathrmhc\nhatmathcalO_5pm2 =phi_pm(hatS_+^2pmhatS_-^2)(3hatS_z^3-(X+6)hatS_z)+mathrmhc\nhatmathcalO_5pm1 =phi_pm(hatS_+pmhatS_-)(21hatS_z^4-14XhatS_z^2+(X^2-X+32))+mathrmhc\nhatmathcalO_50 =63hatS_z^5-(70X-105)hatS_z^3+(15X^2-50X+12)hatS_z\n\nhatmathcalO_6pm6 =phi_pm(hatS_+^6pmhatS_-^6)+mathrmhc\nhatmathcalO_6pm5 =phi_pm(hatS_+^5pmhatS_-^5)hatS_z+mathrmhc\nhatmathcalO_6pm4 =phi_pm(hatS_+^4pmhatS_-^4)(11hatS_z^2-X-38)+mathrmhc\nhatmathcalO_6pm3 =phi_pm(hatS_+^3pmhatS_-^3)(11hatS_z^3-(3X+59)hatS_z)+mathrmhc\nhatmathcalO_6pm2 =phi_pm(hatS_+^2pmhatS_-^2)(33hatS_z^4-(18X+123)hatS_z^2+X^2+10X+102)+mathrmhc\nhatmathcalO_6pm1 =phi_pm(hatS_+pmhatS_-)(33hatS_z^5-(30X-15)hatS_z^3+(5X^2-10X+12)hatS_z)+mathrmhc\nhatmathcalO_60 =231hatS_z^6-(315X-735)hatS_z^4+(105X^2-525X+294)hatS_z^2-5X^3+40X^2-60X\nendalign*","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Computer-generated tables of Stevens operators with k 6 are available from C. Rudowicz and C. Y. Chung, J. Phys.: Condens. Matter 16, 5825 (2004), but these typically do not appear in magnetic simulations.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"The case k=1 gives the dipole operators,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"(hatmathcalO_11 hatmathcalO_10 hatmathcalO_1-1) = (hatS_x hatS_z hatS_y)","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"The case k=2 gives the quadrupole operators,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"(hatmathcalO_22 dots hatmathcalO_2-2) = left(hatS_x^2 - hatS_y^2 frachatS_x hatS_z + hatS_z hatS_x2 2hatS_z^2-hatS_x^2-hatS_y^2 frachatS_y hatS_z + hatS_z hatS_y2 hatS_x hatS_y + hatS_y hatS_xright)","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"For each k value, the set of operators hatmathcalO_kq for q = -k dots k form an irreducible representation of the group of rotations O(3). That is, rotation will transform hatmathcalO_kq into a linear combination of hatmathcalO_kq where q varies but k remains fixed. ","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"In taking the large-S limit, each dipole operator is replaced by its expectation value mathbfs = langle hatmathbfS rangle, and only leading-order terms are retained. The operator hatmathcalO_kq becomes a homogeneous polynomial O_kq(mathbfs) of order k in the spin components. One can see these polynomials by constructing stevens_matrices with the argument S = Inf. Due to the normalization constraint, each dipole can be expressed in polar angles, (theta phi). Then the Stevens functions O_kq(mathbfs) correspond to the spherical harmonic functions Y_l^m(theta phi) where l=k and m=q; this correspondence is valid up to k and q-dependent rescaling factors.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"EditURL = \"../../../examples/fei2_classical.jl\"","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/fei2_classical.html#FeI-at-Finite-Temperature","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"","category":"section"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"using Sunny, LinearAlgebra, GLMakie","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"In our previous Case Study: FeI₂, we used linear spin wave theory (LSWT) to calculate the dynamical structure factor. Here, we perform a similar calculation using classical spin dynamics. Because we are interested in the coupled dynamics of spin dipoles and quadrupoles, we employ a classical dynamics of SU(3) coherent states that generalizes the Landau-Lifshitz equation.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Compared to LSWT, simulations using classical dynamics are much slower, and are limited in k-space resolution. However, they make it is possible to capture nonlinear effects associated with finite temperature fluctuations. Classical dynamics are also appealing for studying out-of-equilibrium systems (e.g., relaxation of spin glasses), or systems with quenched inhomogeneities that require large simulation volumes.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"In this tutorial, we show how to study the finite temperature dynamics of FeI₂ using the classical approach. It is important to stress that the estimation of S(𝐪ω) with classical dynamics is fundamentally a Monte Carlo calculation: sample spin configurations are drawn from thermal equilibrium and used as initial conditions for generating dissipationless trajectories. The correlations of these trajectories are then averaged and used to calculate scattering intensities. It is therefore important to ensure that the initial spin configurations are sampled appropriately and that sufficient statistics are collected. We will demonstrate one approach here.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"As an overview, we will:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Identify the ground state\nMeasure correlation data describing the excitations around that ground state\nUse the correlation data to compute scattering intensities","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"As the implementation of the FeI₂ model is already covered in detail in the LSWT tutorial, we will not repeat it below. Instead, we will assume that you already have defined a sys in the same way with lattice dimensions 444.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"a = b = 4.05012#hide\nc = 6.75214#hide\nlatvecs = lattice_vectors(a, b, c, 90, 90, 120)#hide\npositions = [[0,0,0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]]#hide\ntypes = [\"Fe\", \"I\", \"I\"]#hide\nFeI2 = Crystal(latvecs, positions; types)#hide\ncryst = subcrystal(FeI2, \"Fe\")#hide\nsys = System(cryst, (4,4,4), [SpinInfo(1,S=1,g=2)], :SUN, seed=2)#hide\nJ1pm = -0.236#hide\nJ1pmpm = -0.161#hide\nJ1zpm = -0.261#hide\nJ2pm = 0.026#hide\nJ3pm = 0.166#hide\nJ′0pm = 0.037#hide\nJ′1pm = 0.013#hide\nJ′2apm = 0.068#hide\nJ1zz = -0.236#hide\nJ2zz = 0.113#hide\nJ3zz = 0.211#hide\nJ′0zz = -0.036#hide\nJ′1zz = 0.051#hide\nJ′2azz = 0.073#hide\nJ1xx = J1pm + J1pmpm#hide\nJ1yy = J1pm - J1pmpm#hide\nJ1yz = J1zpm#hide\nset_exchange!(sys, [J1xx 0.0 0.0; 0.0 J1yy J1yz; 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))#hide\nset_exchange!(sys, [J2pm 0.0 0.0; 0.0 J2pm 0.0; 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))#hide\nset_exchange!(sys, [J3pm 0.0 0.0; 0.0 J3pm 0.0; 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))#hide\nset_exchange!(sys, [J′0pm 0.0 0.0; 0.0 J′0pm 0.0; 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))#hide\nset_exchange!(sys, [J′1pm 0.0 0.0; 0.0 J′1pm 0.0; 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))#hide\nset_exchange!(sys, [J′2apm 0.0 0.0; 0.0 J′2apm 0.0; 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))#hide\nD = 2.165#hide\nset_onsite_coupling!(sys, S -> -D*S[3]^2, 1)#hide\nsys","category":"page"},{"location":"examples/fei2_classical.html#Finding-a-ground-state","page":"FeI₂ at Finite Temperature","title":"Finding a ground state","text":"","category":"section"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Sunny uses the Langevin dynamics of SU(N) coherent states to sample spin configurations from the thermal equlibrium. One first constructs a Langevin integrator. This requires a time step, temperature, and a phenomenological damping parameter λ that sets the coupling to the thermal bath.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Δt = 0.05/D # Should be inversely proportional to the largest energy scale\n # in the system. For FeI2, this is the easy-axis anisotropy,\n # `D = 2.165` (meV). The prefactor 0.05 is relatively small,\n # and achieves high accuracy.\nkT = 0.2 # Temperature of the thermal bath (meV).\nλ = 0.1 # This value is typically good for Monte Carlo sampling,\n # independent of system details.\n\nlangevin = Langevin(Δt; kT, λ);\nnothing #hide","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Langevin dynamics can be used to search for a magnetically ordered state. For this, the temperature kT must be below the ordering temperature, but large enough that the dynamical sampling procedure can overcome local energy barriers and eliminate defects.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"randomize_spins!(sys)\nfor _ in 1:20_000\n step!(sys, langevin)\nend","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Although thermal fluctuations are present, the correct antiferromagnetic order (2 up, 2 down) is apparent.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"plot_spins(sys; color=[s[3] for s in sys.dipoles])","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"For other systems, it can be much harder to find the magnetic ordering in an unbiased way, and more complicated sampling procedures may be necessary.","category":"page"},{"location":"examples/fei2_classical.html#Calculating-Thermal-Averaged-Correlations-\\langle-S{\\alpha\\beta}(𝐪,ω)\\rangle","page":"FeI₂ at Finite Temperature","title":"Calculating Thermal-Averaged Correlations langle S^alphabeta(𝐪ω)rangle","text":"","category":"section"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Our aim is to study the classical spin dynamics for states sampled in thermal equilibrium. To minimize finite size effects, and achieve sufficient momentum space resolution, we should significantly enlarge the system volume. The function resize_supercell takes new dimensions as multiples of the unit cell lattice vectors.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"sys_large = resize_supercell(sys, (16,16,4)) # 16x16x4 copies of the original unit cell\nplot_spins(sys_large; color=[s[3] for s in sys_large.dipoles])","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Now we will re-thermalize the system to a configuration just above the ordering temperature. Sunny expects energies in meV by default, so we use meV_per_K to convert from kelvin.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"kT = 3.5 * meV_per_K # 3.5K ≈ 0.30 meV\nlangevin.kT = kT\nfor _ in 1:10_000\n step!(sys_large, langevin)\nend","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"The next step is to collect correlation data S^alphabeta. This will involve sampling spin configurations from thermal equilibrium, and then integrating the Hamiltonian dynamics of SU(N) coherent states to collect Fourier-space information about normal modes. Quantization of these modes yields the magnons, and the associated dynamical spin-spin correlations can be compared with neutron scattering intensities S^alphabeta(qomega). Because this a real-space calculation, data is only available for discrete q modes (the resolution scales like inverse system size).","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"To store the correlation data, we initialize a SampledCorrelations object by calling dynamical_correlations. It requires three keyword arguments: an integration step size, a target number of ωs to retain, and a maximum energy ω to resolve. For the time step, twice the value used for the Langevin integrator is usually a good choice.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"sc = dynamical_correlations(sys_large; Δt=2Δt, nω=120, ωmax=7.5)","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"The function add_sample! will collect data by running a dynamical trajectory starting from the current system configuration.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"add_sample!(sc, sys_large) # Accumulate the sample into `sc`","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"To collect additional data, it is required to re-sample the spin configuration from the thermal distribution. For efficiency, the dynamics should be run long enough that consecutive samples are uncorrelated.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"for _ in 1:2\n for _ in 1:1000 # Enough steps to decorrelate spins\n step!(sys_large, langevin)\n end\n add_sample!(sc, sys_large) # Accumulate the sample into `sc`\nend","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Now, sc has more samples included:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"sc","category":"page"},{"location":"examples/fei2_classical.html#Computing-Scattering-Intensities","page":"FeI₂ at Finite Temperature","title":"Computing Scattering Intensities","text":"","category":"section"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"With the thermally-averaged correlation data langle S^alphabeta(qomega)rangle in hand, we now need to specify how to extract a scattering intensity from this information. This is done by constructing an intensity_formula. By way of example, we will use a formula which computes the trace of the structure factor and applies a classical-to-quantum temperature-dependent rescaling kT.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"formula = intensity_formula(sc, :trace; kT)","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Recall that langle S^alphabeta(qomega)rangle is only available at certain discrete q values, due to the finite lattice size. There are two basic approaches to handling this discreteness. The first approach is to interpolate between the available data using intensities_interpolated. For example, we can plot single-q slices at (0,0,0) and (π,π,π) using this method:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"qs = [[0, 0, 0], [0.5, 0.5, 0.5]]\nis = intensities_interpolated(sc, qs, formula; interpolation = :round)\n\nωs = available_energies(sc)\nfig = lines(ωs, is[1,:]; axis=(xlabel=\"meV\", ylabel=\"Intensity\"), label=\"(0,0,0)\")\nlines!(ωs, is[2,:]; label=\"(π,π,π)\")\naxislegend()\nfig","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"The resolution in energy can be improved by increasing nω (and decreasing Δt), and the general accuracy can be improved by collecting additional samples from the thermal equilibrium.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"For real calculations, one often wants to apply further corrections and more accurate formulas. Here, we apply FormFactor corrections appropriate for Fe2 magnetic ions, and a dipole polarization correction :perp.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"formfactors = [FormFactor(\"Fe2\"; g_lande=3/2)]\nnew_formula = intensity_formula(sc, :perp; kT, formfactors = formfactors)","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Frequently, one wants to extract energy intensities along lines that connect special wave vectors–a so-called \"spaghetti plot\". The function reciprocal_space_path creates an appropriate horizontal axis for this plot by linearly sampling between provided q-points with a given sample density. The number of sample points between two wavevectors q1 and q2 is given by dist*density where dist = norm(cryst.recipvecs * (q1 - q2)) is measured in the global frame.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"points = [[0, 0, 0], # List of wave vectors that define a path\n [1, 0, 0],\n [0, 1, 0],\n [1/2, 0, 0],\n [0, 1, 0],\n [0, 0, 0]]\ndensity = 40\npath, xticks = reciprocal_space_path(cryst, points, density);\nnothing #hide","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Again using intensities_interpolated, we can evaluate the (interpolated) intensity at each point on the path. Since scattering intensities are only available at a certain discrete (Qomega) points, the intensity on the path can be calculated by interpolating between these discrete points:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"is_interpolated = intensities_interpolated(sc, path, new_formula;\n interpolation = :linear, # Interpolate between available wave vectors\n);\n# Add artificial broadening\nis_interpolated_broadened = broaden_energy(sc, is, (ω, ω₀)->lorentzian(ω-ω₀, 0.05));\nnothing #hide","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"The second approach to handle the discreteness of the data is to bin the intensity at the discrete points into the bins of a histogram. First, the five sub-histograms are set up using reciprocal_space_path_bins in analogy to reciprocal_space_path.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"cut_width = 0.3\ndensity = 15\nparamsList, markers, ranges = reciprocal_space_path_bins(sc,points,density,cut_width);\nnothing #hide","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Then, the intensity data is computed using intensities_binned for each sub-histogram:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"total_bins = ranges[end][end]\nenergy_bins = paramsList[1].numbins[4]\nis_binned = zeros(Float64,total_bins,energy_bins)\nintegrated_kernel = integrated_lorentzian(0.05) # Lorentzian broadening\nfor k in eachindex(paramsList)\n bin_data, counts = intensities_binned(sc,paramsList[k], new_formula;\n integrated_kernel = integrated_kernel\n )\n is_binned[ranges[k],:] = bin_data[:,1,1,:] ./ counts[:,1,1,:]\nend","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"The graph produced by interpolating (top) is similar to the one produced by binning (bottom):","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"fig = Figure()\nax_top = Axis(fig[1,1],ylabel = \"meV\",xticklabelrotation=π/8,xticklabelsize=12;xticks)\nax_bottom = Axis(fig[2,1],ylabel = \"meV\",xticks = (markers, string.(points)),xticklabelrotation=π/8,xticklabelsize=12)\n\nheatmap!(ax_top,1:size(is_interpolated,1), ωs, is_interpolated;\n colorrange=(0.0,0.07),\n)\n\nheatmap!(ax_bottom,1:size(is_binned,1), ωs, is_binned;\n colorrange=(0.0,0.05),\n)\n\nfig","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Note that we have clipped the colors in order to make the higher-energy excitations more visible.","category":"page"},{"location":"examples/fei2_classical.html#Unconventional-RLU-Systems-and-Constant-Energy-Cuts","page":"FeI₂ at Finite Temperature","title":"Unconventional RLU Systems and Constant Energy Cuts","text":"","category":"section"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Often it is useful to plot cuts across multiple wave vectors but at a single energy. We'll pick an energy,","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"ωidx = 60\ntarget_ω = ωs[ωidx]","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"and take a constant-energy cut at that energy. The most straightforward way is to make a plot whose axes are aligned with the conventional reciprocal lattice of the crystal. This is accomplished using unit_resolution_binning_parameters:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"params = unit_resolution_binning_parameters(sc)\nparams.binstart[1:2] .= -1 # Expand plot range slightly\n\n# Set energy integration range\nomega_width = 0.3\nparams.binstart[4] = target_ω - (omega_width/2)\nparams.binend[4] = target_ω # `binend` should be inside (e.g. at the center) of the range\nparams.binwidth[4] = omega_width\n\nintegrate_axes!(params, axes = 3) # Integrate out z direction entirely\n\nparams","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"In each of the following plots, black dashed lines represent (direct) lattice vectors. Since these plots are in reciprocal space, direct lattice vectors are represented as covectors (i.e. coordinate grids) instead of as arrows.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"is, counts = intensities_binned(sc,params,new_formula)\n\nfig = Figure()\nax = Axis(fig[1,1];\n title=\"Δω=0.3 meV (Binned)\", aspect=true,\n xlabel = \"[H, 0, 0]\",\n ylabel = \"[0, K, 0]\"\n)\nbcs = axes_bincenters(params)\nhm = heatmap!(ax,bcs[1],bcs[2],is[:,:,1,1] ./ counts[:,:,1,1])\nfunction add_lines!(ax,params)#hide\n bes = Sunny.axes_binedges(params)#hide\n hrange = range(-2,2,length=17)#hide\n linesegments!(ax,[(Point2f(params.covectors[1,1:3] ⋅ [h,-10,0],params.covectors[2,1:3] ⋅ [h,-10,0]),Point2f(params.covectors[1,1:3] ⋅ [h,10,0],params.covectors[2,1:3] ⋅ [h,10,0])) for h = hrange],linestyle=:dash,color=:black)#hide\n krange = range(-2,2,length=17)#hide\n linesegments!(ax,[(Point2f(params.covectors[1,1:3] ⋅ [-10,k,0],params.covectors[2,1:3] ⋅ [-10,k,0]),Point2f(params.covectors[1,1:3] ⋅ [10,k,0],params.covectors[2,1:3] ⋅ [10,k,0])) for k = krange],linestyle=:dash,color=:black)#hide\n xlims!(ax,bes[1][1],bes[1][end])#hide\n ylims!(ax,bes[2][1],bes[2][end])#hide\nend#hide\nadd_lines!(ax,params)\nColorbar(fig[1,2], hm);\nfig","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"In the above plot, the dashed-line (direct) lattice vectors are clearly orthogonal. However, we know that in real space, the lattice vectors a and b are not orthogonal, but rather point along the edges of a hexagon (see lower left corner):","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":" ","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Thus, plotting the direct lattice vectors as orthogonal (even in reciprocal space) is somewhat misleading. Worse yet, the [H,0,0] by [0,K,0] plot apparently loses the 6-fold symmetry of the crystal! Lastly, if one works out the components of the real-space metric with respect to the axes of the plot, one finds that there are non-zero off-diagonal entries,","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"latvecs = sys.crystal.latvecs\nmetric = latvecs' * I(3) * latvecs","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"so real-space rotations and angles map into reciprocal space rotations angles in a complicated way.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"To resolve these important issues, we want to use axes which are orthogonal (i.e. they diagonalize the metric and solve all of the problems just mentioned). The canonical choice is to use the combination frac12a + b of lattice vectors (equiv. a^* - frac12b^*), which is orthogonal to a:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"(latvecs * [1/2,1,0]) ⋅ (latvecs * [1,0,0]) == 0","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"This new vector frac12a+b is visibly orthogonal to a in real space:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"f = Figure()#hide\nax = Axis(f[1,1])#hide\narrows!(ax,[Point2f(0,0),Point2f(latvecs[1:2,1] ./ 2)],[Vec2f(latvecs[1:2,1] ./ 2), Vec2f(latvecs[1:2,2])],arrowcolor = :blue,arrowsize = 30.,linewidth = 5.,linecolor = :blue)#hide\narrows!(ax,[Point2f(0,0)],[Vec2f(latvecs[1:2,:] * [1/2,1,0])],arrowcolor = :red,arrowsize = 30.,linewidth = 5.,linecolor = :red, linestyle = :dash)#hide\nscatter!(ax,[Point2f(latvecs[1:2,:] * [a,b,0]) for a in -1:1, b in -1:1][:],color = :black)#hide\nannotations!(ax,[\"0\",\"0+b\",\"0+a\", \"a/2\", \"b\"],[Point2f(0,-0.3),Point2f(latvecs[1:2,2]) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1]) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1] ./ 4) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1] ./ 2) .+ Vec2f(latvecs[1:2,2] ./ 2) .+ Vec2f(0.3,0.3)],color=[:black,:black,:black,:blue,:blue])#hide\nf#hide","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"To use \"projection onto the new vector\" as a histogram axis, only a single change is needed to the binning parameters. The second covector (previously b) must be swapped out for frac12a + b (recall that reciprocal space covectors, such as those used in BinningParameters correspond to direct space vectors).","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"params.covectors[2,1:3] = [1/2,1,0] # [1/2,1,0] times [a;b;c] is (a/2 + b)\nparams#hide","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"The second axis of the histogram now agrees with what is conventionally labelled as [H,-H/2,0].","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"warning: Length of the new vector\nNote that, although frac12a+b is orthogonal to a, it is not the same length as a. Instead, it is sqrt(3/4) times as long. Note the unsymmetrical axes labels in the plots that follow as a direct result of this!","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"# Zoom out horizontal axis\nparams.binstart[1], params.binend[1] = -2, 2\n\n# Adjust vertical axis bounds to account for\n# length of a/2 + b\nparams.binstart[2], params.binend[2] = -2 * sqrt(3/4), 2 * sqrt(3/4)\n\n# Re-compute in the new coordinate system\nis, counts = intensities_binned(sc,params,new_formula)\n\nfig = Figure(; resolution=(1200,500))#hide\nax_right = Axis(fig[1,3];#hide\n title=\"ω≈$(round(target_ω, digits=2)) meV with Δω=0.3 meV (Binned)\", aspect=true,#hide\n xlabel = \"[H, -1/2H, 0]\"#hide\n)#hide\nbcs = axes_bincenters(params)#hide\nhm_right = heatmap!(ax_right,bcs[1],bcs[2],is[:,:,1,1] ./ counts[:,:,1,1])#hide\nadd_lines!(ax_right,params)\nColorbar(fig[1,4], hm_right);#hide\nnothing #hide","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"For comparison purposes, we will make the same plot using intensities_interpolated to emulate zero-width bins. This time, it's more convenient to think in terms of reciprocal vectors a^* and b^*. Now, our coordinate transformation consists of establishing a new, orthogonal basis to specify our wave vectors: a^* - frac12b^*, b^* and c^*. Writing this in matrix form allows us to sample a rectilinear grid of wave vectors in this frame. Finally, we'll convert these back into the original RLU system for input into Sunny.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"# New basis matrix\nA = [1 0 0\n -1/2 1 0\n 0 0 1]\n\n# Define our grid of wave vectors\nnpoints = 60\nas = range(-2, 2, npoints)\nbs = range(-3/√3, 3/√3, npoints)\nqs_ortho = [[a, b, 0] for a in as, b in bs]\n\n# Convert to original RLU system for input to Sunny\nqs = [A * q for q in qs_ortho]\n\n# Use interpolation to get intensities\nis = intensities_interpolated(sc, qs, new_formula; interpolation=:linear)\n\nax_left = Axis(fig[1,2];#hide\n title=\"ω≈$(round(ωs[ωidx], digits=2)) meV (Interpolated)\", aspect=true,#hide\n xlabel = \"[H, -1/2H, 0]\", ylabel = \"[0, K, 0]\"#hide\n)#hide\nhm_left = heatmap!(ax_left, as, bs, is[:,:,ωidx])#hide\nadd_lines!(ax_left,params)\nColorbar(fig[1,1], hm_left);#hide\nfig","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Now, not only are the dashed-line lattice vectors no longer misleadingly orthogonal, but the six-fold symmetry has been restored as well! Further, the metric has been diagonalized:","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"metric = (latvecs * inv(A'))' * I(3) * (latvecs * inv(A'))","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"Finally, we note that instantaneous structure factor data, 𝒮(𝐪), can be obtained from a dynamic structure factor with instant_intensities_interpolated. Here we'll reuse the grid of wave vectors we generated above.","category":"page"},{"location":"examples/fei2_classical.html","page":"FeI₂ at Finite Temperature","title":"FeI₂ at Finite Temperature","text":"is_static = instant_intensities_interpolated(sc, qs, new_formula; interpolation = :linear)\n\nhm = heatmap(as, bs, is_static;\n axis=(\n title=\"Instantaneous Structure Factor\",\n xlabel = \"[H, -1/2H, 0]\",\n ylabel = \"[0, K, 0]\",\n aspect=true\n )\n)\nColorbar(hm.figure[1,2], hm.plot)\nhm","category":"page"},{"location":"versions.html#Version-History","page":"Version History","title":"Version History","text":"","category":"section"},{"location":"versions.html#v0.5.6","page":"Version History","title":"v0.5.6","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Animated spin dynamics is now possible. Call notify on the result of plot_spins to trigger redrawing of the frame. The argument colorfn to plot_spins supports animation of colors.\nGeneral pair couplings are now supported in set_pair_coupling! and set_pair_coupling_at!. :SUN supports interactions of any order, but :dipole mode is limited to bilinear and biquadratic coupling of the spin.\nTo perform a calculation with dipoles in the large-S limit, use the new mode :dipole_large_S when constructing a System.\nDeprecate the option biquad to set_exchange!. Use instead set_pair_coupling!, which generalizes beyond the scalar biquadratic.\nDeprecate spin_operators, stevens_operators, large_S_spin_operators and large_S_stevens_operators. Use instead spin_matrices and stevens_matrices, which require a specific spin-S label. To infer this, one can use spin_label.\nRemove unused option energy_tol in SpinWaveTheory.","category":"page"},{"location":"versions.html#v0.5.5","page":"Version History","title":"v0.5.5","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"reshape_supercell now allows reshaping to multiples of the primitive unit cell, which can speed up certain calculations. This is illustrated in the CoRh₂O₄ powder averaging tutorial.\nresize_supercell now allows all resizings.\nAdded energy_per_site.\nset_spiral_order_on_sublattice! cannot work on reshaped systems.\nVarious bug fixes. In particular, an intensity_formula with :full will now uniformly calculate a 3x3 matrix of complex numbers.","category":"page"},{"location":"versions.html#v0.5.4","page":"Version History","title":"v0.5.4","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Various enhancements to view_crystal. Atoms are now labeled by index, and bonds support interactive inspection (GLMakie only). Font sizes work correctly on Makie v0.20-beta. If using Makie v0.19 on a high-resolution display, pass rescale=1.5 to enlarge font sizes.\nThe function suggest_magnetic_supercell now requires only a list of wavevectors, and will return a 33 matrix that can be programmatically passed to reshape_supercell. The new tolerance parameter tol allows suggest_magnetic_supercell to approximate incommensurate wavevectors with nearby commensurate ones.\nNew functions set_spiral_order! and set_spiral_order_on_sublattice! can be used to initialize a spiral, single-Q order.\nSunny now retains all constant energy shifts that have been introduced by anisotropy operators.\nFix export_vtk functionality.","category":"page"},{"location":"versions.html#v0.5.3","page":"Version History","title":"v0.5.3","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Add large_S_spin_operators and large_S_stevens_operators to support single-ion anisotropies in dipole mode without renormalization. Set large_S=true in set_exchange! to avoid renormalization of biquadratics.\nview_crystal has been rewritten in Makie.\nplot_spins now expects ghost_radius in physical length units.\nSpinWaveTheory will (currently) error if provided a system with enable_dipole_dipole!.","category":"page"},{"location":"versions.html#v0.5.2","page":"Version History","title":"v0.5.2","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Form factors for 5d transition ions.\nDownload links for notebooks and scripts on each doc example\nVarious bug fixes.","category":"page"},{"location":"versions.html#v0.5.1","page":"Version History","title":"v0.5.1","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Fix binning edge cases.\nplot_spins accepts resolution argument.","category":"page"},{"location":"versions.html#v0.5.0","page":"Version History","title":"v0.5.0","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"New features.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Support for Linear Spin Wave Theory in :dipole and :SUN modes. (Thanks Hao Zhang!)","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"New function minimize_energy! to efficiently find an optimal configuration of spin dipoles or SU(N) coherent states.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Major refactors and enhancements to intensity calculations. This new interface allows unification between LSWT and classical spin dynamics calculations. This interface allows: Custom observables as local quantum operators, better support for linebroadening, and automatic binning to facilitate comparison with experimental data. See intensity_formula for documentation. Use load_nxs to load experimental neutron scattering data.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Breaking changes.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Require Julia 1.9.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Replace set_anisotropy! with a new function set_onsite_coupling! (and similarly set_onsite_coupling_at!). The latter expects an explicit matrix representation for the local Hamiltonian. This can be constructed, e.g., as a linear combination of stevens_operators, or as a polynomial of spin_operators. To understand the mapping between these two, the new function print_stevens_expansion acts on an arbitrary local operator.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Remove set_biquadratic!. Instead, use an optional keyword argument biquad to set_exchange!.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename DynamicStructureFactor to dynamical_correlations. Similarly, replace InstantStructureFactor with instant_correlations. The return type has been renamed SampledCorrelations to emphasize that the object may be holding thermodynamic samples, which are collected using add_sample!. Upon construction, the SampledCorrelations object will be empty (no initial sample).","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Remove intensities function. Instead, use one of intensities_interpolated or intensities_binned. These will require an intensity_formula, which defines a calculator (e.g., LSWT).","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename connected_path to reciprocal_space_path, which now returns an xticks object that can be used in plotting. Replace spherical_shell with reciprocal_space_shell that functions similarly.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename polarize_spin! to set_dipole! for consistency with set_coherent!. The behavior of the former function is unchanged: the spin at a given site will still be polarized along the provided direction.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename all_sites to eachsite consistent with Julia convention for iterators.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename reshape_geometry to reshape_supercell, which is the fundamental reshaping function. Rename resize_periodically to resize_supercell.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The constructor SpinInfo now requires a g-factor or tensor as a named argument.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The constructor FormFactor no longer accepts an atom index. Instead, the form factors are associated with site-symmetry classes in order of appearance.","category":"page"},{"location":"versions.html#v0.4.3","page":"Version History","title":"v0.4.3","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Experimental support for linear SpinWaveTheory, implemented in SU(N) mode. This module may evolve rapidly.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Implement renormalization of single-ion anisotropy and biquadratic interactions when in :dipole mode. This makes the model more faithful to the quantum mechanical Hamiltonian, but is also a breaking change.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Various improvements and bugfixes for to_inhomogeneous. Setting inhomogeneous interactions via set_exchange_at! should now infer the correct bond offset direction, or will report an ambiguity error. Ambiguities can be resolved by passing an explicit offset.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The function remove_periodicity! disables periodicity along specified dimensions.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename StaticStructureFactor to InstantStructureFactor.","category":"page"},{"location":"versions.html#v0.4.2","page":"Version History","title":"v0.4.2","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Introduce LocalSampler, a framework for MCMC sampling with local spin updates.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename print_dominant_wavevectors to print_wrapped_intensities to reduce confusion with the physical instantaneous intensities.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The function spherical_shell now takes a radius in physical units of inverse Å.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"New exported functions global_position, magnetic_moment, all_sites.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Remove all uses of Base.deepcopy which resolves crashes.","category":"page"},{"location":"versions.html#v0.4.1","page":"Version History","title":"v0.4.1","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The function to_inhomogeneous creates a system that supports inhomogeneous interactions, which can be set using set_exchange_at!, etc.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"set_biquadratic! replaces set_exchange_with_biquadratic!.","category":"page"},{"location":"versions.html#v0.4.0","page":"Version History","title":"v0.4.0","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"This update includes many breaking changes, and is missing some features of 0.3.0.","category":"page"},{"location":"versions.html#Creating-a-spin-System","page":"Version History","title":"Creating a spin System","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename SpinSystem to System. Its constructor now has the form,","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"System(crystal, latsize, infos, mode)","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The parameter infos is now a list of SpinInfo objects. Each defines spin angular momentum S = frac12 1 frac32 , and an optional g-factor or tensor.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The parameter mode is one of :SUN or :dipole.","category":"page"},{"location":"versions.html#Setting-interactions","page":"Version History","title":"Setting interactions","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Interactions are now added mutably to an existing System using the following functions: set_external_field!, set_exchange!, set_onsite_coupling!, enable_dipole_dipole!.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"As a convenience, one can use dmvec(D) to convert a DM vector to a 33 antisymmetric exchange matrix.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Fully general single-ion anisotropy is now possible. The function set_onsite_coupling! expects the single ion anisotropy to be expressed as a polynomial in symbolic spin operators 𝒮, or as a linear combination of symbolic Stevens operators 𝒪. For example, an easy axis anisotropy in the direction n may be written D*(𝒮⋅n)^2.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Stevens operators 𝒪[k,q] admit polynomial expression in spin operators 𝒮[α]. Conversely, a polynomial of spin operators can be expressed as a linear combination of Stevens operators. To see this expansion use print_anisotropy_as_stevens.","category":"page"},{"location":"versions.html#Inhomogeneous-field","page":"Version History","title":"Inhomogeneous field","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"An external field can be applied to a single site with set_external_field_at!. ","category":"page"},{"location":"versions.html#Structure-factor-rewrite","page":"Version History","title":"Structure factor rewrite","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The calculation of structure factors has been completely rewritten. For the new interface, see the FeI₂ at Finite Temperature page.","category":"page"},{"location":"versions.html#Various","page":"Version History","title":"Various","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The \"Sampler\" interface is in flux. Langevin replaces both LangevinHeunP and LangevinSampler. Local spin-flip Monte Carlo sampling methods are temporarily broken.\nrepeat_periodically replaces extend_periodically.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Additional related functions include resize_periodically and reshape_geometry, the latter being fundamental.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"print_symmetry_table replaces print_bond_table().","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The new function includes the list of symmetry-allowed single ion anisotropies in addition to exchange interactions.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"When reading CIF files, the field _atom_site_label is now used in place of the field _atom_site_type_symbol.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"This is required for correctness. The field _atom_site_label is guaranteed to be present, and is guaranteed to be a distinct label for each symmetry-inequivalent site. Code that explicitly referred to site labels (e.g. in calls to subcrystal) will need to be updated to use the new label.","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"EditURL = \"../../../../examples/spinw_ports/15_Ba3NbFe3Si2O14.jl\"","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html#Tutorial-15-BaNbFeSiO","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"","category":"section"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"This is a Sunny port of SpinW Tutorial 15, originally authored by Sandor Toth. The goal is to calculate the linear spin wave theory spectrum for Ba₃NbFe₃Si₂O₁₄.","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Load packages","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Build a Crystal for Ba₃NbFe₃Si₂O₁₄ using the crystal structure from Marty et al., Phys. Rev. Lett. 101, 247201 (2008).","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"a = b = 8.539 # (Å)\nc = 5.2414\nlatvecs = lattice_vectors(a, b, c, 90, 90, 120)\ntypes = [\"Fe\",\"Nb\",\"Ba\",\"Si\",\"O\",\"O\",\"O\"]\npositions = [[0.24964,0,0.5],[0,0,0],[0.56598,0,0],[2/3,1/3,0.5220],[2/3,1/3,0.2162],[0.5259,0.7024,0.3536],[0.7840,0.9002,0.7760]]\nlangasite = Crystal(latvecs, positions, 150; types)\ncrystal = subcrystal(langasite, \"Fe\")\nview_crystal(crystal, 7)","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Create a System with a lattice size of (117). The magnetic structure of Ba₃NbFe₃Si₂O₁₄ was determined to have the ordering wavevector 𝐐=(0017) and hence the magnetic unit cell has 7 sites.","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"latsize = (1,1,7)\nS = 5/2\nseed = 5\nsys = System(crystal, latsize, [SpinInfo(1; S, g=2)], :dipole)","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Set exchange interactions as parametrized in Loire et al., Phys. Rev. Lett. 106, 207201 (2011)","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"J₁ = 0.85\nJ₂ = 0.24\nJ₃ = 0.053\nJ₄ = 0.017\nJ₅ = 0.24\nset_exchange!(sys, J₁, Bond(3, 2, [1,1,0]))\nset_exchange!(sys, J₄, Bond(1, 1, [0,0,1]))\nset_exchange!(sys, J₂, Bond(1, 3, [0,0,0]))","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"The final two exchanges define the chirality of the magnetic structure. The crystal chirality, epsilon_T, the chirality of each triangle, ϵ_D and the sense of rotation of the spin helices along c, ϵ_H. The three chiralities are related by ϵ_T=ϵ_D ϵ_H. We now assign J_3 and J_5 according to the crystal chirality.","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"ϵD = -1\nϵH = +1\nϵT = ϵD * ϵH\n\nif ϵT == -1\n set_exchange!(sys, J₃, Bond(2, 3, [-1,-1,1]))\n set_exchange!(sys, J₅, Bond(3, 2, [1,1,1]))\nelseif ϵT == 1\n set_exchange!(sys, J₅, Bond(2, 3, [-1,-1,1]))\n set_exchange!(sys, J₃, Bond(3, 2, [1,1,1]))\nelse\n throw(\"Provide a valid chirality\")\nend","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Whilst Sunny provides tools to optimize the ground state automatically, in this case we already know the model ground state. Set the spiral magnetic order using set_spiral_order_on_sublattice!. It takes an ordering wavevector q, an axis of rotation for the spins axis, and the initial spin S0 for each sublattice.","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"q = [0, 0, 1/7]\naxis = [0,0,1]\nset_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[1, 0, 0])\nset_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[-1/2, -sqrt(3)/2, 0])\nset_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[-1/2, +sqrt(3)/2, 0])\n\nplot_spins(sys; color=[s[1] for s in sys.dipoles])","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Define a path in reciprocal space, 01-1+xi for xi = 0 dots 3.","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"points_rlu = [[0,1,-1],[0,1,-1+1],[0,1,-1+2],[0,1,-1+3]];\ndensity = 100\npath, xticks = reciprocal_space_path(crystal, points_rlu, density);\nnothing #hide","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Calculate broadened intensities","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"swt = SpinWaveTheory(sys)\nγ = 0.15 # width in meV\nbroadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))\nenergies = collect(0:0.01:6) # 0 < ω < 6 (meV).\nis = intensities_broadened(swt, path, energies, broadened_formula);\nnothing #hide","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"Plot","category":"page"},{"location":"examples/spinw/15_Ba3NbFe3Si2O14.html","page":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","title":"Tutorial 15 - Ba₃NbFe₃Si₂O₁₄","text":"fig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nheatmap!(ax, 1:size(is,1), energies, is, colorrange=(0,5))\nfig","category":"page"},{"location":"index.html#Overview","page":"Overview","title":"Overview","text":"","category":"section"},{"location":"index.html","page":"Overview","title":"Overview","text":"Sunny is a Julia package for modeling atomic-scale magnetism. It provides powerful tools to study equilibrium and non-equilibrium magnetic phenomena. In particular, it allows estimation of dynamical structure factor intensities, mathcalS(𝐪ω), to support quantitative modeling of experimental scattering data.","category":"page"},{"location":"index.html","page":"Overview","title":"Overview","text":"Features include:","category":"page"},{"location":"index.html","page":"Overview","title":"Overview","text":"Generalized spin dynamics using SU(N) coherent states.\nAbility to specify a crystal from a .cif file or its spacegroup symmetry.\nInteractive visualizations of the 3D crystals and magnetic ordering.\nSymmetry analysis to classify allowed interaction terms, and to propagate them by symmetry.\nSingle-ion anisotropy at arbitrary order, which can be specified using Stevens operators or as a polynomial of spin operators.\nMonte Carlo sampling of spin configurations in thermal equilibrium, and optimization tools.\nMeasurements of dynamical correlations. At low temperature, one can use linear spin wave theory and its multi-boson generalization. This generalizes to finite temperatures using the classical dynamics, which allows for strongly nonlinear effects.\nLong-range dipole-dipole interactions accelerated with the fast Fourier transform (FFT).\nSupport for comparison with experimental data: form factor, dipole factor, temperature-dependent classical-to-quantum factors, intensity binning, etc.","category":"page"}]
+[{"location":"parallelism.html#Parallelizing-Calculations","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"","category":"section"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Calculating structure factors with classical dynamics is computationally expensive, and Sunny does not currently parallelize these calculations at a fine-grained level. However, Julia provides facilities that allow users to run multiple simulations in parallel with only a little extra effort. We will look at two approaches to doing this: multithreading and Julia's Distributed package. We'll present these approaches in a series of code snippets that can be copied and pasted into your preferred Julia development environment.","category":"page"},{"location":"parallelism.html#Review-of-the-serial-workflow","page":"Parallelizing Calculations","title":"Review of the serial workflow","text":"","category":"section"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"The serial approach to calculating a structure factor, covered in the FeI₂ tutorial, involves thermalizing a spin System and then calling add_sample!. add_sample! uses the state of the System as an initial condition for the calculation of a dynamical trajectory. The correlations of the trajectory are calculated and accumulated into a running average of the 𝒮(𝐪ω). This sequence is repeated to generate additional samples.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"To illustrate, we'll set up a a simple model: a spin-1 antiferromagnet on an FCC crystal. ","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"using Sunny, GLMakie\n\nfunction make_system(cryst; J, dims, seed=nothing)\n sys = System(cryst, dims, [SpinInfo(1, S=1, g=2)], :dipole; seed)\n set_exchange!(sys, J, Bond(1,1,[1,0,0]))\n return sys\nend\n\nJ = 1.0 # meV \ncryst = Sunny.fcc_primitive_crystal()\nsys = make_system(cryst; J, dims=(10,10,2))","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"To thermalize and generate equilibrium samples, we'll need a Langevin integrator. ","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"kT = 0.5 # meV\nΔt = 0.05/J\nintegrator = Langevin(Δt; kT, λ=0.1)","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Finally, call dynamical_correlations to configure a SampledCorrelations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"sc = dynamical_correlations(sys; Δt=0.1, nω=100, ωmax=10.0)","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"The serial calculation can now be performed as follows:","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"nsamples = 10\n\n# Thermalize the system\nfor _ in 1:5000 # Sufficient number of steps to thermalize\n step!(sys, integrator)\nend\n\nfor _ in 1:nsamples\n # Generate new sample by running Langevin dynamics\n for _ in 1:1000 # Sufficient number of steps to decorrelate\n step!(sys, integrator)\n end\n # Add the sample to the correlation data\n add_sample!(sc, sys)\nend","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"This will take a second or two on a modern workstation, resulting in a single SampledCorrelations that contains 10 samples.","category":"page"},{"location":"parallelism.html#Multithreading-approach","page":"Parallelizing Calculations","title":"Multithreading approach","text":"","category":"section"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"To use threads in Julia, you must launch your Julia environment appropriately.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"From the command line, launch Julia with julia --threads=N, where N is the number of threads. If you don't know how many threads you'd like, you can let Julia decide with --threads=auto.\nJupyter notebook users will need to to set up a multithreaded Julia kernel and restart into this kernel. The kernel can be created inside Julia with the following:","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":" using IJulia\n IJulia.installkernel(\"Julia Multithreaded\",\n env=Dict(\"JULIA_NUM_THREADS\" => \"auto\"))","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"VSCode users should open their settings and search for Julia: Additional Args. There will be link called Edit in settings.json. Click on this, add \"--threads=auto\" to the list julia.additionalArgs, and start a new REPL.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Before going further, make sure that Threads.nthreads() returns a number greater than 1.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"We will use multithreading in a very simple way, essentially employing a distributed memory approach to avoid conflicts around memory access. First preallocate a number of systems and correlations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"npar = Threads.nthreads()\nsystems = [make_system(cryst; J, dims=(10,10,2), seed=i) for i in 1:npar]\nscs = [dynamical_correlations(sys; Δt=0.1, nω=100, ωmax=10.0) for _ in 1:npar]","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"warning: Dealing with memory constraints\nIf you have many threads available and are working with a large system, you may not have enough memory to store all these systems and correlations. In that case, simply reduce npar to a small enough value that you can make the necessary allocations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"When the Threads.@threads macro is applied before a for loop, the iterations of the loop will execute in parallel using the available threads. We will put the entire thermalization and sampling process inside the loop, with each thread acting on a unique System and SampledCorrelations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Threads.@threads for id in 1:npar\n integrator = Langevin(Δt; kT, λ=0.1)\n for _ in 1:5000\n step!(systems[id], integrator)\n end\n for _ in 1:nsamples\n for _ in 1:1000\n step!(systems[id], integrator)\n end\n add_sample!(scs[id], systems[id])\n end\nend","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"You may find this takes a little bit longer than the serial example, but, at the end of it, you will have generated npar correlation objects, each with 10 samples. We can merge these into a summary SampledCorrelations with 10*npar samples with merge_correlations:","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"sc = merge_correlations(scs)","category":"page"},{"location":"parallelism.html#Using-Distributed","page":"Parallelizing Calculations","title":"Using Distributed","text":"","category":"section"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Julia also provides a distributed memory approach to parallelism through the standard library package Distributed. This works by launching independent Julia environments on different \"processes.\" An advantage of this approach is that it scales naturally to clusters since the processes are easily distributed across many different compute nodes. A disadvantage, especially when working on a single computer, is the increased memory overhead associated with launching all these environments.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"We begin by importing the package,","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"using Distributed","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"and launching some new processes. It is often sensible to create as many processes as there are cores.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"ncores = length(Sys.cpu_info())\naddprocs(ncores)","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"You can think of each process as a separate computer running a fresh Julia environment, so we'll need to import Sunny and define our function in each of these environments. This is easily achieved with the @everywhere macro.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"@everywhere using Sunny\n\n@everywhere function make_system(cryst; J, dims, seed=nothing)\n sys = System(cryst, dims, [SpinInfo(1, S=1, g=2)], :dipole; seed)\n set_exchange!(sys, J, Bond(1,1,[1,0,0]))\n return sys\nend","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"A simple way to perform work on these processes is to use the parallel map function, pmap. This will apply a function to each element of some iterable, such as a list of numbers, and return a list of the results. It is a parallel map because these function calls may occur at the same time on different Julia processes. The pmap function takes care of distributing the work among the different processes and retrieving the results.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"In the example below, we give pmap a list of RNG seeds to iterate over, and we define the function that will be applied to each of these seeds in a do block. The contents of this block are essentially the same as what we put inside our parallel for loop in the multithreading example. The main difference is that the Systems and SampledCorrelations are not created in advance of the parallelization but are instead created inside each Julia process. The do block returns a SampledCorrelations, and the output of all the parallel computations are collected into list of SampledCorrelations called scs.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"scs = pmap(1:ncores) do seed\n sys = make_system(Sunny.fcc_primitive_crystal(); J=1.0, dims=(10,10,2), seed)\n sc = dynamical_correlations(sys; Δt=0.1, nω=100, ωmax=10.0)\n integrator = Langevin(Δt; kT, λ=0.1)\n\n for _ in 1:5000 # Thermalize\n step!(sys, integrator)\n end\n for _ in 1:nsamples \n for _ in 1:1000 \n step!(sys, integrator)\n end\n add_sample!(sc, sys)\n end\n\n return sc\nend","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"Finally, merge the results into a summary SampledCorrelations.","category":"page"},{"location":"parallelism.html","page":"Parallelizing Calculations","title":"Parallelizing Calculations","text":"sc = merge_correlations(scs)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"EditURL = \"../../../examples/01_LSWT_SU3_FeI2.jl\"","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#.-Multi-flavor-spin-wave-simulations-of-FeI-(Showcase)","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"FeI₂ is an effective spin-1 material with strong single-ion anisotropy. Quadrupolar fluctuations give rise to a single-ion bound state that cannot be described by a dipole-only model. This tutorial illustrates how to use the linear spin wave theory of SU(3) coherent states (i.e. 2-flavor bosons) to model the magnetic behavior in FeI₂. The original study was performed in Bai et al., Nature Physics 17, 467–472 (2021).","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The Fe atoms are arranged in stacked triangular layers. The effective spin Hamiltonian takes the form,","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"mathcalH=sum_(ij) 𝐒_i J_ij 𝐒_j - Dsum_i left(S_i^zright)^2","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"where the set of exchange matrices J_ij between bonded sites (ij) includes competing ferromagnetic and antiferromagnetic interactions. This model also includes a strong easy axis anisotropy, D 0.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"We will formulate this Hamiltonian in Sunny and then calculate its dynamic structure factor.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#Get-Julia-and-Sunny","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"Get Julia and Sunny","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Sunny is implemented in Julia. This is a relatively new programming language that allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users may wish to take a look at our Getting Started with Julia guide. Sunny requires Julia 1.9 or later.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"From the Julia prompt, load Sunny. For plotting, one can choose either GLMakie (a pop-up window) or WGLMakie (inline plots for a Jupyter notebook).","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"If these packages are not yet installed, Julia should offer to install them using its built-in package management system. If old versions are installed, you may need to update them to run this tutorial.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#Crystals","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"Crystals","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"A Crystal describes the crystallographic unit cell and will usually be loaded from a .cif file. Here, we instead build a crystal by listing all atoms and their types.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"a = b = 4.05012 # Lattice constants for triangular lattice\nc = 6.75214 # Spacing in the z-direction\n\nlatvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that\n # define the conventional unit cell\npositions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions\n # of lattice vectors\ntypes = [\"Fe\", \"I\", \"I\"]\nFeI2 = Crystal(latvecs, positions; types)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Observe that Sunny inferred the space group, 'P -3 m 1' (164) and labeled the atoms according to their point group symmetries.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Only the Fe atoms are magnetic, so we discard the I ions using subcrystal.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"cryst = subcrystal(FeI2, \"Fe\")","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Importantly, cryst retains the spacegroup symmetry of the full FeI₂ crystal. This information will be used, for example, to propagate exchange interactions between symmetry-equivalent bonds.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"In a running Julia environment, the crystal can be viewed interactively using view_crystal.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"view_crystal(cryst, 8.0)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#Symmetry-analysis","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"Symmetry analysis","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The command print_symmetry_table provides a list of all the symmetry-allowed interactions up to a cutoff distance.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"print_symmetry_table(cryst, 8.0)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The allowed g-tensor is expressed as a 3×3 matrix in the free coefficients A, B, ... The allowed single-ion anisotropy is expressed as a linear combination of Stevens operators. The latter correspond to polynomials of the spin operators, as we will describe below.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The allowed exchange interactions are given as a 3×3 matrix for representative bonds. The notation Bond(i, j, n) indicates a bond between atom indices i and j, with cell offset n. In the general case, it will be necessary to associate atom indices with their positions in the unit cell; these can be viewed with display(cryst). Note that the order of the pair (i j) is significant if the exchange tensor contains antisymmetric Dzyaloshinskii–Moriya (DM) interactions.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"In the case of FeI₂, Bond(1, 1, [1,0,0]) is one of the 6 nearest-neighbor Fe-Fe bonds on a triangular lattice layer, and Bond(1, 1, [0,0,1]) is an Fe-Fe bond between layers.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#Building-a-spin-System","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"Building a spin System","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"In constructing a spin System, we must provide several additional details about the spins.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"sys = System(cryst, (4, 4, 4), [SpinInfo(1, S=1, g=2)], :SUN, seed=2)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"This system includes 444 unit cells, i.e. 64 Fe atoms, each with spin S=1 and a g-factor of 2. Quantum mechanically, spin S=1 involves a superposition of 2S+1=3 distinct angular momentum states. In :SUN mode, this superposition will be modeled explicitly using the formalism of SU(3) coherent states, which captures both dipolar and quadrupolar fluctuations. For the more traditional dipole dynamics, use :dipole mode instead.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Next we will use set_exchange! to assign interaction to bonds. Sunny will automatically propagate each interaction to all symmetry-equivalent bonds in the unit cell. The FeI₂ interactions below follow Bai et al.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"J1pm = -0.236\nJ1pmpm = -0.161\nJ1zpm = -0.261\nJ2pm = 0.026\nJ3pm = 0.166\nJ′0pm = 0.037\nJ′1pm = 0.013\nJ′2apm = 0.068\n\nJ1zz = -0.236\nJ2zz = 0.113\nJ3zz = 0.211\nJ′0zz = -0.036\nJ′1zz = 0.051\nJ′2azz = 0.073\n\nJ1xx = J1pm + J1pmpm\nJ1yy = J1pm - J1pmpm\nJ1yz = J1zpm\n\nset_exchange!(sys, [J1xx 0.0 0.0;\n 0.0 J1yy J1yz;\n 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))\nset_exchange!(sys, [J2pm 0.0 0.0;\n 0.0 J2pm 0.0;\n 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))\nset_exchange!(sys, [J3pm 0.0 0.0;\n 0.0 J3pm 0.0;\n 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))\nset_exchange!(sys, [J′0pm 0.0 0.0;\n 0.0 J′0pm 0.0;\n 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))\nset_exchange!(sys, [J′1pm 0.0 0.0;\n 0.0 J′1pm 0.0;\n 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))\nset_exchange!(sys, [J′2apm 0.0 0.0;\n 0.0 J′2apm 0.0;\n 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The function set_onsite_coupling! assigns a single-ion anisotropy. The argument can be constructed using spin_matrices or stevens_matrices. Here we use Julia's anonymous function syntax to assign an easy-axis anisotropy along the direction hatz.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"D = 2.165\nset_onsite_coupling!(sys, S -> -D*S[3]^2, 1)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#Calculating-structure-factor-intensities","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"Calculating structure factor intensities","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"In the remainder of this tutorial, we will examine Sunny's tools for calculating the dynamical structure factor using a multi-boson generalization of linear spin wave theory (LSWT). This theory describes non-interacting quasi-particle excitations that hybridize dipolar and quadrupolar modes.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#Finding-the-ground-state","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"Finding the ground state","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Begin with a random configuration and use minimize_energy! to find a configuration of the SU(3) coherent states (i.e. spin dipoles and quadrupoles) that locally minimizes energy.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"randomize_spins!(sys)\nminimize_energy!(sys)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"A positive number above indicates that the procedure has converged to a local energy minimum. The configuration, however, may still have defects. This can be checked by visualizing the spins, colored according to their z-components.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"plot_spins(sys; color=[s[3] for s in sys.dipoles])","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"A different understanding of the magnetic ordering can be obtained by moving to Fourier space. The 'instant' structure factor 𝒮(𝐪) is an experimental observable. To investigate 𝒮(𝐪) as true 3D data, Sunny provides instant_correlations and related functions. Here, however, we will use print_wrapped_intensities, which gives average intensities for the individual Bravais sublattices (in effect, all wavevectors are wrapped to the first Brillouin zone).","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"print_wrapped_intensities(sys)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The result will likely be approximately consistent with the known zero-field energy-minimizing magnetic structure of FeI₂, which is single-Q (two-up, two-down antiferromagnetic order). Mathematically, spontaneous symmetry breaking should select one of Q = 0 -14 14, 14 0 14, or -141414, associated with the three-fold rotational symmetry of the crystal spacegroup. In nature, however, one will frequently encounter competing \"domains\" associated with the three possible orientations of the ground state.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"If the desired ground state is already known, as with FeI₂, it could be entered by hand using set_dipole!. Alternatively, in the case of FeI₂, we could repeatedly employ the above randomization and minimization procedure until a defect-free configuration is found. Some systems will have more complicated ground states, which can be much more challenging to find. For this, Sunny provides experimental support for powerful simulated annealing via parallel tempering, but that is outside the scope of this tutorial.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Here, let's break the three-fold symmetry of FeI₂ by hand. Given one or more desired Q modes, Sunny can suggest a magnetic supercell with appropriate periodicity. Let's arbitrarily select one of the three possible ordering wavevectors, Q = 0 -14 14. Sunny suggests a corresponding magnetic supercell in units of the crystal lattice vectors.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"suggest_magnetic_supercell([[0, -1/4, 1/4]])","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The system returned by reshape_supercell is smaller, and is sheared relative to the original system. This makes it much easier to find the global energy minimum.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"sys_min = reshape_supercell(sys, [1 0 0; 0 2 1; 0 -2 1])\nrandomize_spins!(sys_min)\nminimize_energy!(sys_min);\nnothing #hide","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Plot the system again, now including \"ghost\" spins out to 12Å","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"plot_spins(sys_min; color=[s[3] for s in sys_min.dipoles], ghost_radius=12)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#Linear-spin-wave-theory","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"Linear spin wave theory","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Now that we have found the ground state for a magnetic supercell, we can immediately proceed to perform zero-temperature calculations using linear spin wave theory. We begin by instantiating a SpinWaveTheory type using the supercell.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"swt = SpinWaveTheory(sys_min)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"Select a sequence of wavevectors that will define a piecewise linear interpolation in reciprocal lattice units (RLU).","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"q_points = [[0,0,0], [1,0,0], [0,1,0], [1/2,0,0], [0,1,0], [0,0,0]];\nnothing #hide","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The function reciprocal_space_path will linearly sample a path between the provided q-points with a given density. The xticks return value provides labels for use in plotting.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"density = 50\npath, xticks = reciprocal_space_path(cryst, q_points, density);\nnothing #hide","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The dispersion function defines the quasiparticle excitation energies ω_i(𝐪) for each point 𝐪 along the reciprocal space path.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"disp = dispersion(swt, path);\nnothing #hide","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"In addition to the band energies ω_i(𝐪), Sunny can calculate the inelastic neutron scattering intensity I_i(𝐪) for each band i according to an intensity_formula. We choose to apply a polarization correction (1 - 𝐪𝐪) by setting the mode argument to :perp. Selecting delta_function_kernel specifies that we want the energy and intensity of each band individually.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The function intensities_bands uses linear spin wave theory to calculate both the dispersion and intensity data for the provided path.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"disp, intensity = intensities_bands(swt, path, formula);\nnothing #hide","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"These can be plotted in GLMakie.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"fig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nylims!(ax, 0.0, 7.5)\nxlims!(ax, 1, size(disp, 1))\ncolorrange = extrema(intensity)\nfor i in axes(disp, 2)\n lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange)\nend\nfig","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"To make comparisons with inelastic neutron scattering (INS) data, it is helpful to employ an empirical broadening kernel, e.g., a lorentzian.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"γ = 0.15 # width in meV\nbroadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The intensities_broadened function requires an energy range in addition to the 𝐪-space path.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"energies = collect(0:0.01:10) # 0 < ω < 10 (meV).\nis1 = intensities_broadened(swt, path, energies, broadened_formula);\nnothing #hide","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"A real FeI₂ sample will exhibit competing magnetic domains associated with spontaneous symmetry breaking of the 6-fold rotational symmetry of the triangular lattice. Note that the wavevectors 𝐪 and -𝐪 are equivalent in the structure factor, which leaves three distinct domain orientations, which are related by 120° rotations about the z-axis. Rather than rotating the spin configuration directly, on can rotate the 𝐪-space path. Below, we use rotation_in_rlu to average the intensities over all three possible orientations.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"R = rotation_in_rlu(cryst, [0, 0, 1], 2π/3)\nis2 = intensities_broadened(swt, [R*q for q in path], energies, broadened_formula)\nis3 = intensities_broadened(swt, [R*R*q for q in path], energies, broadened_formula)\nis_averaged = (is1 + is2 + is3) / 3\n\nfig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nheatmap!(ax, 1:size(is_averaged, 1), energies, is_averaged)\nfig","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"This result can be directly compared to experimental neutron scattering data from Bai et al.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"(The publication figure accidentally used a non-standard coordinate system to label the wave vectors.)","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"To get this agreement, the use of SU(3) coherent states is essential. In other words, we needed a theory of multi-flavored bosons. The lower band has large quadrupolar character, and arises from the strong easy-axis anisotropy of FeI₂. By setting mode = :SUN, the calculation captures this coupled dipole-quadrupole dynamics.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"An interesting exercise is to repeat the same study, but using mode = :dipole instead of :SUN. That alternative choice would constrain the coherent state dynamics to the space of dipoles only.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The full dynamical spin structure factor (DSSF) can be retrieved as a 33 matrix with the dssf function, for a given path of 𝐪-vectors.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"disp, is = dssf(swt, path);\nnothing #hide","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The first output disp is identical to that obtained from dispersion. The second output is contains a list of 33 matrix of intensities. For example, is[q,n][2,3] yields the (yz) component of the structure factor intensity for nth mode at the qth wavevector in the path.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html#What's-next?","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"What's next?","text":"","category":"section"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The multi-boson linear spin wave theory, applied above, can be understood as the quantization of a certain generalization of the Landau-Lifshitz spin dynamics. Rather than dipoles, this dynamics takes places on the space of SU(N) coherent states.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The full SU(N) coherent state dynamics, with appropriate quantum correction factors, can be useful to model finite temperature scattering data. In particular, it captures certain anharmonic effects due to thermal fluctuations. See our generalized spin dynamics tutorial.","category":"page"},{"location":"examples/01_LSWT_SU3_FeI2.html","page":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","title":"1. Multi-flavor spin wave simulations of FeI₂ (Showcase)","text":"The classical dynamics is also a good starting point to study non-equilibrium phenomena. Empirical noise and damping terms can be used to model coupling to a thermal bath. This yields a Langevin dynamics of SU(N) coherent states. Our dynamical SU(N) quench tutorial illustrates how a temperature quench can give rise to novel liquid phase of CP² skyrmions.","category":"page"},{"location":"structure-factor.html#Structure-Factor-Calculations","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"","category":"section"},{"location":"structure-factor.html#Overview","page":"Structure Factor Calculations","title":"Overview","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The dynamical structure factor is of fundamental importance for characterizing a magnetic system, and facilitates quantitative comparison between theory and experimental scattering data.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Consider, for example, a two-point dynamical spin correlation function, s^α(𝐱+Δ𝐱 t+Δt) s^β(𝐱 t). Here s^α(𝐱 t) represents the time dynamics of a spin dipole component α at position 𝐱, and brackets represent an average over equilibrium initial conditions and over (𝐱 t). The dynamical structure factor is defined as the Fourier transform of this two-point correlation in both space and time, up to an overall scaling factor. Using the convolution theorem, the result is,","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ(𝐪 ω) = frac1V s^α(𝐪 ω)^ast s^β(𝐪 ω) ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"with V the system volume. We will restrict attention to lattice systems with periodic boundaries.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Consider a crystal unit cell defined by three lattice vectors 𝐚_1 𝐚_2 𝐚_3, and linear system sizes L_1 L_2 L_3 measured in unit cells. The allowed momentum vectors take on discrete values 𝐪 = sum_α=1^3 m_α 𝐛_α L_α, where m_α are an integers and the reciprocal lattice vectors 𝐛_α are defined to satisfy 𝐚_α 𝐛_β = 2π δ_αβ. For a Bravais lattice, 𝐪 will be periodic in the first Brillouin zone, i.e., under any shift 𝐪 𝐪 𝐛_α. More generally, consider a non-Bravais lattice such that each unit cell may contain multiple spins. By partitioning spins s_j(𝐱t) according to their sublattice index j, the relevant momenta 𝐪 remain discretized as above, but now periodicity in the first Brillouin zone is lost. The structure factor may be written as a phase-average over the displacements between sublattices 𝐫_jk,","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ(𝐪 ω) = _jk e^i 𝐫_jk 𝐪 𝒮^αβ_jk(𝐪 ω) ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"From a theoretical perspective, the quantity","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ_jk(𝐪 ω) = frac1V s_j^α(𝐪 ω)^ast s_k^β(𝐪 ω)","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"is fundamental. For each sublattice j, the data s_j^α(𝐪 ω) can be efficiently obtained by fast Fourier tranformation of a real space configuration s_j^α(𝐱 t). Internally, Sunny will calculate and store the discrete 𝒮^αβ_jk(𝐪 ω) correlation data, and use this to construct 𝒮^αβ(𝐪ω) intensities that can be compared with experiment.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Calculating this structure factor involves several steps, with various possible settings. Sunny provides a number of tools to facilitate this calculation and to extract information from the results. These tools are briefly outlined below. Please see the Examples for a \"real life\" use case. Detailed function information is available in the Library API.","category":"page"},{"location":"structure-factor.html#Estimating-stucture-factors-with-classical-dynamics","page":"Structure Factor Calculations","title":"Estimating stucture factors with classical dynamics","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Classical dynamics may be used to estimate structure factor data by analyzing the spin-spin correlations of dynamical trajectories. This is fundamentally a Monte Carlo approach, as the trajectories must be started from an initial spin configuration that is sampled at thermal equilibrium. (Note that it is not possible to estimate a true T=0 dynamical structure factor using this method, but the temperature may be very low.) Samples are accumulated into a SampledCorrelations, from which intensity information may be extracted. The user does not typically build their own SampledCorrelations but instead initializes one by calling either dynamical_correlations or instant_correlations, as described below.","category":"page"},{"location":"structure-factor.html#Estimating-a-dynamical-structure-factor:-𝒮(𝐪,ω)","page":"Structure Factor Calculations","title":"Estimating a dynamical structure factor: 𝒮(𝐪ω)","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A SampledCorrelations for estimating the dynamical structure factor, 𝒮^αβ(𝐪ω), may be created by calling dynamical_correlations. This requires three keyword arguments. These will determine the dynamics used to calculate samples and, consequently, the ω information that will be available. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Δt: Determines the step size used for simulating the dynamics. A smaller number will require proportionally more calculation time. While a smaller Δt will enable the resolution of higher energies, Δt is typically selected to ensure numerical stability rather than to maximize the largest ω value. A safe choice is to use the smaller value of Δt = 0.1/(J* S^2) or Δt = 0.1/(D * S), where S is magnetic moment of the largest local spin (as specified in SpinInfo), J is the parameter governing the largest bilinear interaction (e.g. exchange), and D is the parameter governing the largest single-site term of the Hamiltonian (e.g., anisotropy or Zeeman term).\nωmax: Sets the maximum resolved energy. Note that this is not independent of Δt. If ωmax too large, Sunny will throw an error and ask you to choose a smaller Δt. \nnω: Determines the number of energy bins to resolve. A larger number will require more calculation time.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A sample may be added by calling add_sample!(sc, sys). The input sys must be a spin configuration in good thermal equilibrium, e.g., using the continuous Langevin dynamics or using single spin flip trials with LocalSampler. The statistical quality of the 𝒮^αβ(𝐪ω) can be improved by repeatedly generating decorrelated spin configurations in sys and calling add_sample! on each configuration.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The outline of typical use case might look like this:","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"# Make a `SampledCorrelations`\nsc = dynamical_correlations(sys; Δt=0.05, ωmax=10.0, nω=100) \n\n# Add samples\nfor _ in 1:nsamples\n decorrelate_system(sys) # Perform some type of Monte Carlo simulation\n add_sample!(sc, sys) # Use spins to calculate trajectory and accumulate new sample of 𝒮(𝐪,ω)\nend","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The calculation may be configured in a number of ways; see the dynamical_correlations documentation for a list of all keywords.","category":"page"},{"location":"structure-factor.html#Estimating-an-instantaneous-(\"static\")-structure-factor:-𝒮(𝐪)","page":"Structure Factor Calculations","title":"Estimating an instantaneous (\"static\") structure factor: 𝒮(𝐪)","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Sunny provides two methods for calculating instantaneous, or static, structure factors: 𝒮^αβ(𝐪). The first involves calculating spatial spin-spin correlations at single time slices. The second involves calculating a dynamic structure factor first and integrating out the ω information. The advantage of the latter approach is that it enables application of an ω-dependent classical-to-quantum rescaling of structure factor intensities, a method that should be preferred whenever comparing results to experimental data or spin wave calculations. A disadvantage of this approach is that it is computationally more expensive. There are also many cases when it is not straightforward to calculate a meaningful dynamics, as when working with Ising spins. In this section we will discuss how to calculate instantaneous structure factors from static spin configurations. Information about calculating instantaneous data from a dynamical correlations can be found in the following section.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The basic usage for the instantaneous case is very similar to the dynamic case, except one calls instant_correlations instead of dynamical_correlations to configure a SampledCorrelations. Note that there are no required keywords as there is no need to specify any dynamics. instant_correlations will return a SampledCorrelations containing no data. Samples may be added by calling add_sample!(sc, sys), where sc is the SampledCorrelations. When performing a finite-temperature calculation, it is important to ensure that the spin configuration in the sys represents a good equilibrium sample, as in the dynamical case. Note, however, that we recommend calculating instantaneous correlations at finite temperature calculations by using full dynamics (i.e., using dynamical_correlations) and then integrating out the energy axis. An approach to doing this is described in the next section.","category":"page"},{"location":"structure-factor.html#Extracting-information-from-sampled-correlation-data","page":"Structure Factor Calculations","title":"Extracting information from sampled correlation data","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The basic function for extracting information from a SampledCorrelations at a particular wave vector, 𝐪, is intensities_interpolated. It takes a SampledCorrelations, a list of wave vectors, and an intensity_formula. The intensity_formula specifies how to contract and correct correlation data to arrive at a physical intensity. A simple example is formula = intensity_formula(sc, :perp), which will instruct Sunny apply polarization corrections: sum_αβ(I-q_α q_β) 𝒮^αβ(𝐪ω). An intensity at the wave vector 𝐪 = (𝐛_2 + 𝐛_3)2 may then be retrieved with intensities_interpolated(sf, [[0.0, 0.5, 0.5]], formula) . intensities_interpolated returns a list of nω elements at each wavevector. The corresponding ω values can be retrieved by calling available_energies on sf.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Since Sunny only calculates the structure factor on a finite lattice when performing classical simulations, it is important to realize that exact information is only available at a discrete set of wave vectors. Specifically, for each axis index i, we will get information at q_i = fracnL_i, where n runs from (frac-L_i2+1) to fracL_i2 and L_i is the linear dimension of the lattice used for the calculation. If you request a wave vector that does not fall into this set, Sunny will automatically round to the nearest 𝐪 that is available. If intensities_interpolated is given the keyword argument interpolation=:linear, Sunny will use trilinear interpolation to determine a result at the requested wave vector. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"To retrieve the intensities at all wave vectors for which there is exact data, first call the function available_wave_vectors to generate a list of qs. This takes an optional keyword argument bzsize, which must be given a tuple of three integers specifying the number of Brillouin zones to calculate, e.g., bzsize=(2,2,2). The resulting list of wave vectors may then be passed to intensities_interpolated.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Alternatively, intensities_binned can be used to place the exact data into histogram bins for comparison with experiment.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The convenience function reciprocal_space_path returns a list of wavevectors sampled along a path that connects specified 𝐪 points. This list can be used as an input to intensities. Another convenience method, reciprocal_space_shell will generate points on a sphere of a given radius. This is useful for powder averaging. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A number of arguments for intensity_formula are available which modify the calculation of structure factor intensity. It is generally recommended to provide a value of kT corresponding to the temperature of sampled configurations. Given kT, Sunny will include an energy- and temperature-dependent classical-to-quantum rescaling of intensities in the formula.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"To retrieve intensity data from a instantaneous structure factor, use instant_intensities_interpolated, which accepts similar arguments to intensities_interpolated. This function may also be used to calculate instantaneous information from a dynamical correlation data, i.e. from a SampledCorrelations created with dynamical_correlations. Note that it is important to supply a value to kT to reap the benefits of this approach over simply calculating a static structure factor at the outset. ","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"EditURL = \"../../../examples/02_LSWT_CoRh2O4.jl\"","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html#.-Spin-wave-simulations-of-CoRhO","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"","category":"section"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"This tutorial illustrates the conventional spin wave theory of dipoles. We consider a simple model of the diamond-cubic crystal CoRh₂O₄, with parameters extracted from Ge et al., Phys. Rev. B 96, 064413.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"Construct a diamond Crystal in the conventional (non-primitive) cubic unit cell. Sunny will populate all eight symmetry-equivalent sites when given the international spacegroup number 227 (\"Fd-3m\") and the appropriate setting. For this spacegroup, there are two conventional translations of the unit cell, and it is necessary to disambiguate through the setting keyword argument. (On your own: what happens if setting is omitted?)","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"a = 8.5031 # (Å)\nlatvecs = lattice_vectors(a, a, a, 90, 90, 90)\ncryst = Crystal(latvecs, [[0,0,0]], 227, setting=\"1\")","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"In a running Julia environment, the crystal can be viewed interactively using view_crystal.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"view_crystal(cryst, 8.0)","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"Construct a System with quantum spin S=32 constrained to the space of dipoles. Including an antiferromagnetic nearest neighbor interaction J will favor Néel order. To optimize this magnetic structure, it is sufficient to employ a magnetic lattice consisting of a single crystal unit cell, latsize=(1,1,1). Passing an explicit random number seed will ensure repeatable results.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"latsize = (1, 1, 1)\nS = 3/2\nJ = 7.5413*meV_per_K # (~ 0.65 meV)\nsys = System(cryst, latsize, [SpinInfo(1; S, g=2)], :dipole; seed=0)\nset_exchange!(sys, J, Bond(1, 3, [0,0,0]))","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"In the ground state, each spin is exactly anti-aligned with its 4 nearest-neighbors. Because every bond contributes an energy of -JS^2, the energy per site is -2JS^2. In this calculation, a factor of 1/2 avoids double-counting the bonds. Due to lack of frustration, direct energy minimization is successful in finding the ground state.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"randomize_spins!(sys)\nminimize_energy!(sys)\n\n@assert energy_per_site(sys) ≈ -2J*S^2","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"Plotting the spins confirms the expected Néel order. Note that the overall, global rotation of dipoles is arbitrary.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"s0 = sys.dipoles[1,1,1,1]\nplot_spins(sys; color=[s'*s0 for s in sys.dipoles])","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"For numerical efficiency, it will be helpful to work with the smallest possible magnetic supercell. Here, it is the primitive unit cell, which contains just two sites. The variable shape below defines the primitive lattice vectors cryst.prim_latvecs in units of the conventional lattice vectors. This result is used as input to reshape_supercell. The energy per site remains the same, which verifies that the magnetic supercell is valid.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"shape = cryst.latvecs \\ cryst.prim_latvecs\nsys_prim = reshape_supercell(sys, shape)\n@assert energy_per_site(sys_prim) ≈ -2J*S^2","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"Now estimate 𝒮(𝐪ω) with SpinWaveTheory and an intensity_formula. The mode :perp contracts with a dipole factor to return the unpolarized intensity. The formula also employs lorentzian broadening. The isotropic FormFactor for Cobalt(2+) dampens intensities at large 𝐪.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"swt = SpinWaveTheory(sys_prim)\nη = 0.4 # (meV)\nkernel = lorentzian(η)\nformfactors = [FormFactor(\"Co2\")]\nformula = intensity_formula(swt, :perp; kernel, formfactors)","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"For the \"single crystal\" result, we may use reciprocal_space_path to construct a path that connects high-symmetry points in reciprocal space. The intensities_broadened function collects intensities along this path for the given set of energy values.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"qpoints = [[0, 0, 0], [1/2, 0, 0], [1/2, 1/2, 0], [0, 0, 0]]\npath, xticks = reciprocal_space_path(cryst, qpoints, 50)\nenergies = collect(0:0.01:6)\nis = intensities_broadened(swt, path, energies, formula)\n\nfig = Figure()\nax = Axis(fig[1,1]; aspect=1.4, ylabel=\"ω (meV)\", xlabel=\"𝐪 (r.l.u.)\",\n xticks, xticklabelrotation=π/10)\nheatmap!(ax, 1:size(is, 1), energies, is, colormap=:gnuplot2)\nfig","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"A powder measurement effectively involves an average over all possible crystal orientations. We use the function reciprocal_space_shell to sample n wavevectors on a sphere of a given radius (inverse angstroms), and then calculate the spherically-averaged intensity.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"radii = 0.01:0.02:3 # (1/Å)\noutput = zeros(Float64, length(radii), length(energies))\nfor (i, radius) in enumerate(radii)\n n = 300\n qs = reciprocal_space_shell(cryst, radius, n)\n is = intensities_broadened(swt, qs, energies, formula)\n output[i, :] = sum(is, dims=1) / size(is, 1)\nend\n\nfig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Q (Å⁻¹)\", ylabel=\"ω (meV)\")\nheatmap!(ax, radii, energies, output, colormap=:gnuplot2)\nfig","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"This result can be compared to experimental neutron scattering data from Fig. 5 of Ge et al.","category":"page"},{"location":"examples/02_LSWT_CoRh2O4.html","page":"2. Spin wave simulations of CoRh₂O₄","title":"2. Spin wave simulations of CoRh₂O₄","text":"","category":"page"},{"location":"writevtk.html#ParaView-Rendering","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The 4D correlation data produced by Sunny is too high-dimensional to visualize directly. This page describes how to export 3D slices of correlation data from Sunny to the Visual ToolKit (VTK) format, which is compatible with the ParaView visualization software. ParaView supports volumetric rendering:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html#Simulation-data","page":"ParaView Rendering","title":"Simulation data","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"First, generate some correlation data in Sunny. We will use a 2D lattice, since the correlation data S(Q_xQ_yomega) is 3D and can be exported in its entirety. The following code sets up the system, thermalizes it, and records the correlation data in a SampledCorrelations called dsf.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"using Sunny\n\n# Single layer 12x12 periodic square lattice\nlatsize = (12,12,1)\n\nlatvecs = lattice_vectors(8.,8.,12.,90,100,90)\npositions = [[0,0,0]]\ntypes = [\"Cu\"]\nformfactors = [FormFactor(\"Cu2\")]\nxtal = Crystal(latvecs, positions; types)\n\nsys = System(xtal, latsize, [SpinInfo(1, S=1/2, g=2)], :SUN; seed=1)\n\nJ = 10.\nset_exchange!(sys, J, Bond(1,1,[1,0,0]))\nset_exchange!(sys, J, Bond(1,1,[0,1,0]))\n\nΔt = 0.01\nkT = 0.5\nlangevin = Langevin(Δt; λ=0.5, kT)\nrandomize_spins!(sys)\nfor i in 1:10_000 # Long enough to reach equilibrium\n step!(sys, langevin)\nend \n\nωmax=10.\n\ndsf = dynamical_correlations(sys\n ;Δt=Δt\n ,nω=48\n ,ωmax=ωmax\n ,process_trajectory=:symmetrize)\n\nnsamples = 10\nfor _ in 1:nsamples\n for _ in 1:1000 \n step!(sys, langevin)\n end\n add_sample!(dsf, sys)\nend","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The default histogram BinningParameters are already integrated over the z direction because the system is 2D:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"unit_resolution_binning_parameters(dsf)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 12 bins from -0.042 to +0.958 along [+1.27 dx] (Δ = 0.065)\n⊡ 12 bins from -0.042 to +0.958 along [+1.27 dy] (Δ = 0.065)\n∫ Integrated from +0.000 to +0.000 along [-0.33 dx +1.88 dz] (Δ = 0.524)\n⊡ 48 bins from -0.107 to +10.134 along [+1.00 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The histogram is very oblong; it's approximately 1x1x10. To make it a nicer shape, we will rescale the energy axis to be be fractions of ωmax:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"params = unit_resolution_binning_parameters(dsf)\nscale_factor = ωmax\nparams.binend[4] /= scale_factor\nparams.binstart[4] /= scale_factor\nparams.binwidth[4] /= scale_factor\nparams.covectors[4,:] ./= scale_factor","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Doing this changes the last axis of the histogram to fit in [0,1]:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 49 bins from -0.011 to +1.013 along [+0.10 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that our histogram is a cube, we compute the intensity in the histogram bins using the usual intensities_binned:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"formula = intensity_formula(dsf,:trace)\nsignal, counts = intensities_binned(dsf, params; formula)\nintensity = signal ./ counts","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that we have our intensity data and the binning parameters, we can export to VTK format using export_vtk and move to ParaView for the visualization.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"# Importing WriteVTK enables Sunny's export-to-VTK functions\nimport WriteVTK\n\n# [1,2,4] specifies that the (x,y,z) axes in ParaView are (Qx,Qy,ω)\nexport_vtk(\"square_lattice\", params, intensity; dims_kept = [1,2,4])\n# Writes a file square_lattice.vti in the current directory","category":"page"},{"location":"writevtk.html#Loading-in-ParaView","page":"ParaView Rendering","title":"Loading in ParaView","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In ParaView, use File > Open to open square_lattice.vti. This will add the file to the Pipeline Browser with a closed eye icon, indicating that the data is ready to be loaded.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In the Properties panel, both bin_centers and data will be selected for import by default. Uncheck bin_centers because we don't need that information for the visualization. Click the green Apply button to load the data.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"By default, only the outline of the data is shown in the 3D viewport. Since we adjusted the energy axis, the outline is a 1x1x1 cube. Optionally enable the axes grid under \"View\", and customize using the adjacent edit button.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"To enable the volumetric render:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Select \"Volume\" from the \"Representation\" drop-down menu under \"Display\".\nThe \"Coloring\" drop-down should automatically select data because it's the only data loaded.\nOpen the Color Map Editor to adjust the opacity of the fog, which may be too faint to see by default.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Depending on your computer and your dataset size, the volumetric rendering may be slow, but our dataset is relatively small, so the render should be fast.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"If nothing shows up at first, don't despair. Often, there are Bragg-like peaks in the correlation data which outshine everything else. To see this, enable Display Data Histogram in the Color Map Editor panel. To zoom in on the lower-intensity data, click and drag the right side handle of the opacity transfer function box to the middle a few times.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"After suitable color mapping, the dispersion curve should become visible:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"page"},{"location":"writevtk.html#Experiment-data","page":"ParaView Rendering","title":"Experiment data","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Note that since only the data and binning parameters are required for exporting to VTK, experiment data can be exported in the same way. For example, to visualize S(Q_xQ_yQ_z), do this:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"# Load 4D histogram data from Mantid\nparams, signal = load_nxs(\"experiment_data.nxs\")\n\n# Integrate out the energy axis so we are 3D\nintegrate_axes!(params; axes = 4)\nsignal = sum(signal; dims = 4)\n\n# Export to ParaView\nexport_vtk(\"experiment_data_as_vtk\", params, signal)","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"EditURL = \"../../../examples/04_GSD_FeI2.jl\"","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/04_GSD_FeI2.html#.-Generalized-spin-dynamics-of-FeI-at-finite-*T*","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"","category":"section"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"using Sunny, LinearAlgebra, GLMakie","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"In the previous FeI₂ tutorial, we used multi-flavor spin wave theory to calculate the dynamical structure factor. Here, we perform a similar calculation using classical spin dynamics at finite temperature. Because we are interested in the coupled dynamics of spin dipoles and quadrupoles, we employ a classical dynamics of SU(3) coherent states that generalizes the Landau-Lifshitz equation.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Compared to LSWT, simulations using classical dynamics are much slower, and are limited in k-space resolution. However, they make it is possible to capture nonlinear effects associated with finite temperature fluctuations. Classical dynamics are also appealing for studying out-of-equilibrium systems (e.g., relaxation of spin glasses), or systems with quenched inhomogeneities that require large simulation volumes.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"In this tutorial, we show how to study the finite temperature dynamics of FeI₂ using the classical approach. It is important to stress that the estimation of S(𝐪ω) with classical dynamics is fundamentally a Monte Carlo calculation: sample spin configurations are drawn from thermal equilibrium and used as initial conditions for generating dissipationless trajectories. The correlations of these trajectories are then averaged and used to calculate scattering intensities. It is therefore important to ensure that the initial spin configurations are sampled appropriately and that sufficient statistics are collected. We will demonstrate one approach here.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"As an overview, we will:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Identify the ground state\nMeasure correlation data describing the excitations around that ground state\nUse the correlation data to compute scattering intensities","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"As the implementation of the FeI₂ model is already covered in detail in the LSWT tutorial, we will not repeat it below. Instead, we will assume that you already have defined a sys in the same way with lattice dimensions 444.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"a = b = 4.05012#hide\nc = 6.75214#hide\nlatvecs = lattice_vectors(a, b, c, 90, 90, 120)#hide\npositions = [[0,0,0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]]#hide\ntypes = [\"Fe\", \"I\", \"I\"]#hide\nFeI2 = Crystal(latvecs, positions; types)#hide\ncryst = subcrystal(FeI2, \"Fe\")#hide\nsys = System(cryst, (4,4,4), [SpinInfo(1,S=1,g=2)], :SUN, seed=2)#hide\nJ1pm = -0.236#hide\nJ1pmpm = -0.161#hide\nJ1zpm = -0.261#hide\nJ2pm = 0.026#hide\nJ3pm = 0.166#hide\nJ′0pm = 0.037#hide\nJ′1pm = 0.013#hide\nJ′2apm = 0.068#hide\nJ1zz = -0.236#hide\nJ2zz = 0.113#hide\nJ3zz = 0.211#hide\nJ′0zz = -0.036#hide\nJ′1zz = 0.051#hide\nJ′2azz = 0.073#hide\nJ1xx = J1pm + J1pmpm#hide\nJ1yy = J1pm - J1pmpm#hide\nJ1yz = J1zpm#hide\nset_exchange!(sys, [J1xx 0.0 0.0; 0.0 J1yy J1yz; 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))#hide\nset_exchange!(sys, [J2pm 0.0 0.0; 0.0 J2pm 0.0; 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))#hide\nset_exchange!(sys, [J3pm 0.0 0.0; 0.0 J3pm 0.0; 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))#hide\nset_exchange!(sys, [J′0pm 0.0 0.0; 0.0 J′0pm 0.0; 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))#hide\nset_exchange!(sys, [J′1pm 0.0 0.0; 0.0 J′1pm 0.0; 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))#hide\nset_exchange!(sys, [J′2apm 0.0 0.0; 0.0 J′2apm 0.0; 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))#hide\nD = 2.165#hide\nset_onsite_coupling!(sys, S -> -D*S[3]^2, 1)#hide\nsys","category":"page"},{"location":"examples/04_GSD_FeI2.html#Finding-a-ground-state","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"Finding a ground state","text":"","category":"section"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Sunny uses the Langevin dynamics of SU(N) coherent states to sample spin configurations from the thermal equlibrium. One first constructs a Langevin integrator. This requires a time step, temperature, and a phenomenological damping parameter λ that sets the coupling to the thermal bath.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Δt = 0.05/D # Should be inversely proportional to the largest energy scale\n # in the system. For FeI2, this is the easy-axis anisotropy,\n # `D = 2.165` (meV). The prefactor 0.05 is relatively small,\n # and achieves high accuracy.\nkT = 0.2 # Temperature of the thermal bath (meV).\nλ = 0.1 # This value is typically good for Monte Carlo sampling,\n # independent of system details.\n\nlangevin = Langevin(Δt; kT, λ);\nnothing #hide","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Langevin dynamics can be used to search for a magnetically ordered state. For this, the temperature kT must be below the ordering temperature, but large enough that the dynamical sampling procedure can overcome local energy barriers and eliminate defects.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"randomize_spins!(sys)\nfor _ in 1:20_000\n step!(sys, langevin)\nend","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Although thermal fluctuations are present, the correct antiferromagnetic order (2 up, 2 down) is apparent.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"plot_spins(sys; color=[s[3] for s in sys.dipoles])","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"For other systems, it can be much harder to find the magnetic ordering in an unbiased way, and more complicated sampling procedures may be necessary.","category":"page"},{"location":"examples/04_GSD_FeI2.html#Calculating-Thermal-Averaged-Correlations-\\langle-S{\\alpha\\beta}(𝐪,ω)\\rangle","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"Calculating Thermal-Averaged Correlations langle S^alphabeta(𝐪ω)rangle","text":"","category":"section"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Our aim is to study the classical spin dynamics for states sampled in thermal equilibrium. To minimize finite size effects, and achieve sufficient momentum space resolution, we should significantly enlarge the system volume. The function resize_supercell takes new dimensions as multiples of the unit cell lattice vectors.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"sys_large = resize_supercell(sys, (16,16,4)) # 16x16x4 copies of the original unit cell\nplot_spins(sys_large; color=[s[3] for s in sys_large.dipoles])","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Now we will re-thermalize the system to a configuration just above the ordering temperature. Sunny expects energies in meV by default, so we use meV_per_K to convert from kelvin.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"kT = 3.5 * meV_per_K # 3.5K ≈ 0.30 meV\nlangevin.kT = kT\nfor _ in 1:10_000\n step!(sys_large, langevin)\nend","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"The next step is to collect correlation data S^alphabeta. This will involve sampling spin configurations from thermal equilibrium, and then integrating the Hamiltonian dynamics of SU(N) coherent states to collect Fourier-space information about normal modes. Quantization of these modes yields the magnons, and the associated dynamical spin-spin correlations can be compared with neutron scattering intensities S^alphabeta(qomega). Because this a real-space calculation, data is only available for discrete q modes (the resolution scales like inverse system size).","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"To store the correlation data, we initialize a SampledCorrelations object by calling dynamical_correlations. It requires three keyword arguments: an integration step size, a target number of ωs to retain, and a maximum energy ω to resolve. For the time step, twice the value used for the Langevin integrator is usually a good choice.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"sc = dynamical_correlations(sys_large; Δt=2Δt, nω=120, ωmax=7.5)","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"The function add_sample! will collect data by running a dynamical trajectory starting from the current system configuration.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"add_sample!(sc, sys_large) # Accumulate the sample into `sc`","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"To collect additional data, it is required to re-sample the spin configuration from the thermal distribution. For efficiency, the dynamics should be run long enough that consecutive samples are uncorrelated.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"for _ in 1:2\n for _ in 1:1000 # Enough steps to decorrelate spins\n step!(sys_large, langevin)\n end\n add_sample!(sc, sys_large) # Accumulate the sample into `sc`\nend","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Now, sc has more samples included:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"sc","category":"page"},{"location":"examples/04_GSD_FeI2.html#Computing-Scattering-Intensities","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"Computing Scattering Intensities","text":"","category":"section"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"With the thermally-averaged correlation data langle S^alphabeta(qomega)rangle in hand, we now need to specify how to extract a scattering intensity from this information. This is done by constructing an intensity_formula. By way of example, we will use a formula which computes the trace of the structure factor and applies a classical-to-quantum temperature-dependent rescaling kT.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"formula = intensity_formula(sc, :trace; kT)","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Recall that langle S^alphabeta(qomega)rangle is only available at certain discrete q values, due to the finite lattice size. There are two basic approaches to handling this discreteness. The first approach is to interpolate between the available data using intensities_interpolated. For example, we can plot single-q slices at (0,0,0) and (π,π,π) using this method:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"qs = [[0, 0, 0], [0.5, 0.5, 0.5]]\nis = intensities_interpolated(sc, qs, formula; interpolation = :round)\n\nωs = available_energies(sc)\nfig = lines(ωs, is[1,:]; axis=(xlabel=\"meV\", ylabel=\"Intensity\"), label=\"(0,0,0)\")\nlines!(ωs, is[2,:]; label=\"(π,π,π)\")\naxislegend()\nfig","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"The resolution in energy can be improved by increasing nω (and decreasing Δt), and the general accuracy can be improved by collecting additional samples from the thermal equilibrium.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"For real calculations, one often wants to apply further corrections and more accurate formulas. Here, we apply FormFactor corrections appropriate for Fe2 magnetic ions, and a dipole polarization correction :perp.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"formfactors = [FormFactor(\"Fe2\"; g_lande=3/2)]\nnew_formula = intensity_formula(sc, :perp; kT, formfactors = formfactors)","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Frequently, one wants to extract energy intensities along lines that connect special wave vectors–a so-called \"spaghetti plot\". The function reciprocal_space_path creates an appropriate horizontal axis for this plot by linearly sampling between provided q-points with a given sample density. The number of sample points between two wavevectors q1 and q2 is given by dist*density where dist = norm(cryst.recipvecs * (q1 - q2)) is measured in the global frame.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"points = [[0, 0, 0], # List of wave vectors that define a path\n [1, 0, 0],\n [0, 1, 0],\n [1/2, 0, 0],\n [0, 1, 0],\n [0, 0, 0]]\ndensity = 40\npath, xticks = reciprocal_space_path(cryst, points, density);\nnothing #hide","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Again using intensities_interpolated, we can evaluate the (interpolated) intensity at each point on the path. Since scattering intensities are only available at a certain discrete (Qomega) points, the intensity on the path can be calculated by interpolating between these discrete points:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"is_interpolated = intensities_interpolated(sc, path, new_formula;\n interpolation = :linear, # Interpolate between available wave vectors\n);\n# Add artificial broadening\nis_interpolated_broadened = broaden_energy(sc, is, (ω, ω₀)->lorentzian(ω-ω₀, 0.05));\nnothing #hide","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"The second approach to handle the discreteness of the data is to bin the intensity at the discrete points into the bins of a histogram. First, the five sub-histograms are set up using reciprocal_space_path_bins in analogy to reciprocal_space_path.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"cut_width = 0.3\ndensity = 15\nparamsList, markers, ranges = reciprocal_space_path_bins(sc,points,density,cut_width);\nnothing #hide","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Then, the intensity data is computed using intensities_binned for each sub-histogram:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"total_bins = ranges[end][end]\nenergy_bins = paramsList[1].numbins[4]\nis_binned = zeros(Float64,total_bins,energy_bins)\nintegrated_kernel = integrated_lorentzian(0.05) # Lorentzian broadening\nfor k in eachindex(paramsList)\n bin_data, counts = intensities_binned(sc,paramsList[k], new_formula;\n integrated_kernel = integrated_kernel\n )\n is_binned[ranges[k],:] = bin_data[:,1,1,:] ./ counts[:,1,1,:]\nend","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"The graph produced by interpolating (top) is similar to the one produced by binning (bottom):","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"fig = Figure()\nax_top = Axis(fig[1,1],ylabel = \"meV\",xticklabelrotation=π/8,xticklabelsize=12;xticks)\nax_bottom = Axis(fig[2,1],ylabel = \"meV\",xticks = (markers, string.(points)),xticklabelrotation=π/8,xticklabelsize=12)\n\nheatmap!(ax_top,1:size(is_interpolated,1), ωs, is_interpolated;\n colorrange=(0.0,0.07),\n)\n\nheatmap!(ax_bottom,1:size(is_binned,1), ωs, is_binned;\n colorrange=(0.0,0.05),\n)\n\nfig","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Note that we have clipped the colors in order to make the higher-energy excitations more visible.","category":"page"},{"location":"examples/04_GSD_FeI2.html#Unconventional-RLU-Systems-and-Constant-Energy-Cuts","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"Unconventional RLU Systems and Constant Energy Cuts","text":"","category":"section"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Often it is useful to plot cuts across multiple wave vectors but at a single energy. We'll pick an energy,","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"ωidx = 60\ntarget_ω = ωs[ωidx]","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"and take a constant-energy cut at that energy. The most straightforward way is to make a plot whose axes are aligned with the conventional reciprocal lattice of the crystal. This is accomplished using unit_resolution_binning_parameters:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"params = unit_resolution_binning_parameters(sc)\nparams.binstart[1:2] .= -1 # Expand plot range slightly\n\n# Set energy integration range\nomega_width = 0.3\nparams.binstart[4] = target_ω - (omega_width/2)\nparams.binend[4] = target_ω # `binend` should be inside (e.g. at the center) of the range\nparams.binwidth[4] = omega_width\n\nintegrate_axes!(params, axes = 3) # Integrate out z direction entirely\n\nparams","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"In each of the following plots, black dashed lines represent (direct) lattice vectors. Since these plots are in reciprocal space, direct lattice vectors are represented as covectors (i.e. coordinate grids) instead of as arrows.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"is, counts = intensities_binned(sc,params,new_formula)\n\nfig = Figure()\nax = Axis(fig[1,1];\n title=\"Δω=0.3 meV (Binned)\", aspect=true,\n xlabel = \"[H, 0, 0]\",\n ylabel = \"[0, K, 0]\"\n)\nbcs = axes_bincenters(params)\nhm = heatmap!(ax,bcs[1],bcs[2],is[:,:,1,1] ./ counts[:,:,1,1])\nfunction add_lines!(ax,params)#hide\n bes = Sunny.axes_binedges(params)#hide\n hrange = range(-2,2,length=17)#hide\n linesegments!(ax,[(Point2f(params.covectors[1,1:3] ⋅ [h,-10,0],params.covectors[2,1:3] ⋅ [h,-10,0]),Point2f(params.covectors[1,1:3] ⋅ [h,10,0],params.covectors[2,1:3] ⋅ [h,10,0])) for h = hrange],linestyle=:dash,color=:black)#hide\n krange = range(-2,2,length=17)#hide\n linesegments!(ax,[(Point2f(params.covectors[1,1:3] ⋅ [-10,k,0],params.covectors[2,1:3] ⋅ [-10,k,0]),Point2f(params.covectors[1,1:3] ⋅ [10,k,0],params.covectors[2,1:3] ⋅ [10,k,0])) for k = krange],linestyle=:dash,color=:black)#hide\n xlims!(ax,bes[1][1],bes[1][end])#hide\n ylims!(ax,bes[2][1],bes[2][end])#hide\nend#hide\nadd_lines!(ax,params)\nColorbar(fig[1,2], hm);\nfig","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"In the above plot, the dashed-line (direct) lattice vectors are clearly orthogonal. However, we know that in real space, the lattice vectors a and b are not orthogonal, but rather point along the edges of a hexagon (see lower left corner):","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":" ","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Thus, plotting the direct lattice vectors as orthogonal (even in reciprocal space) is somewhat misleading. Worse yet, the [H,0,0] by [0,K,0] plot apparently loses the 6-fold symmetry of the crystal! Lastly, if one works out the components of the real-space metric with respect to the axes of the plot, one finds that there are non-zero off-diagonal entries,","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"latvecs = sys.crystal.latvecs\nmetric = latvecs' * I(3) * latvecs","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"so real-space rotations and angles map into reciprocal space rotations angles in a complicated way.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"To resolve these important issues, we want to use axes which are orthogonal (i.e. they diagonalize the metric and solve all of the problems just mentioned). The canonical choice is to use the combination frac12a + b of lattice vectors (equiv. a^* - frac12b^*), which is orthogonal to a:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"(latvecs * [1/2,1,0]) ⋅ (latvecs * [1,0,0]) == 0","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"This new vector frac12a+b is visibly orthogonal to a in real space:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"f = Figure()#hide\nax = Axis(f[1,1])#hide\narrows!(ax,[Point2f(0,0),Point2f(latvecs[1:2,1] ./ 2)],[Vec2f(latvecs[1:2,1] ./ 2), Vec2f(latvecs[1:2,2])],arrowcolor = :blue,arrowsize = 30.,linewidth = 5.,linecolor = :blue)#hide\narrows!(ax,[Point2f(0,0)],[Vec2f(latvecs[1:2,:] * [1/2,1,0])],arrowcolor = :red,arrowsize = 30.,linewidth = 5.,linecolor = :red, linestyle = :dash)#hide\nscatter!(ax,[Point2f(latvecs[1:2,:] * [a,b,0]) for a in -1:1, b in -1:1][:],color = :black)#hide\nannotations!(ax,[\"0\",\"0+b\",\"0+a\", \"a/2\", \"b\"],[Point2f(0,-0.3),Point2f(latvecs[1:2,2]) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1]) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1] ./ 4) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1] ./ 2) .+ Vec2f(latvecs[1:2,2] ./ 2) .+ Vec2f(0.3,0.3)],color=[:black,:black,:black,:blue,:blue])#hide\nf#hide","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"To use \"projection onto the new vector\" as a histogram axis, only a single change is needed to the binning parameters. The second covector (previously b) must be swapped out for frac12a + b (recall that reciprocal space covectors, such as those used in BinningParameters correspond to direct space vectors).","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"params.covectors[2,1:3] = [1/2,1,0] # [1/2,1,0] times [a;b;c] is (a/2 + b)\nparams#hide","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"The second axis of the histogram now agrees with what is conventionally labelled as [H,-H/2,0].","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"warning: Length of the new vector\nNote that, although frac12a+b is orthogonal to a, it is not the same length as a. Instead, it is sqrt(3/4) times as long. Note the unsymmetrical axes labels in the plots that follow as a direct result of this!","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"# Zoom out horizontal axis\nparams.binstart[1], params.binend[1] = -2, 2\n\n# Adjust vertical axis bounds to account for\n# length of a/2 + b\nparams.binstart[2], params.binend[2] = -2 * sqrt(3/4), 2 * sqrt(3/4)\n\n# Re-compute in the new coordinate system\nis, counts = intensities_binned(sc,params,new_formula)\n\nfig = Figure(; resolution=(1200,500))#hide\nax_right = Axis(fig[1,3];#hide\n title=\"ω≈$(round(target_ω, digits=2)) meV with Δω=0.3 meV (Binned)\", aspect=true,#hide\n xlabel = \"[H, -1/2H, 0]\"#hide\n)#hide\nbcs = axes_bincenters(params)#hide\nhm_right = heatmap!(ax_right,bcs[1],bcs[2],is[:,:,1,1] ./ counts[:,:,1,1])#hide\nadd_lines!(ax_right,params)\nColorbar(fig[1,4], hm_right);#hide\nnothing #hide","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"For comparison purposes, we will make the same plot using intensities_interpolated to emulate zero-width bins. This time, it's more convenient to think in terms of reciprocal vectors a^* and b^*. Now, our coordinate transformation consists of establishing a new, orthogonal basis to specify our wave vectors: a^* - frac12b^*, b^* and c^*. Writing this in matrix form allows us to sample a rectilinear grid of wave vectors in this frame. Finally, we'll convert these back into the original RLU system for input into Sunny.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"# New basis matrix\nA = [1 0 0\n -1/2 1 0\n 0 0 1]\n\n# Define our grid of wave vectors\nnpoints = 60\nas = range(-2, 2, npoints)\nbs = range(-3/√3, 3/√3, npoints)\nqs_ortho = [[a, b, 0] for a in as, b in bs]\n\n# Convert to original RLU system for input to Sunny\nqs = [A * q for q in qs_ortho]\n\n# Use interpolation to get intensities\nis = intensities_interpolated(sc, qs, new_formula; interpolation=:linear)\n\nax_left = Axis(fig[1,2];#hide\n title=\"ω≈$(round(ωs[ωidx], digits=2)) meV (Interpolated)\", aspect=true,#hide\n xlabel = \"[H, -1/2H, 0]\", ylabel = \"[0, K, 0]\"#hide\n)#hide\nhm_left = heatmap!(ax_left, as, bs, is[:,:,ωidx])#hide\nadd_lines!(ax_left,params)\nColorbar(fig[1,1], hm_left);#hide\nfig","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Now, not only are the dashed-line lattice vectors no longer misleadingly orthogonal, but the six-fold symmetry has been restored as well! Further, the metric has been diagonalized:","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"metric = (latvecs * inv(A'))' * I(3) * (latvecs * inv(A'))","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"Finally, we note that instantaneous structure factor data, 𝒮(𝐪), can be obtained from a dynamic structure factor with instant_intensities_interpolated. Here we'll reuse the grid of wave vectors we generated above.","category":"page"},{"location":"examples/04_GSD_FeI2.html","page":"4. Generalized spin dynamics of FeI₂ at finite T","title":"4. Generalized spin dynamics of FeI₂ at finite T","text":"is_static = instant_intensities_interpolated(sc, qs, new_formula; interpolation = :linear)\n\nhm = heatmap(as, bs, is_static;\n axis=(\n title=\"Instantaneous Structure Factor\",\n xlabel = \"[H, -1/2H, 0]\",\n ylabel = \"[0, K, 0]\",\n aspect=true\n )\n)\nColorbar(hm.figure[1,2], hm.plot)\nhm","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"EditURL = \"../../../examples/06_CP2_Skyrmions.jl\"","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html#.-Dynamical-quench-into-CP-skyrmion-liquid","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"","category":"section"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"This example demonstrates Sunny's ability to simulate the out-of-equilibrium dynamics of generalized spin systems. We will implement the model Hamiltonian of Zhang et al., Nature Communications 14, 3626 (2023), which supports a novel type of topological defect, a CP² skyrmion, that involves both the dipolar and quadrupolar parts of a quantum spin.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"Beginning from an initial high-temperature state, a disordered gas of CP² skyrmions can be formed by rapidly quenching to low temperature. To model the coupled dynamics of dipoles and quadrupoles, Sunny uses a recently developed generalization of the Landau-Lifshitz spin dynamics, Dahlbom et al., Phys. Rev. B 106, 235154 (2022).","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"The Hamiltonian we will implement,","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"mathcalH = sum_langle ij rangle J_ij( hatS_i^x hatS_j^x + hatS_i^y hatS_j^y + DeltahatS_i^z hatS_j^z) - hsum_ihatS_i^z + Dsum_i(hatS_i^z)^2","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"contains competing ferromagnetic nearest-neightbor and antiferromagnetic next-nearest-neighbor exchange terms on a triangular lattice. Both exchanges exhibit anisotropy on the z-term. Additionally, there is an external magnetic field, h, and easy-plane single-ion anisotropy, D 0. We begin by implementing the Crystal.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"lat_vecs = lattice_vectors(1.0, 1.0, 2.0, 90, 90, 120)\nbasis_vecs = [[0,0,0]]\ncryst = Crystal(lat_vecs, basis_vecs)","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"The crystal is then used to create a spin System. All parameters in this model system are dimensionless, so we select \"theory\" units and set the g-factor to one.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"L = 40\ndims = (L, L, 1)\nsys = System(cryst, dims, [SpinInfo(1, S=1, g=1)], :SUN; seed=101, units=Units.theory)","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"We proceed to implement each term of the Hamiltonian, selecting our parameters so that the system occupies a region of the phase diagram that supports skyrmions. The exchange interactions are set as follows.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"J1 = -1 # Nearest-neighbor ferromagnetic\nJ2 = (2.0/(1+√5)) # Tune competing exchange to set skyrmion scale length\nΔ = 2.6 # Exchange anisotropy\n\nex1 = J1 * [1.0 0.0 0.0;\n 0.0 1.0 0.0;\n 0.0 0.0 Δ]\nex2 = J2 * [1.0 0.0 0.0;\n 0.0 1.0 0.0;\n 0.0 0.0 Δ]\nset_exchange!(sys, ex1, Bond(1, 1, [1, 0, 0]))\nset_exchange!(sys, ex2, Bond(1, 1, [1, 2, 0]))","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"Next we add the external field,","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"h = 15.5\nfield = set_external_field!(sys, [0.0 0.0 h])","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"and finally an easy-plane single-ion anisotropy,","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"D = 19.0\nset_onsite_coupling!(sys, S -> D*S[3]^2, 1)","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"Initialize system to an infinite temperature (fully randomized) initial condition.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"randomize_spins!(sys)","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"We are now ready to simulate the quenching process using a generalized Langevin spin dynamics. If we were working with spin dipoles only, then Langevin dynamics would be the usual Landau-Lifshitz spin dynamics, augmented with damping and noise terms. In the present study, we are instead working with quantum spin-1 (an (N=3)-level system that includes both dipoles and quadrupoles). Here, Langevin captures the coupled dipole-quadrupole dynamics using the formalism of SU(N) coherent states.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"Selecting kT = 0 in the Langevin dynamics will effective disable the noise term. Then the parameter λ effectively determines the damping time-scale.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"Δt = 0.2/D # Integration time step (inverse meV). Typically this will be\n # inversely proportional to the largest energy scale in the\n # system. We can use a fairly large time-step here because\n # accuracy isn't critical.\nkT = 0 # Target equilibrium temperature (meV)\nλ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)\nintegrator = Langevin(Δt; kT, λ)","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"Finally we run the dynamics. We will record the state of the system at three different times during the quenching process by copying the coherents field of the System.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"τs = [4., 16, 256] # Times to record snapshots\nframes = [] # Empty array to store snapshots\nfor i in eachindex(τs)\n dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate\n numsteps = round(Int, dur/Δt)\n for _ in 1:numsteps # Perform the integration\n step!(sys, integrator)\n end\n push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration\nend","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"To visualize the state of the system contained in each snapshot, we will calculate and plot the skyrmion density on each plaquette of our lattice. The function plot_triangular_plaquettes is not part of the core Sunny package, but rather something you could define yourself. We are using the definition in plotting2d.jl from the Sunny examples/extra directory.","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"include(pkgdir(Sunny, \"examples\", \"extra\", \"Plotting\", \"plotting2d.jl\"))\n\nfunction sun_berry_curvature(z₁, z₂, z₃)\n z₁, z₂, z₃ = normalize.((z₁, z₂, z₃))\n n₁ = z₁ ⋅ z₂\n n₂ = z₂ ⋅ z₃\n n₃ = z₃ ⋅ z₁\n return angle(n₁ * n₂ * n₃)\nend\n\nplot_triangular_plaquettes(sun_berry_curvature, frames; resolution=(1800,600),\n offset_spacing=10, texts = [\"\\tt = \"*string(τ) for τ in τs], text_offset = (0.0, 6.0)\n)","category":"page"},{"location":"examples/06_CP2_Skyrmions.html","page":"6. Dynamical quench into CP² skyrmion liquid","title":"6. Dynamical quench into CP² skyrmion liquid","text":"The times are given in hbarJ_1. The white background corresponds to a quantum paramagnetic state, where the local spin exhibits a strong quadrupole moment and little or no dipole moment. Observe that the process has generated a number of well-formed skyrmions of both positive (red) and negative (blue) charge in addition to a number of other metastable spin configurations. A full-sized version of this figure is available in Dahlbom et al..","category":"page"},{"location":"library.html#Library-API","page":"Library API","title":"Library API","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"This page describes the public types and functions exported by Sunny. This documentation can be also be accessed using the Julia help system (enter ? at the Julia command prompt).","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"Sunny.plot_spins\nSunny.view_crystal\nSunny.export_vtk","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"Modules = [Sunny]\nPrivate = false","category":"page"},{"location":"library.html#Sunny.Site","page":"Library API","title":"Sunny.Site","text":"(cell1, cell2, cell3, i) :: Site\n\nFour indices identifying a single site in a System. The first three indices select the lattice cell and the last selects the sublattice (i.e., the atom within the unit cell).\n\nThis object can be used to index dipoles and coherents fields of a System. A Site is also required to specify inhomogeneous interactions via functions such as set_external_field_at! or set_exchange_at!.\n\nNote that the definition of a cell may change when a system is reshaped. In this case, it is convenient to construct the Site using position_to_site, which always takes a position in fractional coordinates of the original lattice vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Units","page":"Library API","title":"Sunny.Units","text":"Units.meV\nUnits.theory\n\nThe unit system is implicitly determined by the definition of two physical constants: the vacuum permeability μ₀ and the Bohr magneton μ_B. Temperatures are effectively measured in units of energy (k_B = 1) and time is effectively measured in units of inverse energy (ħ = 1). The default unit system, Units.meV, employs (meV, Å, tesla). Select alternatively Units.theory for a units system defined so that μ₀ = μ_B = 1.\n\nSee also meV_per_K\n\n\n\n\n\n","category":"constant"},{"location":"library.html#Sunny.meV_per_K","page":"Library API","title":"Sunny.meV_per_K","text":"meV_per_K = 0.086173332621451774\n\nA physical constant. Useful for converting kelvin into the default energy units, meV.\n\n\n\n\n\n","category":"constant"},{"location":"library.html#Sunny.BinningParameters","page":"Library API","title":"Sunny.BinningParameters","text":"BinningParameters(binstart,binend,binwidth;covectors = I(4))\nBinningParameters(binstart,binend;numbins,covectors = I(4))\n\nDescribes a 4D parallelepided histogram in a format compatible with experimental Inelasitic Neutron Scattering data. See generate_mantid_script_from_binning_parameters to convert BinningParameters to a format understandable by the Mantid software, or load_nxs to load BinningParameters from a Mantid .nxs file.\n\nThe coordinates of the histogram axes are specified by multiplication of (q,ω) with each row of the covectors matrix, with q given in [R.L.U.]. Since the default covectors matrix is the identity matrix, the default axes are (qx,qy,qz,ω) in absolute units.\n\nThe convention for the binning scheme is that:\n\nThe left edge of the first bin starts at binstart\nThe bin width is binwidth\nThe last bin contains binend\nThere are no \"partial bins;\" the last bin may contain values greater than binend. C.f. count_bins.\n\nA value can be binned by computing its bin index:\n\ncoords = covectors * value\nbin_ix = 1 .+ floor.(Int64,(coords .- binstart) ./ binwidth)\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Bond","page":"Library API","title":"Sunny.Bond","text":"Bond(i, j, n)\n\nRepresents a bond between atom indices i and j. n is a vector of three integers specifying unit cell displacement in terms of lattice vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Crystal","page":"Library API","title":"Sunny.Crystal","text":"An object describing a crystallographic unit cell and its space group symmetry. Constructors are as follows:\n\nCrystal(filename; symprec=1e-5)\n\nReads the crystal from a .cif file located at the path filename. The optional parameter symprec controls the precision tolerance for spacegroup symmetries.\n\nCrystal(latvecs, positions; types=nothing, symprec=1e-5)\n\nConstructs a crystal from the complete list of atom positions positions, with coordinates (between 0 and 1) in units of lattice vectors latvecs. Spacegroup symmetry information is automatically inferred. The optional parameter types is a list of strings, one for each atom, and can be used to break symmetry-equivalence between atoms.\n\nCrystal(latvecs, positions, spacegroup_number; types=nothing, setting=nothing, symprec=1e-5)\n\nBuilds a crystal by applying symmetry operators for a given international spacegroup number. For certain spacegroups, there are multiple possible unit cell settings; in this case, a warning message will be printed, and a list of crystals will be returned, one for every possible setting. Alternatively, the optional setting string will disambiguate between unit cell conventions.\n\nCurrently, crystals built using only the spacegroup number will be missing some symmetry information. It is generally preferred to build a crystal from a .cif file or from the full specification of the unit cell.\n\nExamples\n\n# Read a Crystal from a .cif file\nCrystal(\"filename.cif\")\n\n# Build an FCC crystal using the primitive unit cell. The spacegroup number\n# 225 is inferred.\nlatvecs = [1 1 0;\n 1 0 1;\n 0 1 1] / 2\npositions = [[0, 0, 0]]\nCrystal(latvecs, positions)\n\n# Build a CsCl crystal (two cubic sublattices). By providing distinct type\n# strings, the spacegroup number 221 is inferred.\nlatvecs = lattice_vectors(1, 1, 1, 90, 90, 90)\npositions = [[0,0,0], [0.5,0.5,0.5]]\ntypes = [\"Na\", \"Cl\"]\ncryst = Crystal(latvecs, positions; types)\n\n# Build a diamond cubic crystal from its spacegroup number 227. This\n# spacegroup has two possible settings (\"1\" or \"2\"), which determine an\n# overall unit cell translation.\nlatvecs = lattice_vectors(1, 1, 1, 90, 90, 90)\npositions = [[1, 1, 1] / 4]\ncryst = Crystal(latvecs, positions, 227; setting=\"1\")\n\nSee also lattice_vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.FormFactor-Tuple{String}","page":"Library API","title":"Sunny.FormFactor","text":"FormFactor(ion::String; g_lande=2)\n\nThe magnetic form factor for a given magnetic ion and charge state. When passed to an intensity_formula, determines a 𝐪-dependent scaling of the structure factor.\n\nThe parameter ion must be one of the following strings:\n\nAm2, Am3, Am4, Am5, Am6, Am7, Au1, Au2, Au3, Au4, Au5, Ce2, Co0, Co1, Co2, Co3,\nCo4, Cr0, Cr1, Cr2, Cr3, Cr4, Cu0, Cu1, Cu2, Cu3, Cu4, Dy2, Dy3, Er2, Er3, Eu2,\nEu3, Fe0, Fe1, Fe2, Fe3, Fe4, Gd2, Gd3, Hf2, Hf3, Ho2, Ho3, Ir0a, Ir0b, Ir0c,\nIr1a, Ir1b, Ir2, Ir3, Ir4, Ir5, Ir6, Mn0, Mn1, Mn2, Mn3, Mn4, Mo0, Mo1, Nb0,\nNb1, Nd2, Nd3, Ni0, Ni1, Ni2, Ni3, Ni4, Np3, Np4, Np5, Np6, Os0a, Os0b, Os0c,\nOs1a, Os1b, Os2, Os3, Os4, Os5, Os6, Os7, Pd0, Pd1, Pr3, Pt1, Pt2, Pt3, Pt4,\nPt5, Pt6, Pu3, Pu4, Pu5, Pu6, Re0a, Re0b, Re0c, Re1a, Re1b, Re2, Re3, Re4, Re5,\nRe6, Rh0, Rh1, Ru0, Ru1, Sc0, Sc1, Sc2, Sm2, Sm3, Ta2, Ta3, Ta4, Tb2, Tb3, Tc0,\nTc1, Ti0, Ti1, Ti2, Ti3, Tm2, Tm3, U3, U4, U5, V0, V1, V2, V3, V4, W0a, W0b,\nW0c, W1a, W1b, W2c, W3, W4, W5, Y0, Yb2, Yb3, Zr0, Zr1\n\nThe trailing number denotes ionization state. For example, \"Fe0\" denotes a neutral iron atom, while \"Fe2\" denotes Fe²⁺. If multiple electronic configurations are possible, they will be distinguished by a trailing letter (a, b, ...). Omitting this letter will print an informative error,\n\nFormFactor(\"Ir0\")\n\nERROR: Disambiguate form factor according to electronic configuration:\n \"Ir0a\" -- 6s⁰5d⁹\n \"Ir0b\" -- 6s¹5d⁸\n \"Ir0c\" -- 6s²5d⁷\n\nThe form factor is approximated as\n\nF(s) = j_0(s) + frac2-gg j_2(s) s^2,\n\ninvolving the Landé g-factor. The j_l(s) are radial integrals associated with the lth Bessel function of the magnetic dipole, where s = k4π, and k is the magnitude of momentum transfer. \n\nThe radial integrals have been calculated using Hartree-Fock for transition metals, or Dirac-Fock for the rare earths and actinide series [1–3]. Sunny uses approximate fits as a sum of Gaussians,\n\nj_0(s) = A e^-as^2 + B e^-bs^2 + C e^-cs^2 + D e^-ds^2 + E \nj_l(s) = (A e^-as^2 + B e^-bs^2 + C e^-cs^2 + D e^-ds^2 + E) s^2\n\nReferences:\n\nhttps://www.ill.eu/sites/ccsl/ffacts/ffachtml.html\nJ. Brown, The Neutron Data Booklet, 2nd ed., Sec. 2.5 Magnetic Form Factors (2003)\nK. Kobayashi, T. Nagao, M. Ito, Acta Cryst. A, 67 pp 473–480 (2011)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.ImplicitMidpoint","page":"Library API","title":"Sunny.ImplicitMidpoint","text":"ImplicitMidpoint(Δt::Float64; atol=1e-12) where N\n\nEnergy-conserving spin dynamics. One call to the step! function will advance a System by Δt units of time.\n\nUses the spherical midpoint integration scheme for dipole systems and the Schrödinger midpoint integration scheme for SU(N) spin systems. Both integration schemes are symplectic, and therefore avoid energy drift over long periods of simulation time.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Langevin","page":"Library API","title":"Sunny.Langevin","text":"Langevin(Δt::Float64; λ::Float64, kT::Float64)\n\nSpin dynamics with coupling to a Langevin thermostat, which includes damping and noise terms. One call to the step! function will advance a System by Δt units of time.\n\nAssuming ergodicity, the Langevin dynamics will sample from thermal equilibrium for the target temperature kT. The empirical parameter λ determines the strength of the coupling to the thermal bath. In other words, 1/λ is the decorrelation time-scale. If λ = 0, then the spin dynamics coincides with ImplicitMidpoint.\n\nAn alternative approach to sampling is LocalSampler, which may be preferred when the allowed spin values become effective discrete (e.g. Ising spins).\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.LocalSampler","page":"Library API","title":"Sunny.LocalSampler","text":"LocalSampler(; kT, nsweeps=1.0, propose=propose_uniform)\n\nMonte Carlo simulation involving Metropolis updates to individual spins. One call to the step! function will perform nsweeps of MCMC sampling for a provided System. The default value of 1.0 means that step! performs, on average, one trial update per spin.\n\nAssuming ergodicity, the LocalSampler will sample from thermal equilibrium for the target temperature kT. \n\nThe trial spin updates are sampled using the propose function. Built-in options include propose_uniform, propose_flip, and propose_delta. Multiple proposals can be mixed with the macro @mix_proposals.\n\nThe returned object stores fields ΔE and Δs, which represent the cumulative change to the net energy and dipole, respectively.\n\nAn alternative approach to sampling is Langevin, which may be preferred for simulating continuous spins, especially in the presence of long-range dipole-dipole interactions (cf. enable_dipole_dipole!).\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SampledCorrelations","page":"Library API","title":"Sunny.SampledCorrelations","text":"SampledCorrelations\n\nBasic data type for storing sampled correlation data. A SampleCorrelations is initialized by calling either dynamical_correlations or instant_correlations.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SpinInfo","page":"Library API","title":"Sunny.SpinInfo","text":"SpinInfo(atom::Int; S, g=2)\n\nCharacterizes the spin at a given atom index within the crystal unit cell. S is an integer multiple of 1/2 and gives the spin angular momentum in units of ħ. g is the g-factor or tensor, such that an angular momentum dipole s produces a magnetic moment g s in units of the Bohr magneton.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SpinWaveTheory","page":"Library API","title":"Sunny.SpinWaveTheory","text":"SpinWaveTheory(sys, energy_ϵ::Float64=1e-8)\n\nConstructs an object to perform linear spin wave theory. Use it with dispersion and dssf functions.\n\nThe optional parameter energy_ϵ adds a small positive shift to the diagonal of the dynamical matrix D to avoid numerical issues with zero-energy quasi-particle modes.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.System-Tuple{Crystal, Tuple{Int64, Int64, Int64}, Vector{SpinInfo}, Symbol}","page":"Library API","title":"Sunny.System","text":"System(crystal::Crystal, latsize, infos, mode; units=Units.meV, seed::Int)\n\nConstruct a System of spins for a given Crystal symmetry. The latsize parameter determines the number of unit cells in each lattice vector direction. The infos parameter is a list of SpinInfo objects, which determine the magnitude S and g-tensor of each spin.\n\nThe two primary options for mode are :SUN and :dipole. In the former, each spin-S degree of freedom is described as an SU(N) coherent state, i.e. a quantum superposition of N = 2S + 1 levels. This formalism can be useful to capture multipolar spin fluctuations or local entanglement effects. \n\nMode :dipole projects the SU(N) dynamics onto the restricted space of pure dipoles. In practice this means that Sunny will simulate Landau-Lifshitz dynamics, but single-ion anisotropy and biquadratic exchange interactions will be renormalized to improve accuracy. To disable this renormalization, use the mode :dipole_large_S which applies the S classical limit. For details, see the documentation page: Interaction Strength Renormalization.\n\nThe default units system of (meV, Å, tesla) can be overridden by with the units parameter; see Units. \n\nAn optional seed may be provided to achieve reproducible random number generation.\n\nAll spins are initially polarized in the z-direction.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.add_sample!-Tuple{SampledCorrelations, System}","page":"Library API","title":"Sunny.add_sample!","text":"add_sample!(sc::SampledCorrelations, sys::System)\n\nadd_trajectory uses the spin configuration contained in the System to generate a correlation data and accumulate it into sc. For static structure factors, this involves analyzing the spin-spin correlations of the spin configuration provided. For a dynamic structure factor, a trajectory is calculated using the given spin configuration as an initial condition. The spin-spin correlations are then calculating in time and accumulated into sc. \n\nThis function will change the state of sys when calculating dynamical structure factor data. To preserve the initial state of sys, it must be saved separately prior to calling add_sample!. Alternatively, the initial spin configuration may be copied into a new System and this new System can be passed to add_sample!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.available_energies-Tuple{SampledCorrelations}","page":"Library API","title":"Sunny.available_energies","text":"available_energies(sc::SampledCorrelations; negative_energies=false)\n\nReturn the ω values for the energy index of a SampledCorrelations. By default, only returns values for non-negative energies, which corresponds to the default output of intensities. Set negative_energies to true to retrieve all ω values.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.available_wave_vectors-Tuple{SampledCorrelations}","page":"Library API","title":"Sunny.available_wave_vectors","text":"available_wave_vectors(sc::SampledCorrelations; bzsize=(1,1,1))\n\nReturns all wave vectors for which sc contains exact values. bsize specifies the number of Brillouin zones to be included.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.axes_bincenters-Tuple{Any, Any, Any}","page":"Library API","title":"Sunny.axes_bincenters","text":"axes_bincenters(params::BinningParameters)\n\nReturns tick marks which label the bins of the histogram described by BinningParameters by their bin centers.\n\nThe following alternative syntax can be used to compute bin centers for a single axis:\n\naxes_bincenters(binstart,binend,binwidth)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.broaden_energy-Tuple{SampledCorrelations, Any, Function}","page":"Library API","title":"Sunny.broaden_energy","text":"broaden_energy(sc::SampledCorrelations, vals, kernel::Function; negative_energies=false)\n\nPerforms a real-space convolution along the energy axis of an array of intensities. Assumes the format of the intensities array corresponds to what would be returned by intensities_interpolated. kernel must be a function that takes two numbers: kernel(ω, ω₀), where ω is a frequency, and ω₀ is the center frequency of the kernel. Sunny provides lorentzian for the most common use case:\n\nnewvals = broaden_energy(sc, vals, (ω, ω₀) -> lorentzian(ω-ω₀, 0.2))\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.count_bins-Tuple{Any, Any, Any}","page":"Library API","title":"Sunny.count_bins","text":"count_bins(binstart,binend,binwidth)\n\nReturns the number of bins in the binning scheme implied by binstart, binend, and binwidth. To count the bins in a BinningParameters, use params.numbins.\n\nThis function defines how partial bins are handled, so it should be used preferentially over computing the number of bins manually.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dispersion-Tuple{SpinWaveTheory, Any}","page":"Library API","title":"Sunny.dispersion","text":"dispersion(swt::SpinWaveTheory, qs)\n\nComputes the spin excitation energy dispersion relations given a SpinWaveTheory and an array of wave vectors qs. Each element q of qs must be a 3-vector in units of reciprocal lattice units. I.e., qᵢ is given in 2πaᵢ with aᵢ the lattice constant of the original chemical lattice.\n\nThe first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of the array is an energy.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dmvec-Tuple{Any}","page":"Library API","title":"Sunny.dmvec","text":"dmvec(D)\n\nAntisymmetric matrix representation of the Dzyaloshinskii-Moriya pseudo-vector,\n\n [ 0 D[3] -D[2]\n -D[3] 0 D[1]\n D[2] -D[1] 0 ]\n\nUseful in the context of set_exchange!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dssf-Tuple{SpinWaveTheory, Any}","page":"Library API","title":"Sunny.dssf","text":"dssf(swt::SpinWaveTheory, qs)\n\nGiven a SpinWaveTheory object, computes the dynamical spin structure factor,\n\n 𝒮^αβ(𝐤 ω) = 1(2πN)dt _𝐫 expi(ωt - 𝐤𝐫) S^α(𝐫 t)S^β(0 0)\n\nusing the result from linear spin-wave theory,\n\n 𝒮^αβ(𝐤 ω) = _n A_n^αβ(𝐤)^2 δω-ω_n(𝐤)\n\nqs is an array of wave vectors of arbitrary dimension. Each element q of qs must be a 3-vector in reciprocal lattice units (RLU), i.e., in the basis of reciprocal lattice vectors.\n\nThe first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of this array is a tensor (3×3 matrix) corresponding to the indices α and β.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dynamical_correlations-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.dynamical_correlations","text":"dynamical_correlations(sys::System; Δt, nω, ωmax, \n process_trajectory=:none, observables=nothing, correlations=nothing)\n\nCreates an empty SampledCorrelations object for calculating and storing dynamical structure factor intensities 𝒮(𝐪ω). Call add_sample! to accumulate data for the given configuration of a spin system. Internally, this will run a dynamical trajectory and measure time correlations. The 𝒮(𝐪ω) data can be retrieved by calling intensities_interpolated. Alternatively, instant_intensities_interpolated will integrate out ω to obtain 𝒮(𝐪), optionally applying classical-to-quantum correction factors.\n\nThree keywords are required to specify the dynamics used for the trajectory calculation.\n\nΔt: The time step used for calculating the trajectory from which dynamic spin-spin correlations are calculated. The trajectories are calculated with an ImplicitMidpoint integrator.\nωmax: The maximum energy, ω, that will be resolved.\nnω: The number of energy bins to calculated between 0 and ωmax.\n\nAdditional keyword options are the following:\n\nobservables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.\ncorrelations: Specify which correlation functions are calculated, i.e. which matrix elements αβ of 𝒮^αβ(qω) are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.eachsite-Tuple{System}","page":"Library API","title":"Sunny.eachsite","text":"eachsite(sys::System)\n\nAn iterator over all Sites in the system. \n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.enable_dipole_dipole!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.enable_dipole_dipole!","text":"enable_dipole_dipole!(sys::System)\n\nEnables long-range dipole-dipole interactions,\n\n -(μ_04π) _ij (3 (𝐌_j𝐫_ij)(𝐌_i𝐫_ij) - 𝐌_i𝐌_j) 𝐫_ij^3\n\nwhere the sum is over all pairs of spins (singly counted), including periodic images, regularized using the Ewald summation convention. The magnetic moments are 𝐌_i = μ_B g 𝐒_i where g is the g-factor or g-tensor, and 𝐒_i is the spin angular momentum dipole in units of ħ. The Bohr magneton μ_B and vacuum permeability μ_0 are physical constants, with numerical values determined by the unit system.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.energy-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.energy","text":"energy(sys::System)\n\nThe total system energy. See also energy_per_site.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.energy_per_site-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.energy_per_site","text":"energy_per_site(sys::System)\n\nThe total system energy divided by the number of sites.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.generate_mantid_script_from_binning_parameters-Tuple{Any}","page":"Library API","title":"Sunny.generate_mantid_script_from_binning_parameters","text":"generate_mantid_script_from_binning_parameters(params::BinningParameters)\n\nGenerate a Mantid script which bins data according to the given BinningParameters.\n\nwarning: Units\nTake care to ensure the units are correct (R.L.U. or absolute). You may want to call Sunny.bin_rlu_as_absolute_units! or Sunny.bin_absolute_units_as_rlu! first.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.global_position-Tuple{System, Any}","page":"Library API","title":"Sunny.global_position","text":"global_position(sys::System, site::Site)\n\nPosition of a Site in global coordinates.\n\nTo precompute a full list of positions, one can use eachsite as below:\n\npos = [global_position(sys, site) for site in eachsite(sys)]\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.instant_correlations-Tuple{System}","page":"Library API","title":"Sunny.instant_correlations","text":"instant_correlations(sys::System; process_trajectory=:none, observables=nothing, correlations=nothing)\n\nCreates an empty SampledCorrelations object for calculating and storing instantaneous structure factor intensities 𝒮(𝐪). Call add_sample! to accumulate data for the given configuration of a spin system. Call instant_intensities_interpolated to retrieve averaged 𝒮(𝐪) data.\n\nImportant note: When dealing with continuous (non-Ising) spins, consider creating using dynamical_correlations instead of instant_correlations. The former will provide full 𝒮(𝐪ω) data, from which 𝒮(𝐪) can be obtained by integrating out ω. During this integration step, Sunny can incorporate temperature- and ω-dependent classical-to-quantum correction factors to produce more accurate 𝒮(𝐪) estimates. See instant_intensities_interpolated for more information.\n\nThe following optional keywords are available:\n\nobservables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.\ncorrelations: Specify which correlation functions are calculated, i.e. which matrix elements αβ of 𝒮^αβ(qω) are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.instant_intensities_interpolated-Tuple{SampledCorrelations, Any, Any}","page":"Library API","title":"Sunny.instant_intensities_interpolated","text":"instant_intensities_interpolated(sc::SampledCorrelations, qs; kwargs...)\n\nReturn 𝒮(𝐪) intensities at wave vectors qs. The functionality is very similar to intensities_interpolated, except the returned array has dimensions identical to qs. If called on a SampledCorrelations with dynamical information, i.e., 𝒮(𝐪ω), the ω information is integrated out.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.integrate_axes!-Tuple{BinningParameters}","page":"Library API","title":"Sunny.integrate_axes!","text":"integrate_axes!(params::BinningParameters; axes)\n\nIntegrate over one or more axes of the histogram by setting the number of bins in that axis to 1. Examples:\n\nintegrate_axes!(params; axes = [2,3])\nintegrate_axes!(params; axes = 2)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.integrated_lorentzian-Tuple{Float64}","page":"Library API","title":"Sunny.integrated_lorentzian","text":"integrated_lorentzian(η)\n\nReturns x mapsto atan(xη)π for use with intensities_binned.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_bands-Tuple{SpinWaveTheory, Any, Sunny.SpinWaveIntensityFormula}","page":"Library API","title":"Sunny.intensities_bands","text":"dispersion, intensities = intensities_bands(swt::SpinWaveTheory, ks, formula::SpinWaveIntensityFormula)\n\nComputes the scattering intensities at each energy band for each momentum transfer k in ks, according to Linear Spin Wave Theory and the given intensity formula. The formula must have a delta-function kernel, e.g.:\n\nformula = intensity_formula(swt, :perp, formula; kernel = delta_function_kernel)\n\nor else the bands will be broadened, and their intensity can not be computed.\n\nThe outputs will be arrays with indices identical to ks, with the last index giving the band index. dispersions reports the energy of each band, while intensities reports the scattering intensity.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_binned-Tuple{SampledCorrelations, BinningParameters, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.intensities_binned","text":"intensity, counts = intensities_binned(sc::SampledCorrelations, params::BinningParameters, formula; integrated_kernel)\n\nGiven correlation data contained in a SampledCorrelations and BinningParameters describing the shape of a histogram, compute the intensity and normalization for each histogram bin using a given intensity_formula.\n\nThe BinningParameters are expected to accept (q,ω) in R.L.U. for the (possibly reshaped) crystal associated with sc.\n\nThis is an alternative to intensities_interpolated which bins the scattering intensities into a histogram instead of interpolating between them at specified qs values. See unit_resolution_binning_parameters for a reasonable default choice of BinningParameters which roughly emulates intensities_interpolated with interpolation = :round.\n\nIf a function integrated_kernel(Δω) is passed, it will be used as the CDF of a kernel function for energy broadening. For example, integrated_kernel = Δω -> atan(Δω/η)/pi (c.f. integrated_lorentzian implements Lorentzian broadening with parameter η. Energy-dependent energy broadening can be achieved by providing an integrated_kernel(ω,Δω) whose first argument is the energy transfer ω.\n\nCurrently, energy broadening is only supported if the BinningParameters are such that the first three axes are purely spatial and the last (energy) axis is [0,0,0,1].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_broadened-Tuple{SpinWaveTheory, Any, Any, Any}","page":"Library API","title":"Sunny.intensities_broadened","text":"intensities_broadened(swt::SpinWaveTheory, ks, ωvals, formula)\n\nComputes the scattering intensities at each (Q,ω) according to Linear Spin Wave Theory and the given intensity formula. The required formula must have a non-delta-function kernel, e.g.:\n\nformula = intensity_formula(swt, :perp; kernel = lorentzian(0.05))\n\nor else the intensity at ωvals which are not exactly on the dispersion curve can not be calculated.\n\nThe intensity is computed at each wave vector in ks and each energy in ωvals. The output will be an array with indices identical to ks, with the last index matching ωvals.\n\nNote that ks is an array of wave vectors of arbitrary dimension. Each element k of ks must be a 3-wavevector in absolute units.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_interpolated-Tuple{SampledCorrelations, Any, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.intensities_interpolated","text":"intensities_interpolated(sc::SampledCorrelations, qs, formula:ClassicalIntensityFormula; interpolation=nothing, negative_energies=false)\n\nThe basic function for retrieving 𝒮(𝐪ω) information from a SampledCorrelations. Maps an array of wave vectors qs to an array of structure factor intensities, including an additional energy index. The values of ω associated with the energy index can be retrieved by calling available_energies. The three coordinates of each wave vector are measured in reciprocal lattice units, i.e., multiples of the reciprocal lattice vectors.\n\ninterpolation: Since 𝒮(𝐪 ω) is calculated on a finite lattice, data is only available at discrete wave vectors. By default, Sunny will round a requested q to the nearest available wave vector. Linear interpolation can be applied by setting interpolation=:linear.\nnegative_energies: If set to true, Sunny will return the periodic extension of the energy axis. Most users will not want this.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SampledCorrelations, AbstractVector{Int64}}","page":"Library API","title":"Sunny.intensity_formula","text":"formula = intensity_formula(sc::SampledCorrelations)\n\nEstablish a formula for computing the intensity of the discrete scattering modes (q,ω) using the correlation data 𝒮^αβ(qω) stored in the SampledCorrelations. The formula returned from intensity_formula can be passed to intensities_interpolated or intensities_binned.\n\nintensity_formula(sc,...; kT = Inf, formfactors = ...)\n\nThere are keyword arguments providing temperature and form factor corrections:\n\nkT: If a temperature is provided, the intensities will be rescaled by a temperature- and ω-dependent classical-to-quantum factor. kT should be specified when making comparisons with spin wave calculations or experimental data. If kT is not specified, infinite temperature (no correction) is assumed.\nformfactors: To apply form factor corrections, provide this keyword with a list of FormFactors, one for each symmetry-distinct site in the crystal. The order of FormFactors must correspond to the order of site symmetry classes, e.g., as they appear when printed in display(crystal).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SampledCorrelations, Any}","page":"Library API","title":"Sunny.intensity_formula","text":"A custom intensity formula can be specifed by providing a function intensity = f(q,ω,correlations) and specifying which correlations it requires:\n\nintensity_formula(f,sc::SampledCorrelations, required_correlations; kwargs...)\n\nThe function is intended to be specified using do notation. For example, this custom formula sums the off-diagonal correlations:\n\nrequired = [(:Sx,:Sy),(:Sy,:Sz),(:Sx,:Sz)]\nintensity_formula(sc,required,return_type = ComplexF64) do k, ω, off_diagonal_correlations\n sum(off_diagonal_correlations)\nend\n\nIf your custom formula returns a type other than Float64, use the return_type keyword argument to flag this.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SpinWaveTheory, AbstractVector{Int64}}","page":"Library API","title":"Sunny.intensity_formula","text":"formula = intensity_formula(swt::SpinWaveTheory; kernel = ...)\n\nEstablish a formula for computing the scattering intensity by diagonalizing the hamiltonian H(q) using Linear Spin Wave Theory.\n\nIf kernel = delta_function_kernel, then the resulting formula can be used with intensities_bands.\n\nIf kernel is an energy broadening kernel function, then the resulting formula can be used with intensities_broadened. Energy broadening kernel functions can either be a function of Δω only, e.g.:\n\nkernel = Δω -> ...\n\nor a function of both the energy transfer ω and of Δω, e.g.:\n\nkernel = (ω,Δω) -> ...\n\nThe integral of a properly normalized kernel function over all Δω is one.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{SpinWaveTheory, Symbol}","page":"Library API","title":"Sunny.intensity_formula","text":"intensity_formula([swt or sc], contraction_mode::Symbol)\n\nSunny has several built-in formulas that can be selected by setting contraction_mode to one of these values:\n\n:trace (default), which yields operatornametr 𝒮(qω) = _α 𝒮^αα(qω)\n:perp, which contracts 𝒮^αβ(qω) with the dipole factor δ_αβ - q_αq_β, returning the unpolarized intensity.\n:full, which will return all elements 𝒮^αβ(𝐪ω) without contraction.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lattice_params-Tuple{StaticArraysCore.SMatrix{3, 3, Float64, 9}}","page":"Library API","title":"Sunny.lattice_params","text":"lattice_params(latvecs::Mat3)\n\nCompute the lattice parameters (a b c α β γ) for the three lattice vectors provided as columns of latvecs. The inverse mapping is lattice_vectors.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lattice_vectors-NTuple{6, Any}","page":"Library API","title":"Sunny.lattice_vectors","text":"lattice_vectors(a, b, c, α, β, γ)\n\nReturn the lattice vectors, as columns of the 33 output matrix, that correspond to the conventional unit cell defined by the lattice constants (a b c) and the angles (α β γ) in degrees. The inverse mapping is lattice_params.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.load_nxs-Tuple{Any}","page":"Library API","title":"Sunny.load_nxs","text":"params, signal = load_nxs(filename)\n\nGiven the name of a Mantid-exported MDHistoWorkspace file, load the BinningParameters and the signal from that file.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lorentzian-Tuple{Any, Any}","page":"Library API","title":"Sunny.lorentzian","text":"lorentzian(x, η)\n\nReturns η(π(x^2 + η^2)).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.magnetic_moment-Tuple{System, Any}","page":"Library API","title":"Sunny.magnetic_moment","text":"magnetic_moment(sys::System, site::Site)\n\nGet the magnetic moment for a Site. This is the spin dipole multiplied by the Bohr magneton and the local g-tensor.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.merge_correlations-Union{Tuple{Array{SampledCorrelations{N}, 1}}, Tuple{N}} where N","page":"Library API","title":"Sunny.merge_correlations","text":"merge_correlations(scs::Vector{SampledCorrelations)\n\nAccumulate a list of SampledCorrelations into a single, summary SampledCorrelations. Useful for reducing the results of parallel computations.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.minimize_energy!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.minimize_energy!","text":"minimize_energy!(sys::System{N}; maxiters=100, subiters=20,\n method=Optim.ConjugateGradient(), kwargs...) where N\n\nOptimizes the spin configuration in sys to minimize energy. A total of maxiters iterations will be attempted, with restarts after every subiters iterations. The remaining kwargs will be forwarded to the optimize method of the Optim.jl package.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.polarize_spins!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.polarize_spins!","text":"polarize_spins!(sys::System, dir)\n\nPolarize all spins in the system along the direction dir.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.position_to_site-Tuple{System, Any}","page":"Library API","title":"Sunny.position_to_site","text":"position_to_site(sys::System, r)\n\nConverts a position r to four indices of a Site. The coordinates of r are given in units of the lattice vectors for the original crystal. This function can be useful for working with systems that have been reshaped using reshape_supercell.\n\nExample\n\n# Find the `site` at the center of a unit cell which is displaced by four\n# multiples of the first lattice vector\nsite = position_to_site(sys, [4.5, 0.5, 0.5])\n\n# Print the dipole at this site\nprintln(sys.dipoles[site])\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.powder_average_binned-Tuple{SampledCorrelations, Any, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.powder_average_binned","text":"powder_average_binned(sc::SampledCorrelations, radial_binning_parameters; formula\n ω_binning_parameters, integrated_kernel = nothing, bzsize = nothing)\n\nThis function emulates the experimental situation of \"powder averaging,\" where only the magnitude (and not the direction) of the momentum transfer is resolvable. The intensities are binned similarly to intensities_binned, but the histogram x-axis is |k| in absolute units, which is a nonlinear function of kx,ky,kz. The y-axis is energy.\n\nRadial binning parameters are specified as tuples (start,end,bin_width), e.g. radial_binning_parameters = (0,6π,6π/55).\n\nEnergy broadening is supported in the same way as intensities_binned, and this function accepts the same kind of intensity_formula.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.primitive_cell_shape-Tuple{Crystal}","page":"Library API","title":"Sunny.primitive_cell_shape","text":"primitive_cell_shape(cryst::Crystal)\n\nReturns the shape of the primitive cell as a 3×3 matrix, in fractional coordinates of the conventional lattice vectors. May be useful for constructing inputs to reshape_supercell.\n\nExamples\n\n# Valid if `cryst` has not been reshaped\n@assert cryst.prim_latvecs ≈ cryst.latvecs * primitive_cell_shape(cryst)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_bond-Tuple{Crystal, Bond}","page":"Library API","title":"Sunny.print_bond","text":"print_bond(cryst::Crystal, bond::Bond; b_ref::Bond)\n\nPrints symmetry information for bond bond. A symmetry-equivalent reference bond b_ref can optionally be provided to fix the meaning of the coefficients A, B, ...\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_site-Tuple{Any, Any}","page":"Library API","title":"Sunny.print_site","text":"print_site(cryst, i; R=I)\n\nPrint symmetry information for the site i, including allowed g-tensor and allowed anisotropy operator. An optional rotation matrix R can be provided to define the reference frame for expression of the anisotropy.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_stevens_expansion-Tuple{AbstractMatrix}","page":"Library API","title":"Sunny.print_stevens_expansion","text":"function print_stevens_expansion(op)\n\nPrints a local Hermitian operator as a linear combination of Stevens operators. The operator op may be a finite-dimensional matrix or an abstract spin polynomial in the large-S limit.\n\nExamples\n\nS = spin_matrices(2)\nprint_stevens_expansion(S[1]^4 + S[2]^4 + S[3]^4)\n# Prints: (1/20)𝒪₄₀ + (1/4)𝒪₄₄ + 102/5\n\nS = spin_matrices(Inf)\nprint_stevens_expansion(S[1]^4 + S[2]^4 + S[3]^4)\n# Prints: (1/20)𝒪₄₀ + (1/4)𝒪₄₄ + (3/5)𝒮⁴\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_suggested_frame-Tuple{Crystal, Int64}","page":"Library API","title":"Sunny.print_suggested_frame","text":"print_suggested_frame(cryst, i; digits=4)\n\nPrint a suggested reference frame, as a rotation matrix R, that can be used as input to print_site(). The purpose is to simplify the description of allowed anisotropies.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_symmetry_table-Tuple{Crystal, Any}","page":"Library API","title":"Sunny.print_symmetry_table","text":"print_symmetry_table(cryst::Crystal, max_dist)\n\nPrint symmetry information for all equivalence classes of sites and bonds, up to a maximum bond distance of max_dist. Equivalent to calling print_bond(cryst, b) for every bond b in reference_bonds(cryst, max_dist), where Bond(i, i, [0,0,0]) refers to a single site i.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_wrapped_intensities-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.print_wrapped_intensities","text":"print_wrapped_intensities(sys::System; nmax=10)\n\nFor Bravais lattices: Prints up to nmax wavevectors according to their instantaneous (static) structure factor intensities, listed in descending order. For non-Bravais lattices: Performs the same analysis for each spin sublattice independently; the output weights are naïvely averaged over sublattices, without incorporating phase shift information. This procedure therefore wraps all wavevectors into the first Brillouin zone. Each wavevector coordinate is given between -12 and 12 in reciprocal lattice units (RLU). The output from this function will typically be used as input to suggest_magnetic_supercell.\n\nBecause this function does not incorporate phase information in its averaging over sublattices, the printed weights are not directly comparable with experiment. For that purpose, use instant_correlations instead.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_delta-Tuple{Any}","page":"Library API","title":"Sunny.propose_delta","text":"propose_delta(magnitude)\n\nGenerate a proposal function that adds a Gaussian perturbation to the existing spin state. In :dipole mode, the procedure is to first introduce a random three-vector perturbation 𝐬 = 𝐬 + 𝐬 ξ and then return the properly normalized spin 𝐬 (𝐬𝐬). Each component of the random vector ξ is Gaussian distributed with a standard deviation of magnitude; the latter is dimensionless and typically smaller than one. \n\nIn :SUN mode, the procedure is analogous, but now involving Gaussian perturbations to each of the N complex components of an SU(N) coherent state.\n\nIn the limit of very large magnitude, this function coincides with propose_uniform.\n\nFor use with LocalSampler.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_flip-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.propose_flip","text":"propose_flip\n\nFunction to propose pure spin flip updates in the context of a LocalSampler. Dipoles are flipped as 𝐬 -𝐬. SU(N) coherent states are flipped using the time-reversal operator.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_uniform","page":"Library API","title":"Sunny.propose_uniform","text":"propose_uniform\n\nFunction to propose a uniformly random spin update in the context of a LocalSampler. In :dipole mode, the result is a random three-vector with appropriate normalization. In :SUN mode, the result is a random SU(N) coherent state with appropriate normalization.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.randomize_spins!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.randomize_spins!","text":"randomize_spins!(sys::System)\n\nRandomizes all spins under appropriate the uniform distribution.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_path-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.reciprocal_space_path","text":"reciprocal_space_path(cryst::Crystal, qs, density)\n\nReturns a pair (path, xticks). The path return value is a list of wavevectors that samples linearly between the provided wavevectors qs. The xticks return value can be used to label the special 𝐪 values on the x-axis of a plot.\n\nSpecial note about units: the wavevectors qs must be provided in reciprocal lattice units (RLU) for the given crystal, but the sampling density must be specified in the global frame. Specifically, the density is given as number of sample points per unit of radian inverse length, where the unit of length is the same as that used to specify the lattice vectors of the Crystal. The path will therefore include more samples between q-points that are further apart in absolute Fourier distance.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_path_bins-Tuple{Any, Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.reciprocal_space_path_bins","text":"reciprocal_space_path_bins(sc,qs,density,args...;kwargs...)\n\nTakes a list of wave vectors, qs in R.L.U., and builds a series of histogram BinningParameters whose first axis traces a path through the provided points. The second and third axes are integrated over according to the args and kwargs, which are passed through to slice_2D_binning_parameters.\n\nAlso returned is a list of marker indices corresponding to the input points, and a list of ranges giving the indices of each histogram x-axis within a concatenated histogram. The density parameter is given in samples per reciprocal lattice unit (R.L.U.).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_shell-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.reciprocal_space_shell","text":"reciprocal_space_shell(cryst::Crystal, radius, n)\n\nSample n points on the reciprocal space sphere with a given radius (units of inverse length).\n\nExamples\n\n# Sample wavevectors on the sphere at fixed density\nreciprocal_space_shell(cryst, r, 4π*r^2*density)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reference_bonds-Tuple{Crystal, Float64}","page":"Library API","title":"Sunny.reference_bonds","text":"reference_bonds(cryst::Crystal, max_dist)\n\nReturns a full list of bonds, one for each symmetry equivalence class, up to distance max_dist. The reference bond b for each equivalence class is selected according to a scoring system that prioritizes simplification of the elements in basis_for_symmetry_allowed_couplings(cryst, b).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.remove_periodicity!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.remove_periodicity!","text":"remove_periodicity!(sys::System, dims)\n\nRemove periodic interactions along the dimensions where dims is true. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nExample\n\n# Remove periodic boundaries along the 1st and 3rd dimensions\nremove_periodicity!(sys::System, (true, false, true))\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.repeat_periodically-Union{Tuple{N}, Tuple{System{N}, Tuple{Int64, Int64, Int64}}} where N","page":"Library API","title":"Sunny.repeat_periodically","text":"repeat_periodically(sys::System{N}, counts::NTuple{3,Int}) where N\n\nCreates a System identical to sys but repeated a given number of times in each dimension, specified by the tuple counts.\n\nSee also reshape_supercell.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reshape_supercell-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.reshape_supercell","text":"reshape_supercell(sys::System, shape)\n\nMaps an existing System to a new one that has the shape and periodicity of a requested supercell. The columns of the 33 integer matrix shape represent the supercell lattice vectors measured in units of the original crystal lattice vectors.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.resize_supercell-Union{Tuple{N}, Tuple{System{N}, Tuple{Int64, Int64, Int64}}} where N","page":"Library API","title":"Sunny.resize_supercell","text":"resize_supercell(sys::System{N}, latsize::NTuple{3,Int}) where N\n\nCreates a System with a given number of conventional unit cells in each lattice vector direction. Interactions and other settings will be inherited from sys.\n\nConvenience function for:\n\nreshape_supercell(sys, [latsize[1] 0 0; 0 latsize[2] 0; 0 0 latsize[3]])\n\nSee also reshape_supercell.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.rotate_operator-Tuple{LinearAlgebra.Hermitian{ComplexF64, Matrix{ComplexF64}}, Any}","page":"Library API","title":"Sunny.rotate_operator","text":"rotate_operator(A, R)\n\nRotates the local quantum operator A according to the 33 rotation matrix R.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.rotation_in_rlu-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.rotation_in_rlu","text":"rotation_in_rlu(cryst::Crystal, axis, angle)\n\nReturns a 33 matrix that rotates wavevectors in reciprocal lattice units (RLU). The axis vector is a real-space direction in absolute units (but arbitrary magnitude), and the angle is in radians.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_coherent!-Union{Tuple{N}, Tuple{System{N}, Any, Any}} where N","page":"Library API","title":"Sunny.set_coherent!","text":"set_coherent!(sys::System, Z, site::Site)\n\nSet a coherent spin state at a Site using the N complex amplitudes in Z.\n\nFor a standard SpinInfo, these amplitudes will be interpreted in the eigenbasis of 𝒮ᶻ. That is, Z[1] represents the amplitude for the basis state fully polarized along the z-direction, and subsequent components represent states with decreasing angular momentum along this axis (m = S S-1 -S).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_dipole!-Union{Tuple{N}, Tuple{System{N}, Any, Any}} where N","page":"Library API","title":"Sunny.set_dipole!","text":"set_dipole!(sys::System, dir, site::Site)\n\nPolarize the spin at a Site along the direction dir.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_exchange!-Union{Tuple{N}, Tuple{System{N}, Any, Bond}} where N","page":"Library API","title":"Sunny.set_exchange!","text":"set_exchange!(sys::System, J, bond::Bond)\n\nSets a 3×3 spin-exchange matrix J along bond, yielding a pairwise interaction energy 𝐒_iJ 𝐒_j. This interaction will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells.\n\nThe parameter J may be scalar or matrix-valued. As a convenience, dmvec(D) can be used to construct the antisymmetric part of the exchange, where D is the Dzyaloshinskii-Moriya pseudo-vector. The resulting interaction will be 𝐃(𝐒_i𝐒_j).\n\nFor more general interactions, such as biquadratic, use set_pair_coupling! instead.\n\nExamples\n\n# An explicit exchange matrix\nJ1 = [2 3 0;\n -3 2 0;\n 0 0 2]\nset_exchange!(sys, J1, bond)\n\n# An equivalent Heisenberg + DM exchange \nJ2 = 2*I + dmvec([0,0,3])\nset_exchange!(sys, J2, bond)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_exchange_at!-Union{Tuple{N}, Tuple{System{N}, Any, Union{NTuple{4, Int64}, CartesianIndex{4}}, Union{NTuple{4, Int64}, CartesianIndex{4}}}} where N","page":"Library API","title":"Sunny.set_exchange_at!","text":"set_exchange_at!(sys::System, J, site1::Site, site2::Site; offset=nothing)\n\nSets an exchange interaction `𝐒_i⋅J 𝐒_j along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nIf the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.\n\nFor more general interactions, such as biquadratic, use set_pair_coupling_at! instead.\n\nSee also set_exchange!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_external_field!-Tuple{System, Any}","page":"Library API","title":"Sunny.set_external_field!","text":"set_external_field!(sys::System, B::Vec3)\n\nSets the external field B that couples to all spins.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_external_field_at!-Tuple{System, Any, Any}","page":"Library API","title":"Sunny.set_external_field_at!","text":"set_external_field_at!(sys::System, B::Vec3, site::Site)\n\nSets a Zeeman coupling between a field B and a single spin. Site includes a unit cell and a sublattice index.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_onsite_coupling!-Tuple{System, Any, Int64}","page":"Library API","title":"Sunny.set_onsite_coupling!","text":"set_onsite_coupling!(sys::System, op, i::Int)\n\nSet the single-ion anisotropy for the ith atom of every unit cell, as well as all symmetry-equivalent atoms. The operator op may be provided as an abstract function of the local spin operators, as a polynomial of spin_matrices, or as a linear combination of stevens_matrices.\n\nExamples\n\n# An easy axis anisotropy in the z-direction\nset_onsite_coupling!(sys, S -> -D*S[3]^3, i)\n\n# The unique quartic single-ion anisotropy for a site with cubic point group\n# symmetry\nset_onsite_coupling!(sys, S -> 20*(S[1]^4 + S[2]^4 + S[3]^4), i)\n\n# An equivalent expression of this quartic anisotropy, up to a constant shift\nO = stevens_matrices(spin_label(sys, i))\nset_onsite_coupling!(sys, O[4,0] + 5*O[4,4], i)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_onsite_coupling_at!-Tuple{System, Any, Union{NTuple{4, Int64}, CartesianIndex{4}}}","page":"Library API","title":"Sunny.set_onsite_coupling_at!","text":"set_onsite_coupling_at!(sys::System, op, site::Site)\n\nSets the single-ion anisotropy operator op for a single Site, ignoring crystal symmetry. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nSee also set_onsite_coupling!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_pair_coupling!-Union{Tuple{N}, Tuple{System{N}, AbstractMatrix, Any}} where N","page":"Library API","title":"Sunny.set_pair_coupling!","text":"set_pair_coupling!(sys::System, op, bond)\n\nSets an arbitrary coupling op along bond. This coupling will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites.\n\nExamples\n\n# Bilinear+biquadratic exchange involving 3×3 matrices J1 and J2\nset_pair_coupling!(sys, (Si, Sj) -> Si'*J1*Sj + (Si'*J2*Sj)^2, bond)\n\n# Equivalent expression using an appropriate fixed matrix representation\nS = spin_matrices(1/2)\nSi, Sj = to_product_space(S, S)\nset_pair_coupling!(sys, Si'*J1*Sj + (Si'*J2*Sj)^2, bond)\n\nSee also spin_matrices, to_product_space.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_pair_coupling_at!-Union{Tuple{N}, Tuple{System{N}, AbstractMatrix, Union{NTuple{4, Int64}, CartesianIndex{4}}, Union{NTuple{4, Int64}, CartesianIndex{4}}}} where N","page":"Library API","title":"Sunny.set_pair_coupling_at!","text":"set_pair_coupling_at!(sys::System, op, bond)\n\nSets an arbitrary coupling along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites. The documentation for set_pair_coupling! provides examples constructing op.\n\nThe system must support inhomogeneous interactions via to_inhomogeneous.\n\nIf the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_spiral_order!-Tuple{Any}","page":"Library API","title":"Sunny.set_spiral_order!","text":"set_spiral_order!(sys; q, axis, S0)\n\nInitializes the system with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0 at the real-space origin. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.\n\nExample\n\n# Spiral order for a wavevector propagating in the direction of the first\n# reciprocal lattice vector (i.e., orthogonal to the lattice vectors ``𝐚_2``\n# and ``𝐚_3``), repeating with a period of 10 lattice constants, and spiraling\n# about the ``ẑ``-axis. The spin at the origin will point in the direction\n# ``𝐒_0 = ŷ + ẑ``. Here, ``(x̂, ŷ, ẑ)`` are the axes of Cartesian coordinate\n# system in the global frame.\nset_spiral_order!(sys; q=[1/10, 0, 0], axis=[0, 0, 1], S0=[0, 1, 1])\n\nSee also set_spiral_order_on_sublattice!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_spiral_order_on_sublattice!-Tuple{Any, Any}","page":"Library API","title":"Sunny.set_spiral_order_on_sublattice!","text":"set_spiral_order_on_sublattice!(sys, i; q, axis, S0)\n\nInitializes sublattice i with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0. The phase is selected such that the spin at sys.dipole[1,1,1,i] will point in the direction of S0. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.\n\nThis function is not available for systems with reshaped unit cells.\n\nSee also set_spiral_order!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_vacancy_at!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.set_vacancy_at!","text":"set_vacancy_at!(sys::System, site::Site)\n\nMake a single site nonmagnetic. Site includes a unit cell and a sublattice index.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.slice_2D_binning_parameters-Tuple{Vector{Float64}, Any, Any, Int64, Any}","page":"Library API","title":"Sunny.slice_2D_binning_parameters","text":"slice_2D_binning_parameter(sc::SampledCorrelations, cut_from_q, cut_to_q, cut_bins::Int64, cut_width::Float64; plane_normal = [0,0,1],cut_height = cutwidth)\n\nCreates BinningParameters which make a cut along one dimension of Q-space.\n\nThe x-axis of the resulting histogram consists of cut_bins-many bins ranging from cut_from_q to cut_to_q. The width of the bins in the transverse direciton is controlled by cut_width and cut_height.\n\nThe binning in the transverse directions is defined in the following way, which sets their normalization and orthogonality properties:\n\ncut_covector = normalize(cut_to_q - cut_from_q)\ntransverse_covector = normalize(plane_normal × cut_covector)\ncotransverse_covector = normalize(transverse_covector × cut_covector)\n\nIn other words, the axes are orthonormal with respect to the Euclidean metric.\n\nIf the cut is too narrow, there will be very few scattering vectors per bin, or the number per bin will vary substantially along the cut. If the output appears under-resolved, try increasing cut_width.\n\nThe four axes of the resulting histogram are:\n\nAlong the cut\nFist transverse Q direction\nSecond transverse Q direction\nEnergy\n\nThis function can be used without reference to a SampledCorrelations using this alternate syntax to manually specify the bin centers for the energy axis:\n\nslice_2D_binning_parameter(ω_bincenters, cut_from, cut_to,...)\n\nwhere ω_bincenters specifies the energy axis, and both cut_from and cut_to are arbitrary covectors, in any units.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.spin_label-Tuple{System, Int64}","page":"Library API","title":"Sunny.spin_label","text":"spin_label(sys::System, i::Int)\n\nIf atom i carries a single spin-S moment, then returns the half-integer label S. Otherwise, throws an error.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.spin_matrices-Tuple{Any}","page":"Library API","title":"Sunny.spin_matrices","text":"spin_matrices(S)\n\nReturns a triple of NN spin matrices, where N = 2S+1. These are the generators of SU(2) in the spin-S representation.\n\nIf S == Inf, then the return values are abstract symbols denoting infinite-dimensional matrices that commute. These can be useful for repeating historical studies, or modeling micromagnetic systems. A technical discussion appears in the Sunny documentation page: Interaction Strength Renormalization.\n\nExample\n\nS = spin_matrices(3/2)\n@assert S'*S ≈ (3/2)*(3/2+1)*I\n@assert S[1]*S[2] - S[2]*S[1] ≈ im*S[3]\n\nS = spin_matrices(Inf)\n@assert S[1]*S[2] - S[2]*S[1] == 0\n\nSee also print_stevens_expansion.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.step!","page":"Library API","title":"Sunny.step!","text":"step!(sys::System, dynamics)\n\nAdvance the spin configuration one dynamical time-step. The dynamics object may be a continuous spin dynamics, such as Langevin or ImplicitMidpoint, or it may be a discrete Monte Carlo sampling scheme such as LocalSampler.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.stevens_matrices-Tuple{Any}","page":"Library API","title":"Sunny.stevens_matrices","text":"stevens_matrices(S)\n\nReturns a generator of Stevens operators in the spin-S representation. The return value O can be indexed as O[k,q], where 0 k 6 labels an irrep of SO(3) and -k q k. This will produce an NN matrix where ``N = 2S\n\n1``. Linear combinations of Stevens operators can be used as a \"physical\n\nbasis\" for decomposing local observables. To see this decomposition, use print_stevens_expansion.\n\nIf S == Inf, then symbolic operators will be returned. In this infinite dimensional limit, the Stevens operators become homogeneous polynomials of commuting spin operators.\n\nExample\n\nO = stevens_matrices(2)\nS = spin_matrices(2)\n\nA = (1/20)O[4,0] + (1/4)O[4,4] + (102/5)I\nB = S[1]^4 + S[2]^4 + S[3]^4\n@assert A ≈ B\n\nSee also spin_matrices and Interaction Strength Renormalization.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.subcrystal-Union{Tuple{N}, Tuple{Crystal, Vararg{String, N}}} where N","page":"Library API","title":"Sunny.subcrystal","text":"subcrystal(cryst, types) :: Crystal\n\nFilters sublattices of a Crystal by atom types, keeping the space group unchanged.\n\nsubcrystal(cryst, classes) :: Crystal\n\nFilters sublattices of Crystal by equivalence classes, keeping the space group unchanged.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.suggest_magnetic_supercell-Tuple{Any}","page":"Library API","title":"Sunny.suggest_magnetic_supercell","text":"suggest_magnetic_supercell(qs; tol=1e-12, maxsize=100)\n\nSuggests a magnetic supercell, in units of the crystal lattice vectors, that is consistent with periodicity of the wavevectors qs in RLU. If the wavevectors are incommensurate (with respect to the maximum supercell size maxsize), one can select a larger error tolerance tol to find a supercell that is almost commensurate.\n\nPrints a 33 matrix of integers that is suitable for use in reshape_supercell.\n\nExamples\n\n# A magnetic supercell for a single-Q structure. Will print\nq1 = [0, -1/4, 1/4]\nsuggest_magnetic_supercell([q1]) # [1 0 0; 0 2 1; 0 -2 1]\n\n# A larger magnetic supercell for a double-Q structure\nq2 = [1/4, 0, 1/4]\nsuggest_magnetic_supercell([q1, q2]) # [1 2 2; -1 2 -2; -1 2 2]\n\n# If given incommensurate wavevectors, find an approximate supercell that\n# is exactly commensurate for nearby wavevectors.\nsuggest_magnetic_supercell([[0, 0, 1/√5], [0, 0, 1/√7]]; tol=1e-2)\n\n# This prints [1 0 0; 0 1 0; 0 0 16], which becomes commensurate under the\n# approximations `1/√5 ≈ 7/16` and `1/√7 ≈ 3/8`.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.symmetry_equivalent_bonds-Tuple{System, Bond}","page":"Library API","title":"Sunny.symmetry_equivalent_bonds","text":"symmetry_equivalent_bonds(sys::System, bond::Bond)\n\nGiven a Bond for the original (unreshaped) crystal, return all symmetry equivalent bonds in the System. Each returned bond is represented as a pair of Sites, which may be used as input to set_exchange_at!. Reverse bonds are not included (no double counting).\n\nExample\n\nfor (site1, site2, offset) in symmetry_equivalent_bonds(sys, bond)\n @assert site1 < site2\n set_exchange_at!(sys, J, site1, site2; offset)\nend\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.to_inhomogeneous-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.to_inhomogeneous","text":"to_inhomogeneous(sys::System)\n\nReturns a copy of the system that allows for inhomogeneous interactions, which can be set using set_onsite_coupling_at!, set_exchange_at!, and set_vacancy_at!.\n\nInhomogeneous systems do not support symmetry-propagation of interactions or system reshaping.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.to_product_space-Tuple{Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.to_product_space","text":"to_product_space(A, B, Cs...)\n\nGiven lists of operators acting on local Hilbert spaces individually, return the corresponding operators that act on the tensor product space. In typical usage, the inputs will represent local physical observables and the outputs will be used to define quantum couplings.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.unit_resolution_binning_parameters-Tuple{Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.unit_resolution_binning_parameters","text":"unit_resolution_binning_parameters(sc::SampledCorrelations)\n\nCreate BinningParameters which place one histogram bin centered at each possible (q,ω) scattering vector of the crystal. This is the finest possible binning without creating bins with zero scattering vectors in them.\n\nThis function can be used without reference to a SampledCorrelations using an alternate syntax to manually specify the bin centers for the energy axis and the lattice size:\n\nunit_resolution_binning_parameters(ω_bincenters,latsize,[reciprocal lattice vectors])\n\nThe last argument may be a 3x3 matrix specifying the reciprocal lattice vectors, or a Crystal.\n\nLastly, binning parameters for a single axis may be specifed by their bin centers:\n\n(binstart,binend,binwidth) = unit_resolution_binning_parameters(bincenters::Vector{Float64})\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.@mix_proposals-Tuple","page":"Library API","title":"Sunny.@mix_proposals","text":"@mix_proposals weight1 propose1 weight2 propose2 ...\n\nMacro to generate a proposal function that randomly selects among the provided functions according to the provided probability weights. For use with LocalSampler.\n\nExample\n\n# A proposal function that proposes a spin flip 40% of the time, and a\n# Gaussian perturbation 60% of the time.\n@mix_proposals 0.4 propose_flip 0.6 propose_delta(0.2)\n\n\n\n\n\n","category":"macro"},{"location":"library.html#Optional-Makie-extensions","page":"Library API","title":"Optional Makie extensions","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"The following will be enabled through a package extension if either GLMakie or WGLMakie is loaded.","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"plot_spins\nview_crystal","category":"page"},{"location":"library.html#Sunny.plot_spins","page":"Library API","title":"Sunny.plot_spins","text":"plot_spins(sys::System; arrowscale=1.0, color=:red, colorfn=nothing,\n colormap=:viridis, colorrange=nothing, show_cell=true, orthographic=false,\n ghost_radius=0, dims=3\n\nPlot the spin configuration defined by sys. Optional parameters are:\n\narrowscale: Scale all arrows by dimensionless factor.\ncolor: Arrow colors. May be symbolic or numeric. If scalar, will be shared among all sites.\ncolorfn: Function that dynamically maps from a site index to a numeric color value. Useful for animations.\ncolormap, colorrange: Used to populate colors from numbers following Makie conventions.\nshow_cell: Show original crystallographic unit cell.\northographic: Use camera with orthographic projection.\nghost_radius: Show translucent periodic images up to a given distance (length units).\ndims: Spatial dimensions of system (1, 2, or 3).\n\nCalling notify on the return value will animate the figure.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.view_crystal","page":"Library API","title":"Sunny.view_crystal","text":"view_crystal(crystal::Crystal, max_dist::Real; show_axis=true, orthographic=false)\n\nAn interactive crystal viewer, with bonds up to max_dist.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Optional-WriteVTK-extensions","page":"Library API","title":"Optional WriteVTK extensions","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"The following will be enabled through a package extension if WriteVTK is loaded.","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"export_vtk","category":"page"},{"location":"library.html#Sunny.export_vtk","page":"Library API","title":"Sunny.export_vtk","text":"export_vtk(filename,params::BinningParameters,data)\n\nExport a VTK-compatible file to filename (do not include file extension when specifying the file name) which contains the data as VTK Cell Data on a grid parameterized by params.\n\nAt least one axis of the BinningParameters must be integrated over, since VTK does not support 4D data. See integrate_axes!.\n\n\n\n\n\n","category":"function"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"EditURL = \"../../../examples/05_MC_Ising.jl\"","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/05_MC_Ising.html#.-Monte-Carlo-sampling-of-the-Ising-model","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"","category":"section"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"This tutorial illustrates simulation of the classical 2D Ising model.","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"Sunny expects a 3D Crystal unit cell. To model a square lattice, we create an orthogonal unit cell where the z-spacing is distinct from the x and y spacing.","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"a = 1\nlatvecs = lattice_vectors(a,a,10a,90,90,90)\ncrystal = Crystal(latvecs, [[0,0,0]])","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"Create a System of spins with linear size L in the x and y directions, and only one layer in the z direction. The option :dipole means that the system will store Heisenberg spins, as opposed to SU(N) coherent states. Polarize the initial spin configuration using polarize_spins!. Following the Ising convention, we will restrict these spins to the z-axis and give them magnitude S=1.","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"By default, Sunny uses physical units, e.g. magnetic field in tesla. Here we specify an alternative Units system, so that the Zeeman coupling between the spin dipole 𝐬 and an external field 𝐁 has the dimensionless form -𝐁𝐬.","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"L = 128\nsys = System(crystal, (L,L,1), [SpinInfo(1, S=1, g=1)], :dipole, units=Units.theory, seed=0)\npolarize_spins!(sys, (0,0,1))","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"Use set_exchange! to include a ferromagnetic Heisenberg interaction along nearest-neighbor bonds. The Bond below connects two spins displaced by one lattice constant in the x-direction. This interaction will be propagated to all nearest-neighbors bonds in the system, consistent with the symmetries of the square lattice.","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"set_exchange!(sys, -1.0, Bond(1,1,(1,0,0)))","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"If an external field is desired, it can be set using set_external_field!.","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"B = 0\nset_external_field!(sys, (0,0,B))","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"The critical temperature for the Ising model is known analytically.","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"Tc = 2/log(1+√2)","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"Use a LocalSampler to perform nsweeps Monte Carlo sweeps. A sweep consists of, on average, one trial update per spin in the system. Each proposed update is accepted or rejected according to the Metropolis acceptance probability. As its name suggests, the propose_flip function will only propose pure spin flips, 𝐬 rightarrow -𝐬.","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"nsweeps = 4000\nsampler = LocalSampler(kT=Tc, propose=propose_flip)\nfor i in 1:nsweeps\n step!(sys, sampler)\nend","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"Plot the Ising spins by extracting the z-component of the dipoles","category":"page"},{"location":"examples/05_MC_Ising.html","page":"5. Monte Carlo sampling of the Ising model","title":"5. Monte Carlo sampling of the Ising model","text":"heatmap(reshape([s.z for s in sys.dipoles], (L,L)))","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Powder-Averaged-CoRhO-at-Finite-Temperature","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Inspired by: Ge et al., Phys. Rev. B 96, 064413 (2017)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Authors: Martin Mourigal, David Dahlbom","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Date: October 28, 2023 (Sunny 0.5.5)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Goal: This script is to calculate the temperature dependence of the magnon excitations in the spin-3/2 Heisenberg Diamond Antiferromagnet and compare to powder-averaged results obtained for the compound CoRh₂O₄","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Load-pacakges.","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Load pacakges.","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"using Sunny, GLMakie, ProgressMeter, Statistics, Random, Brillouin\ncif_path = pkgdir(Sunny, \"examples\", \"longer_examples\", \"CoRh2O4_#109301.cif\");","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Define-custom-functions.","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Define custom functions.","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"The function quench! randomizes the spins of a given System, fixes a target temperature, and lets the system relax at this temperature for nrelax integration steps.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function quench!(sys, integrator; kTtarget, nrelax)\n randomize_spins!(sys);\n integrator.kT = kTtarget;\n prog = Progress(nrelax; dt=10.0, desc=\"Quenched and now relaxing: \", color=:green);\n for _ in 1:nrelax\n step!(sys, integrator)\n next!(prog)\n end\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"quench! (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"dwell! takes a System, sets a target temperature, and has the system dwell at this temperature for ndwell integration steps.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function dwell!(sys, integrator; kTtarget, ndwell)\n integrator.kT = kTtarget;\n prog = Progress(ndwell; dt=10.0, desc=\"Dwelling: \", color=:green);\n for _ in 1:ndwell\n step!(sys, integrator)\n next!(prog)\n end\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"dwell! (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"anneal! takes a temperature schedule and cools the System through it, with ndwell steps of the integrator at each temperature in the schedule. Returns the energy at the end of the dwell for each scheduled temperature.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function anneal!(sys, integrator; kTschedule, ndwell)\n nspins = prod(size(sys.dipoles));\n ensys = zeros(length(kTschedule))\n prog = Progress(ndwell*length(kTschedule); dt=10.0, desc=\"Annealing: \", color=:red);\n for (i, kT) in enumerate(kTschedule)\n integrator.kT = kT\n for _ in 1:ndwell\n step!(sys, integrator)\n next!(prog)\n end\n ensys[i] = energy(sys)\n end\n return ensys/nspins\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"anneal! (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"sample_sf! samples a structure factor, which may be either an instant or dynamical structure factor. The integrator is run ndecorr times before each one of the samples is taken.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function sample_sf!(sf, sys, integrator; nsamples, ndecorr)\n prog = Progress(nsamples*ndecorr; dt=10.0, desc=\"Sampling SF: \", color=:red);\n for _ in 1:nsamples\n for _ in 1:ndecorr\n step!(sys, integrator)\n next!(prog)\n end\n add_sample!(sf, sys) # Accumulate the newly sampled structure factor into `sf`\n end\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"sample_sf! (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"powder_average powder averages a structure factor. Works for both instant and dynamical structure factors. To prevent smearing, removes Bragg peaks before introducing energy broadening. Bragg peaks are added back at ω=0 after broadening.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"function powder_average(sc, rs, npts, formula; η=0.1)\n prog = Progress(length(rs); dt=10., desc=\"Powder Averaging: \", color=:blue)\n ωs = available_energies(sc)\n output = zeros(Float64, length(rs), length(ωs))\n for (i, r) in enumerate(rs)\n qs = reciprocal_space_shell(sc.crystal, r, npts)\n vals = intensities_interpolated(sc, qs, formula)\n bragg_idxs = findall(x -> x > maximum(vals)*0.9, vals)\n bragg_vals = vals[bragg_idxs]\n vals[bragg_idxs] .= 0\n vals = broaden_energy(sc, vals, (ω,ω₀)->lorentzian(ω-ω₀, η))\n vals[bragg_idxs] .= bragg_vals\n output[i,:] .= mean(vals, dims=1)[1,:]\n next!(prog)\n end\n return output\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"powder_average (generic function with 1 method)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#System-Definition-for-CoRhO","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"System Definition for CoRh₂O₄","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Define the crystal structure of CoRh₂O₄ in the conventional cell.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"xtal = Crystal(cif_path; symprec=1e-4)\nmagxtal = subcrystal(xtal,\"Co1\")\nview_crystal(magxtal, 6.0)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Print the symmetry-allowed interactions.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"print_symmetry_table(magxtal, 4.0)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Atom 1\nType 'Co1', position [0, 0, 0], multiplicity 8\nAllowed g-tensor: [A 0 0\n 0 A 0\n 0 0 A]\nAllowed anisotropy in Stevens operators:\n c₁*(𝒪[4,0]+5𝒪[4,4]) +\n c₂*(𝒪[6,0]-21𝒪[6,4])\n\nSunny.Bond(1, 3, [0, 0, 0])\nDistance 3.6784429025744, coordination 4\nConnects 'Co1' at [0, 0, 0] to 'Co1' at [1/4, 1/4, 1/4]\nAllowed exchange matrix:[A B B\n B A B\n B B A]\n\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Assign local Hilbert space","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"S = 3/2\nlhs = [SpinInfo(1; S, g=2)]\nformfactors = [FormFactor(\"Co2\")];","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Create System and randomize it","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"sunmode = :dipole\nlatsize = (10,10,10)\nsys = System(magxtal, latsize, lhs, sunmode; seed=1)\nrandomize_spins!(sys)\nplot_spins(sys)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Define exchange interactions.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"scaleJ = 0.63\nvalJ1 = 1.00*scaleJ\nset_exchange!(sys, valJ1, Bond(1, 3, [0, 0, 0]));","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Thermalize-system-to-an-ordered,-yet-finite-temperature,-state","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Thermalize system to an ordered, yet finite temperature, state","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Define Langevin Integrator and Initialize it","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Δt0 = 0.05/abs(scaleJ*S); ## Time steps in Langevin\nλ0 = 0.1; ## Langevin damping, usually 0.05 or 0.1 is good.\nkT0 = 0.01*abs(scaleJ*S); ## Initialize at some temperature\nintegrator = Langevin(Δt0; λ=λ0, kT=kT0);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Thermalization Option 1: Quench the system from infinite temperature to a target temperature. Note: this may lead to a poorly thermalized sample","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"quench!(sys, integrator; kTtarget=kT0, nrelax=10000);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Option 2: Anneal (according to a temperature schedule) then dwell once we've reach base temperature. (Uncomment to execute.)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"# kTs = [abs(scaleJ)*valS*100 * 0.9^k for k in 0:100]\n# anneal!(sys,integrator;kTschedule=kTs,ndwell=500)\n# dwell!(sys,integrator;kTtarget=kTs[end],ndwell=2000)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot the resulting spin system to check ordering in real space","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"plot_spins(sys)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Calculation-of-Neutron-Scattering-Responses","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Calculation of Neutron Scattering Responses","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Fourier-transformed-instantaneous-two-point-correlation-functions","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Fourier transformed instantaneous two-point correlation functions","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Calculate the instantaneous/equal-time structure factor.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"eqsf = instant_correlations(sys)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"SampledCorrelations (6.252 MiB)\n[S(q) | 0 sample]\nLattice: (10, 10, 10)×8\n6 correlations in Dipole mode:\n╔ ⬤ ⬤ ⬤ Sx\n║ ⋅ ⬤ ⬤ Sy\n╚ ⋅ ⋅ ⬤ Sz\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"If desired, add additional samples by decorrelating and then re-calculating the eqsf.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"nsamples = 1\nndecorr = 1000\n@time sample_sf!(eqsf, sys, integrator; nsamples=nsamples, ndecorr=ndecorr);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":" 0.966485 seconds (1.89 M allocations: 118.815 MiB, 6.25% gc time, 50.12% compilation time)\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Project onto a constant Q-Slice in momentum space.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"nQpts = 200\nQxpts = range(-10.0, 10.0, length=nQpts)\nQypts = range(-10.0, 10.0, length=nQpts)\nqz = 1.0\nQpts = [[qx, qy, qz] for qx in Qxpts, qy in Qypts]\ninstant_formula = intensity_formula(eqsf, :perp; formfactors)\niq = instant_intensities_interpolated(eqsf, Qpts, instant_formula);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot the resulting I(Q)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"heatmap(Qxpts, Qypts, iq;\n colorrange = (0, maximum(iq)/20),\n axis = (\n xlabel=\"Momentum Transfer Qx (r.l.u)\", xlabelsize=16,\n ylabel=\"Momentum Transfer Qy (r.l.u)\", ylabelsize=16,\n aspect=true,\n )\n)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Dynamical-and-energy-integrated-two-point-correlation-functions","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Dynamical and energy-integrated two-point correlation functions","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Calculate the time traces and Fourier transform: Dynamical Structure Factor (first sample).","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"ωmax = 6.0 # Maximum energy to resolve\nnω = 100 # Number of energies to resolve\nsc = dynamical_correlations(sys; Δt=Δt0, nω=nω, ωmax=ωmax, process_trajectory=:symmetrize)\n@time add_sample!(sc, sys) # Add a sample trajectory","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":" 5.471269 seconds (14.33 M allocations: 291.526 MiB, 0.75% gc time, 0.02% compilation time)\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"If desired, add additional decorrelated samples.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"nsamples = 10\nndecorr = 1000\n@time sample_sf!(sc, sys, integrator; nsamples=nsamples, ndecorr=ndecorr);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Sampling SF: 27%|██████████▎ | ETA: 0:00:33\u001b[KSampling SF: 49%|██████████████████▊ | ETA: 0:00:25\u001b[KSampling SF: 68%|█████████████████████████▉ | ETA: 0:00:17\u001b[KSampling SF: 84%|████████████████████████████████ | ETA: 0:00:09\u001b[KSampling SF: 100%|██████████████████████████████████████| Time: 0:00:53\u001b[K\n 59.070784 seconds (143.58 M allocations: 2.866 GiB, 0.56% gc time, 0.24% compilation time)\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Can use the Brillouin package for help on determining high symmetry points","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"kp = irrfbz_path(227,[[1,0,0], [0,1,0], [0,0,1]])\nkpc = cartesianize(kp)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"KPath{3} (6 points, 2 paths, 8 points in paths):\n points: :U => [1.570796, 6.283185, 1.570796]\n :W => [3.141593, 6.283185, 0.0]\n :K => [4.712389, 4.712389, 0.0]\n :Γ => [0.0, 0.0, 0.0]\n :L => [3.141593, 3.141593, 3.141593]\n :X => [0.0, 6.283185, 0.0]\n paths: [:Γ, :X, :U]\n [:K, :Γ, :L, :W, :X]\n basis: [-6.283185, 6.283185, 6.283185]\n [6.283185, -6.283185, 6.283185]\n [6.283185, 6.283185, -6.283185]","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Project onto a constant QE-Slice in momentum-energy space.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"densQpts = 50\nsymQpts = [[0.75, 0.75, 0.00], # List of wave vectors that define a path\n [0.00, 0.00, 0.00],\n [0.50, 0.50, 0.50],\n [0.50, 1.00, 0.00],\n [0.00, 1.00, 0.00],\n [0.25, 1.00, 0.25],\n [0.00, 1.00, 0.00],\n [0.00,-4.00, 0.00]]\n(Qpts, xticks) = reciprocal_space_path(magxtal, symQpts, densQpts)\nformula = intensity_formula(sc, :perp; formfactors, kT=integrator.kT)\niqw = intensities_interpolated(sc, Qpts, formula);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"If desired, broaden the sc in energy.","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"η = 0.1 ## Lorentzian energy broadening parameter\niqwc = broaden_energy(sc, iqw, (ω, ω₀) -> lorentzian(ω-ω₀, η));","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"If desired, calculated the energy-integrated structure factor","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"iqt = instant_intensities_interpolated(sc, Qpts, formula);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot the resulting I(Q,W).","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"ωs = available_energies(sc)\nheatmap(1:size(iqwc, 1), ωs, iqwc;\n colorrange = (0, maximum(iqwc)/20000.0),\n axis = (;\n xlabel=\"Momentum Transfer (r.l.u)\",\n ylabel=\"Energy Transfer (meV)\",\n xticks,\n xticklabelrotation=π/5,\n aspect = 1.4,\n )\n)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Projection into a powder-averaged neutron scattering intensity .","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Qmax = 3.5\nnQpts = 100\nQpow = range(0, Qmax, nQpts)\nnpoints = 100\npqw = powder_average(sc, Qpow, npoints, formula; η);","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Powder Averaging: 78%|█████████████████████████▊ | ETA: 0:00:03\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot resulting Ipow(|Q|,W).","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"heatmap(Qpow, ωs, pqw;\n axis = (\n xlabel=\"|Q| (Å⁻¹)\",\n ylabel=\"Energy Transfer (meV)\",\n aspect = 1.4,\n ),\n colorrange = (0, 40.0)\n)","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html#Calculation-of-temperature-dependent-powder-average-spectrum","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Calculation of temperature-dependent powder average spectrum","text":"","category":"section"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Define a temperature schedule","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"kTs = [60 40 25 20 15 12 10 4] * Sunny.meV_per_K\npqw_res = []\nfor kT in kTs\n dwell!(sys, integrator; kTtarget=kT, ndwell=1000);\n sc_loc = dynamical_correlations(sys; Δt=Δt0, nω, ωmax, process_trajectory=:symmetrize);\n add_sample!(sc_loc, sys)\n formula = intensity_formula(sc, :perp; formfactors, kT)\n push!(pqw_res, powder_average(sc_loc, Qpow, npoints, formula; η))\nend","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"┌ Warning: Assignment to `formula` in soft scope is ambiguous because a global variable by the same name exists: `formula` will be treated as a new local. Disambiguate by using `local formula` to suppress this warning or `global formula` to assign to the existing global variable.\n└ @ ~/Research/SunnyContributed/contributed-docs/build/CoRh2O4-tutorial.md:7\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 81%|██████████████████████████▊ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 83%|███████████████████████████▍ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 81%|██████████████████████████▊ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:12\u001b[K\nPowder Averaging: 87%|████████████████████████████▊ | ETA: 0:00:02\u001b[KPowder Averaging: 100%|█████████████████████████████████| Time: 0:00:11\u001b[K\n","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"Plot the resulting Ipow(|Q|,W) as a function of temperature, to compare with Fig.6 of https://arxiv.org/abs/1706.05881","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"fig = Figure(; resolution=(1200,600))\nfor i in 1:8\n r, c = fldmod1(i, 4)\n ax = Axis(fig[r, c];\n title = \"kT = \"*string(round(kTs[9-i], digits=3))*\" (meV)\",\n xlabel = r == 2 ? \"|Q| (Å⁻¹)\" : \"\",\n ylabel = c == 1 ? \"Energy Transfer (meV)\" : \"\",\n aspect = 1.4,\n )\n heatmap!(ax, Qpow, ωs, pqw_res[9-i]; colorrange = (0, 20.0))\nend\nfig","category":"page"},{"location":"examples/contributed/CoRh2O4-tutorial.html","page":"Powder Averaged CoRh₂O₄ at Finite Temperature","title":"Powder Averaged CoRh₂O₄ at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#MgCr2O4-at-Finite-Temperature","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Author: Martin Mourigal Date: September 9, 2022 (Updated by October 28, 2023 using Sunny 0.5.5)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"In this tutorial, we will walk through an example in Sunny and calculate the spin dynamical properties of the Heisenberg pyrochlore antiferromagnet and apply this knowledge to MgCr2O4 and ZnCr2O4, which are known to approximate this model. Relevant publications include:","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"[1] P. H. Conlon and J. T. Chalker, Phys. Rev. Lett. 102, 237206 (2009)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"[2] P. H. Conlon and J. T. Chalker, Phys. Rev. B 81, 224413 (2010)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"[3] X. Bai, J. A. M. Paddison, et al. Phys. Rev. Lett. 122, 097201 (2019)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Setting-up-Julia","page":"MgCr2O4 at Finite Temperature","title":"Setting up Julia","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To run the examples in the tutorial, you will need a working installation of the Julia programming language and the Sunny package. Some useful references for getting started are:","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Getting started with Julia for Sunny\nSunny Docs","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We will begin by loading the relevant packages.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"using Sunny # The main package\nusing GLMakie # Plotting package","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Setting-up-the-crystal-structure","page":"MgCr2O4 at Finite Temperature","title":"Setting up the crystal structure","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Before specifying the interactions of our system, we first must set up the crystal. We will begin by specifying the pyrochlore lattice (illustrated below) in the manner that is typical of theorists.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Picture Credits: Theory of Quantum Matter Unit, OIST","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#\"Theorist\"-Method","page":"MgCr2O4 at Finite Temperature","title":"\"Theorist\" Method","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"In this approach, we directly define the lattice vectors and specify the position of each atom in fractional coordinates.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"latvecs = lattice_vectors(8.3342, 8.3342, 8.3342, 90, 90, 90)\npositions = [[0.875, 0.625, 0.375],\n [0.625, 0.125, 0.625],\n [0.875, 0.875, 0.125],\n [0.625, 0.875, 0.375],\n [0.875, 0.125, 0.875],\n [0.625, 0.625, 0.125],\n [0.875, 0.375, 0.625],\n [0.625, 0.375, 0.875],\n [0.375, 0.625, 0.875],\n [0.125, 0.125, 0.125],\n [0.375, 0.875, 0.625],\n [0.125, 0.875, 0.875],\n [0.375, 0.125, 0.375],\n [0.125, 0.625, 0.625],\n [0.375, 0.375, 0.125],\n [0.125, 0.375, 0.375]];\ntypes = [\"B\" for _ in positions]\nxtal_pyro = Crystal(latvecs, positions; types) # We will call this crystal the Theoretical Pyrochlore","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'F d -3 m' (227)\nLattice params a=8.334, b=8.334, c=8.334, α=90°, β=90°, γ=90°\nCell volume 578.9\nType 'B', Wyckoff 16c (point group '.-3m'):\n 1. [7/8, 5/8, 3/8]\n 2. [5/8, 1/8, 5/8]\n 3. [7/8, 7/8, 1/8]\n 4. [5/8, 7/8, 3/8]\n 5. [7/8, 1/8, 7/8]\n 6. [5/8, 5/8, 1/8]\n 7. [7/8, 3/8, 5/8]\n 8. [5/8, 3/8, 7/8]\n 9. [3/8, 5/8, 7/8]\n 10. [1/8, 1/8, 1/8]\n 11. [3/8, 7/8, 5/8]\n 12. [1/8, 7/8, 7/8]\n 13. [3/8, 1/8, 3/8]\n 14. [1/8, 5/8, 5/8]\n 15. [3/8, 3/8, 1/8]\n 16. [1/8, 3/8, 3/8]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To examine the result interactively, we can call view_crystal.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"view_crystal(xtal_pyro, 3.2)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#\"Experimentalist\"-Method-#1-(Incorrect)","page":"MgCr2O4 at Finite Temperature","title":"\"Experimentalist\" Method #1 (Incorrect)","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"A real crystal is more complicated than this, however, and we will now construct the system using the actual CIF file of MgCr2O4 from ICSD. This can be done by copying over the data from a CIF file by hand, but this can be dangerous, as shown below.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"latvecs = lattice_vectors(8.3342, 8.3342, 8.3342, 90, 90, 90)\npositions = [[0.1250, 0.1250, 0.1250],\n [0.5000, 0.5000, 0.5000],\n [0.2607, 0.2607, 0.2607]]\ntypes = [\"Mg\",\"Cr\",\"O\"]\nxtal_mgcro_1 = Crystal(latvecs, positions; types)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'R 3 m' (160)\nLattice params a=8.334, b=8.334, c=8.334, α=90°, β=90°, γ=90°\nCell volume 578.9\nType 'Mg', Wyckoff 3a (point group '3m'):\n 1. [1/8, 1/8, 1/8]\nType 'Cr', Wyckoff 3a (point group '3m'):\n 2. [1/2, 1/2, 1/2]\nType 'O', Wyckoff 3a (point group '3m'):\n 3. [0.2607, 0.2607, 0.2607]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Sunny returned a valid crystal, but it did get right space group for MgCr2O4. This can be fixed by modifying the input to include the space group and the setting.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#\"Experimentalist\"-Method-#2-(Correct)","page":"MgCr2O4 at Finite Temperature","title":"\"Experimentalist\" Method #2 (Correct)","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"As above, we will define the crystal structure of MgCr2O4 by copying the info from a CIF file, but we will also specify the space group and setting explicitly.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"latvecs = lattice_vectors(8.3342, 8.3342, 8.3342, 90, 90, 90)\npositions = [[0.1250, 0.1250, 0.1250],\n [0.5000, 0.5000, 0.5000],\n [0.2607, 0.2607, 0.2607]]\ntypes = [\"Mg\",\"Cr\",\"O\"]\nspacegroup = 227 # Space Group Number\nsetting = \"2\" # Space Group setting\nxtal_mgcro_2 = Crystal(latvecs, positions, spacegroup; types, setting)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'F d -3 m' (227)\nLattice params a=8.334, b=8.334, c=8.334, α=90°, β=90°, γ=90°\nCell volume 578.9\nType 'Mg', Wyckoff 8b (point group '-43m'):\n 1. [1/8, 1/8, 1/8]\n 2. [5/8, 5/8, 1/8]\n 3. [7/8, 3/8, 3/8]\n 4. [3/8, 7/8, 3/8]\n 5. [5/8, 1/8, 5/8]\n 6. [1/8, 5/8, 5/8]\n 7. [3/8, 3/8, 7/8]\n 8. [7/8, 7/8, 7/8]\nType 'Cr', Wyckoff 16c (point group '.-3m'):\n 9. [1/2, 0, 0]\n 10. [3/4, 1/4, 0]\n 11. [0, 1/2, 0]\n 12. [1/4, 3/4, 0]\n 13. [3/4, 0, 1/4]\n 14. [1/2, 1/4, 1/4]\n 15. [1/4, 1/2, 1/4]\n 16. [0, 3/4, 1/4]\n 17. [0, 0, 1/2]\n 18. [1/4, 1/4, 1/2]\n 19. [1/2, 1/2, 1/2]\n 20. [3/4, 3/4, 1/2]\n 21. [1/4, 0, 3/4]\n 22. [0, 1/4, 3/4]\n 23. [3/4, 1/2, 3/4]\n 24. [1/2, 3/4, 3/4]\nType 'O', Wyckoff 32e (point group '.3m'):\n 25. [0.7393, 0.0107, 0.0107]\n 26. [0.5107, 0.2393, 0.0107]\n 27. [0.2393, 0.5107, 0.0107]\n 28. [0.0107, 0.7393, 0.0107]\n 29. [0.5107, 0.0107, 0.2393]\n 30. [0.7393, 0.2393, 0.2393]\n 31. [0.0107, 0.5107, 0.2393]\n 32. [0.2393, 0.7393, 0.2393]\n 33. [0.2607, 0.2607, 0.2607]\n 34. [0.4893, 0.4893, 0.2607]\n 35. [0.7607, 0.7607, 0.2607]\n 36. [0.9893, 0.9893, 0.2607]\n 37. [0.4893, 0.2607, 0.4893]\n 38. [0.2607, 0.4893, 0.4893]\n 39. [0.9893, 0.7607, 0.4893]\n 40. [0.7607, 0.9893, 0.4893]\n 41. [0.2393, 0.0107, 0.5107]\n 42. [0.0107, 0.2393, 0.5107]\n 43. [0.7393, 0.5107, 0.5107]\n 44. [0.5107, 0.7393, 0.5107]\n 45. [0.0107, 0.0107, 0.7393]\n 46. [0.2393, 0.2393, 0.7393]\n 47. [0.5107, 0.5107, 0.7393]\n 48. [0.7393, 0.7393, 0.7393]\n 49. [0.7607, 0.2607, 0.7607]\n 50. [0.9893, 0.4893, 0.7607]\n 51. [0.2607, 0.7607, 0.7607]\n 52. [0.4893, 0.9893, 0.7607]\n 53. [0.9893, 0.2607, 0.9893]\n 54. [0.7607, 0.4893, 0.9893]\n 55. [0.4893, 0.7607, 0.9893]\n 56. [0.2607, 0.9893, 0.9893]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"This result is correct, but at this point we might as well import the CIF file directly, which we now proceed to do.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#\"Experimentalist\"-Method-#3-(Correct-–-if-your-CIF-file-is)","page":"MgCr2O4 at Finite Temperature","title":"\"Experimentalist\" Method #3 (Correct – if your CIF file is)","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To import a CIF file, simply give the path to Crystal. One may optionally specify a precision parameter to apply to the symmetry analysis.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"cif = pkgdir(Sunny, \"examples\", \"longer_examples\", \"MgCr2O4_160953_2009.cif\")\nxtal_mgcro_3 = Crystal(cif; symprec=0.001)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'F d -3 m' (227)\nLattice params a=8.333, b=8.333, c=8.333, α=90°, β=90°, γ=90°\nCell volume 578.6\nType 'Mg1', Wyckoff 8b (point group '-43m'):\n 1. [1/8, 1/8, 1/8]\n 2. [5/8, 5/8, 1/8]\n 3. [7/8, 3/8, 3/8]\n 4. [3/8, 7/8, 3/8]\n 5. [5/8, 1/8, 5/8]\n 6. [1/8, 5/8, 5/8]\n 7. [3/8, 3/8, 7/8]\n 8. [7/8, 7/8, 7/8]\nType 'Cr1', Wyckoff 16c (point group '.-3m'):\n 9. [1/2, 0, 0]\n 10. [3/4, 1/4, 0]\n 11. [0, 1/2, 0]\n 12. [1/4, 3/4, 0]\n 13. [3/4, 0, 1/4]\n 14. [1/2, 1/4, 1/4]\n 15. [1/4, 1/2, 1/4]\n 16. [0, 3/4, 1/4]\n 17. [0, 0, 1/2]\n 18. [1/4, 1/4, 1/2]\n 19. [1/2, 1/2, 1/2]\n 20. [3/4, 3/4, 1/2]\n 21. [1/4, 0, 3/4]\n 22. [0, 1/4, 3/4]\n 23. [3/4, 1/2, 3/4]\n 24. [1/2, 3/4, 3/4]\nType 'O1', Wyckoff 32e (point group '.3m'):\n 25. [0.7388, 0.0112, 0.0112]\n 26. [0.5112, 0.2388, 0.0112]\n 27. [0.2388, 0.5112, 0.0112]\n 28. [0.0112, 0.7388, 0.0112]\n 29. [0.5112, 0.0112, 0.2388]\n 30. [0.7388, 0.2388, 0.2388]\n 31. [0.0112, 0.5112, 0.2388]\n 32. [0.2388, 0.7388, 0.2388]\n 33. [0.2612, 0.2612, 0.2612]\n 34. [0.4888, 0.4888, 0.2612]\n 35. [0.7612, 0.7612, 0.2612]\n 36. [0.9888, 0.9888, 0.2612]\n 37. [0.4888, 0.2612, 0.4888]\n 38. [0.2612, 0.4888, 0.4888]\n 39. [0.9888, 0.7612, 0.4888]\n 40. [0.7612, 0.9888, 0.4888]\n 41. [0.2388, 0.0112, 0.5112]\n 42. [0.0112, 0.2388, 0.5112]\n 43. [0.7388, 0.5112, 0.5112]\n 44. [0.5112, 0.7388, 0.5112]\n 45. [0.0112, 0.0112, 0.7388]\n 46. [0.2388, 0.2388, 0.7388]\n 47. [0.5112, 0.5112, 0.7388]\n 48. [0.7388, 0.7388, 0.7388]\n 49. [0.7612, 0.2612, 0.7612]\n 50. [0.9888, 0.4888, 0.7612]\n 51. [0.2612, 0.7612, 0.7612]\n 52. [0.4888, 0.9888, 0.7612]\n 53. [0.9888, 0.2612, 0.9888]\n 54. [0.7612, 0.4888, 0.9888]\n 55. [0.4888, 0.7612, 0.9888]\n 56. [0.2612, 0.9888, 0.9888]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Finally, we wish to restrict attention to the magnetic atoms in the unit cell while maintaining symmetry information for the full crystal, which is required to determine the correct exchange and g-factor anisotropies. This can be achieved with the subcrystal function.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"xtal_mgcro = subcrystal(xtal_mgcro_2,\"Cr\")","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Crystal\nHM symbol 'F d -3 m' (227)\nLattice params a=8.334, b=8.334, c=8.334, α=90°, β=90°, γ=90°\nCell volume 578.9\nType 'Cr', Wyckoff 16c (point group '.-3m'):\n 1. [1/2, 0, 0]\n 2. [3/4, 1/4, 0]\n 3. [0, 1/2, 0]\n 4. [1/4, 3/4, 0]\n 5. [3/4, 0, 1/4]\n 6. [1/2, 1/4, 1/4]\n 7. [1/4, 1/2, 1/4]\n 8. [0, 3/4, 1/4]\n 9. [0, 0, 1/2]\n 10. [1/4, 1/4, 1/2]\n 11. [1/2, 1/2, 1/2]\n 12. [3/4, 3/4, 1/2]\n 13. [1/4, 0, 3/4]\n 14. [0, 1/4, 3/4]\n 15. [3/4, 1/2, 3/4]\n 16. [1/2, 3/4, 3/4]\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Making-a-System-and-assigning-interactions","page":"MgCr2O4 at Finite Temperature","title":"Making a System and assigning interactions","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Making-a-System","page":"MgCr2O4 at Finite Temperature","title":"Making a System","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Before assigning any interactions, we first have to set up a System using our Crystal.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"dims = (6, 6, 6) # Supercell dimensions\nspininfos = [SpinInfo(1, S=3/2, g=2)] # Specify spin information, note that all sites are symmetry equivalent\nsys_pyro = System(xtal_pyro, dims, spininfos, :dipole) # Make a system in dipole (Landau-Lifshitz) mode on pyrochlore lattice\nsys_mgcro = System(xtal_mgcro, dims, spininfos, :dipole); # Same on MgCr2O4 crystal","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To understand what interactions we may assign to this system, we have to understand the the symmetry properties of the crystal, which we turn to next.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Symmetry-analysis-for-exchange-and-single-ion-anisotropies","page":"MgCr2O4 at Finite Temperature","title":"Symmetry analysis for exchange and single-ion anisotropies","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"print_symmetry_table reports all the exchange interactions, single-site anisotropies, and g-factors allowed on our crystal. It takes a Cyrstal and a distance. We'll look at both the \"theorist's\" pyrochlore lattice,","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"print_symmetry_table(xtal_pyro, 5.9)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Atom 1\nType 'B', position [7/8, 5/8, 3/8], multiplicity 16\nAllowed g-tensor: [ A B -B\n B A B\n -B B A]\nAllowed anisotropy in Stevens operators:\n c₁*(𝒪[2,-2]+2𝒪[2,-1]-2𝒪[2,1]) +\n c₂*(7𝒪[4,-3]+2𝒪[4,-2]-𝒪[4,-1]+𝒪[4,1]+7𝒪[4,3]) + c₃*(𝒪[4,0]+5𝒪[4,4]) +\n c₄*(-11𝒪[6,-6]-8𝒪[6,-3]+𝒪[6,-2]-8𝒪[6,-1]+8𝒪[6,1]-8𝒪[6,3]) + c₅*(𝒪[6,0]-21𝒪[6,4]) + c₆*((9/5)𝒪[6,-6]+(24/5)𝒪[6,-5]+𝒪[6,-2]+(8/5)𝒪[6,-1]-(8/5)𝒪[6,1]-(24/5)𝒪[6,5])\n\nSunny.Bond(1, 3, [0, 0, 0])\nDistance 2.94658466788246, coordination 6\nConnects 'B' at [7/8, 5/8, 3/8] to 'B' at [7/8, 7/8, 1/8]\nAllowed exchange matrix:[ A -D D\n D B C\n -D C B]\nAllowed DM vector: [0 -D -D]\n\nSunny.Bond(1, 2, [0, 0, 0])\nDistance 5.10363435357589, coordination 12\nConnects 'B' at [7/8, 5/8, 3/8] to 'B' at [5/8, 1/8, 5/8]\nAllowed exchange matrix:[ A C-E D-F\n C+E B -C+E\n D+F -C-E A]\nAllowed DM vector: [E F -E]\n\nSunny.Bond(2, 6, [0, 0, 0])\nDistance 5.89316933576492, coordination 6\nConnects 'B' at [5/8, 1/8, 5/8] to 'B' at [5/8, 5/8, 1/8]\nAllowed exchange matrix:[A D D\n D B C\n D C B]\n\nSunny.Bond(1, 5, [0, 0, 0])\nDistance 5.89316933576492, coordination 6\nConnects 'B' at [7/8, 5/8, 3/8] to 'B' at [7/8, 1/8, 7/8]\nAllowed exchange matrix:[ A D -D\n D B C\n -D C B]\n\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"and for the the MgCrO4 crystal,","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"print_symmetry_table(xtal_mgcro, 6.0)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Atom 1\nType 'Cr', position [1/2, 0, 0], multiplicity 16\nAllowed g-tensor: [A B B\n B A B\n B B A]\nAllowed anisotropy in Stevens operators:\n c₁*(𝒪[2,-2]+2𝒪[2,-1]+2𝒪[2,1]) +\n c₂*(-7𝒪[4,-3]-2𝒪[4,-2]+𝒪[4,-1]+𝒪[4,1]+7𝒪[4,3]) + c₃*(𝒪[4,0]+5𝒪[4,4]) +\n c₄*(-11𝒪[6,-6]-8𝒪[6,-3]+𝒪[6,-2]-8𝒪[6,-1]-8𝒪[6,1]+8𝒪[6,3]) + c₅*(𝒪[6,0]-21𝒪[6,4]) + c₆*((9/5)𝒪[6,-6]+(24/5)𝒪[6,-5]+𝒪[6,-2]+(8/5)𝒪[6,-1]+(8/5)𝒪[6,1]+(24/5)𝒪[6,5])\n\nSunny.Bond(1, 2, [0, 0, 0])\nDistance 2.94658466788246, coordination 6\nConnects 'Cr' at [1/2, 0, 0] to 'Cr' at [3/4, 1/4, 0]\nAllowed exchange matrix:[A C -D\n C A -D\n D D B]\nAllowed DM vector: [-D D 0]\n\nSunny.Bond(1, 7, [0, 0, 0])\nDistance 5.10363435357589, coordination 12\nConnects 'Cr' at [1/2, 0, 0] to 'Cr' at [1/4, 1/2, 1/4]\nAllowed exchange matrix:[ A C-E D-F\n C+E B -C+E\n D+F -C-E A]\nAllowed DM vector: [E F -E]\n\nSunny.Bond(1, 3, [0, 0, 0])\nDistance 5.89316933576492, coordination 6\nConnects 'Cr' at [1/2, 0, 0] to 'Cr' at [0, 1/2, 0]\nAllowed exchange matrix:[A D C\n D A C\n C C B]\n\nSunny.Bond(1, 3, [1, 0, 0])\nDistance 5.89316933576492, coordination 6\nConnects 'Cr' at [1/2, 0, 0] to 'Cr' at [1, 1/2, 0]\nAllowed exchange matrix:[A D C\n D A C\n C C B]\n\n","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Note that the exchange anisotropies allowed on the the pyrochlore lattice are slightly different from those on the MgCr2O4 cyrstal due to the role of Mg and O in the bonds. Also note that Sunny has correctly identified the two inequivalent bonds 3a and 3b having the same length. A question may arises to know which bond is J3a and which is J3b, let's plot the structure.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"view_crystal(xtal_mgcro, 5.9)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"The crystal viewer shows that the second interaction – cyan color with distance of 5.89Å – is in fact the one hopping through a chromium site, meaning it is J3a! We will need to be careful with that later.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Building-the-exchange-interactions-for-our-system","page":"MgCr2O4 at Finite Temperature","title":"Building the exchange interactions for our system","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We begin by setting the scale of our exchange interactions on each bond.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"J1 = 3.27 # value of J1 in meV from Bai's PRL paper\nJ_pyro = [1.00,0.000,0.000,0.000]*J1 # pure nearest neighbor pyrochlore\nJ_mgcro = [1.00,0.0815,0.1050,0.085]*J1; # further neighbor pyrochlore relevant for MgCr2O4\n# val_J_mgcro = [1.00,0.000,0.025,0.025]*val_J1; # this is a funny setting from Conlon-Chalker","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"These values are then assigned to their corresponding bonds with set_exchange!.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"# === Assign exchange interactions to pyrochlore system ===\nset_exchange!(sys_pyro, J_pyro[1], Bond(1, 3, [0,0,0])) # J1\nset_exchange!(sys_pyro, J_pyro[2], Bond(1, 2, [0,0,0])) # J2\nset_exchange!(sys_pyro, J_pyro[3], Bond(2, 6, [0,0,0])) # J3a\nset_exchange!(sys_pyro, J_pyro[4], Bond(1, 5, [0,0,0])) # J3b\n\n# === Assign exchange interactions to MgCr2O4 system ===\nset_exchange!(sys_mgcro, J_mgcro[1], Bond(1, 2, [0,0,0])) # J1\nset_exchange!(sys_mgcro, J_mgcro[2], Bond(1, 7, [0,0,0])) # J2\nset_exchange!(sys_mgcro, J_mgcro[3], Bond(1, 3, [0,0,0])) # J3a -- Careful here!\nset_exchange!(sys_mgcro, J_mgcro[4], Bond(1, 3, [1,0,0])); # J3b -- And here!","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We will not be assigning any single-ion anisotropies, so we have finished specifying our models. For good measure, we will set both systems to a random (infinite temperature) initial condition.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"randomize_spins!(sys_pyro)\nrandomize_spins!(sys_mgcro);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Cooling-our-System-amd-calculating-the-instantaneous-and-dynamic-structure-factors-at-the-final-temperature","page":"MgCr2O4 at Finite Temperature","title":"Cooling our System amd calculating the instantaneous and dynamic structure factors at the final temperature","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We begin by thermalizing our system at a particular temperature. We will accomplish this by running Langevin dynamics. To do this, we must set up a Langevin integrator.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Δt = 0.05 # Integration time step in meV^-1\nλ = 0.1 # Phenomenological damping parameter\nkT = 1.8 # Desired temperature in meV\nlangevin = Langevin(Δt; λ, kT); # Construct integrator","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We can now thermalize our systems by running the integrator.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"for _ in 1:2000\n step!(sys_pyro, langevin)\n step!(sys_mgcro, langevin)\nend","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"As a sanity check, we'll plot the real-space spin configurations of both systems after themalization. First the pyrochlore,","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"plot_spins(sys_pyro)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"and then the MgCr2O4,","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"plot_spins(sys_mgcro)","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Instantaneous-Structure-Factor","page":"MgCr2O4 at Finite Temperature","title":"Instantaneous Structure Factor","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Next we can examine the instantaneous structure factor.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"isf_pyro = instant_correlations(sys_pyro)\nisf_mgcro = instant_correlations(sys_mgcro);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"These are currently empty. Let's add correlation data from 10 trajectories.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"for _ in 1:10\n # Run dynamics to decorrelate\n for _ in 1:500\n step!(sys_pyro, langevin)\n step!(sys_mgcro, langevin)\n end\n # Add samples\n add_sample!(isf_pyro, sys_pyro)\n add_sample!(isf_mgcro, sys_mgcro)\nend","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"To retrieve the intensities, we set up a formula and call intensities_interpolated on an array of wave vectors.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"qvals = -4.0:0.025:4.0\nqs = [(qa, qb, 0) for qa in qvals, qb in qvals] # Wave vectors to query\n\nformula_pyro = intensity_formula(isf_pyro, :perp)\nformula_mgcro = intensity_formula(isf_mgcro, :perp)\nSq_pyro = instant_intensities_interpolated(isf_pyro, qs, formula_pyro)\nSq_mgcro = instant_intensities_interpolated(isf_mgcro, qs, formula_mgcro);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Finally, we can plot the results.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"fig = Figure(; resolution=(1200,500))\naxparams = (aspect = true, xticks=-4:4, yticks=-4:4, titlesize=20,\n xlabel = \"𝐪a (2π a⁻¹)\", ylabel = \"𝐪b (2π b⁻¹)\", xlabelsize = 18, ylabelsize=18,)\nax_pyro = Axis(fig[1,1]; title=\"Pyrochlore\", axparams...)\nax_mgcro = Axis(fig[1,3]; title=\"MgCr2O4\", axparams...)\nhm = heatmap!(ax_pyro, qvals, qvals, Sq_pyro)\nColorbar(fig[1,2], hm)\nhm = heatmap!(ax_mgcro, qvals, qvals, Sq_mgcro)\nColorbar(fig[1,4], hm)\nfig","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Dynamical-Structure-Factor","page":"MgCr2O4 at Finite Temperature","title":"Dynamical Structure Factor","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We can also estimate the dynamical structure factor.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"sc_pyro = dynamical_correlations(sys_pyro; Δt, ωmax = 10.0, nω = 100)\nsc_mgcro = dynamical_correlations(sys_mgcro; Δt, ωmax = 10.0, nω = 100);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Next we add some sample trajectories.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"for _ in 1:3\n # Run dynamics to decorrelate\n for _ in 1:500\n step!(sys_pyro, langevin)\n step!(sys_mgcro, langevin)\n end\n # Add samples\n add_sample!(sc_pyro, sys_pyro)\n add_sample!(sc_mgcro, sys_mgcro)\nend","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We can now examine the structure factor intensities along a path in momentum space.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"fig = Figure(; resolution=(1200,900))\naxsqw = (xticks=-4:4, yticks=0:2:10, ylabel=\"E (meV)\", ylabelsize=18, xlabelsize=18, )\n\nqbs = 0.0:0.5:1.5 # Determine q_b for each slice\nfor (i, qb) in enumerate(qbs)\n path, _ = reciprocal_space_path(xtal_pyro, [[-4.0, qb, 0.0],[4.0, qb, 0.0]], 40) # Generate a path of wave\n # vectors through the BZ\n formula = intensity_formula(sc_pyro, :perp; kT) # Temperature keyword enables intensity rescaling\n Sqω_pyro = intensities_interpolated(sc_pyro, path, formula)\n\n ax = Axis(fig[fldmod1(i,2)...]; xlabel = \"q = (x, $qb, 0)\", axsqw...)\n ωs = available_energies(sc_pyro)\n heatmap!(ax, [p[1] for p in path], ωs, Sqω_pyro; colorrange=(0.0, 4.0))\nend\nfig","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"And let's take a look at the same slices for MgCr2O4.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"fig = Figure(; resolution=(1200,900))\n\nqbs = 0.0:0.5:1.5\nfor (i, qb) in enumerate(qbs)\n path, _ = reciprocal_space_path(xtal_mgcro, [[-4.0, qb, 0.0],[4.0, qb, 0.0]], 40) # Generate a path of wave\n # vectors through the BZ\n formula = intensity_formula(sc_mgcro, :perp; kT) # Temperature keyword enables intensity rescaling\n Sqω_mgcro = intensities_interpolated(sc_mgcro, path, formula)\n\n ax = Axis(fig[fldmod1(i,2)...]; xlabel = \"q = (x, $qb, 0)\", axsqw...)\n ωs = available_energies(sc_mgcro)\n heatmap!(ax, [p[1] for p in path], ωs, Sqω_mgcro; colorrange=(0.0, 4.0))\nend\nfig","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html#Instantaneous-structure-factor-from-a-dynamical-structure-factor","page":"MgCr2O4 at Finite Temperature","title":"Instantaneous structure factor from a dynamical structure factor","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Finally, we note that the instant structure factor can be calculated from the dynamical structure factor. We simply call instant_intensities rather than intensities. This will calculate the instantaneous structure factor from from 𝒮(𝐪ω) by integrating out ω . An advantage of doing this (as opposed to using instant_correlations) is that Sunny is able to apply a temperature- and energy-dependent intensity rescaling before integrating out the dynamical information. The results of this approach are more suitable for comparison with experimental data.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"qvals = -4.0:0.05:4.0\nqs = [(qa, qb, 0) for qa in qvals, qb in qvals] # Wave vectors to query\n\nformula_pyro = intensity_formula(sc_pyro, :perp; kT) # Temperature keyword enables intensity rescaling\nformula_mgcro = intensity_formula(sc_mgcro, :perp; kT) # Temperature keyword enables intensity rescaling\n\nSq_pyro = instant_intensities_interpolated(sc_pyro, qs, formula_pyro)\nSq_mgcro = instant_intensities_interpolated(sc_mgcro, qs, formula_mgcro);","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"We can plot the results below. It is useful to compare these to the plot above generated with an instant_correlations.","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"fig = Figure(; resolution=(1200,500))\nax_pyro = Axis(fig[1,1]; title=\"Pyrochlore\", axparams...)\nax_mgcro = Axis(fig[1,3]; title=\"MgCr2O4\", axparams...)\nhm = heatmap!(ax_pyro, qvals, qvals, Sq_pyro)\nColorbar(fig[1,2], hm)\nhm = heatmap!(ax_mgcro, qvals, qvals, Sq_mgcro)\nColorbar(fig[1,4], hm)\nfig","category":"page"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"(Image: )","category":"page"},{"location":"renormalization.html#Interaction-Strength-Renormalization","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"A unique feature of Sunny is its support for building classical models where quantum spin is represented as an N-level system, rather than just an expected dipole. This generalization can be important when modeling quantum spin Hamiltonians that include, e.g., a single-ion anisotropy, or a biquadratic coupling between sites. Sunny also supports constraining quantum spin to the space of pure dipoles; in this case, Sunny will automatically perform an interaction strength renormalization that maximizes accuracy.","category":"page"},{"location":"renormalization.html#Local-operators","page":"Interaction Strength Renormalization","title":"Local operators","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"A quantum spin-S state has N = 2S + 1 levels. Each local spin operator hatS^xyz is faithfully represented as an NN matrix. These matrices can be accessed using spin_matrices for a given label S. For example, the Pauli matrices are associated with S = 12.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"When S 12, it is possible to construct multipole moments beyond the spin-dipole. For example,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"S = spin_matrices(3/2)\n@assert S[3] ≈ diagm([3/2, 1/2, -1/2, -3/2])\n@assert S[3]^2 ≈ diagm([9/4, 1/4, 1/4, 9/4])","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"If the operator -S[3]^2 is passed to set_onsite_coupling!, it would set an easy-axis anisotropy in the hatz direction.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Any Hermitian operator can be expanded in the basis of Stevens operators hatmathcalO_kq up to a constant shift. To see this expansion, use print_stevens_expansion:","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"print_stevens_expansion((S[1]^2 + S[2]^2)) # Prints -(1/3)𝒪₂₀ + 5/2","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Alternatively, the same operator could have been constructed directly from stevens_matrices:","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"O = stevens_matrices(3/2)\n@assert S[1]^2 + S[2]^2 ≈ -O[2, 0]/3 + (5/2)*I","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"See below for an explicit definition of Stevens operators as polynomials of the spin operators.","category":"page"},{"location":"renormalization.html#Renormalization-procedure-for-:dipole-mode","page":"Interaction Strength Renormalization","title":"Renormalization procedure for :dipole mode","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Sunny will typically operate in one of two modes: :SUN or :dipole. The former faithfully represents quantum spin as an SU(N) coherent-state which, for our purposes, is an N-component complex vector. In contrast, :dipole mode constrains the coherent-state to the space of pure dipoles. Here, Sunny will automatically renormalize the magnitude of each Stevens operator to achieve maximal consistency with :SUN mode. This procedure was derived in D. Dahlbom et al., [arXiv:2304.03874].","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"By way of illustration, consider a quantum operator hatmathcalH_mathrmlocal giving a single-ion anisotropy for one site. In Stevens operators,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"hatmathcal H_mathrmlocal = sum_k q A_kq hatmathcalO_kq","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"for some coefficients A_kq.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"In :SUN mode, Sunny will faithfully represent hatmathcal H_mathrmlocal as an NN matrix. In :dipole mode, the expected energy langle hatmathcal H_mathrmlocal rangle must somehow be approximated as a function of the expected dipole mathbfs.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"One approach is to formally take S to infty, and this yields the traditional classical limit of a spin system. In this, limit spin operators commute, and expectation values of polynomials become polynomials of expectation values. For example, langle hatS^alpha hatS^betarangle to langle hatS^alpha rangle langle hatS^betarangle, because any corrections are damped by the factor S^-1 to 0. The expectation of a Stevens operator langle hatmathcalO_kq rangle becomes a Stevens function mathcalO_kq(mathbfs), i.e., a polynomial of expected dipole mathbfs = langle hatmathbfS rangle.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"In a real magnetic compound, however, the spin magnitude S is not necessarily large. To obtain a better approximation, one should avoid the assumption S to infty. Our approach is to start with the full dynamics of SU(N) coherent states, and then constrain it to the space of pure dipole states mathbfsrangle. The latter are defined such that expectation values,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"langle mathbfs hatmathbfS mathbfsrangle = mathbfs","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"have magnitude mathbfs = S, which is maximal.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"For pure dipole states, expectations can be computed exactly,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"langle mathbfs hatmathcalO mathbfsrangle = c_k mathcalO_kq(mathbfs)","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"The right-hand side involves a renormalization of the Stevens functions, where","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"beginalign*\nc_1 = 1 \nc_2 = 1-frac12S^-1 \nc_3 = 1-frac32S^-1+frac12S^-2 \nc_4 = 1-3S^-1+frac114S^-2-frac34S^-3 \nc_5 = 1-5S^-1+frac354S^-2-frac254S^-3+frac32S^-4 \nc_6 = 1-frac152S^-1+frac854S^-2-frac2258S^-3+frac1378S^-4-frac154S^-5 \nvdots\nendalign*","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Constrained to the space of dipoles, the expected local energy becomes","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"E_mathrmlocal(mathbfs) = langle mathbfs hatmathcal H_mathrmlocal mathbfsrangle = sum_k q c_k A_kq mathcalO_kq(mathbfs)","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"It can be shown that SU(N) dynamics reduces to the usual Landau-Lifshitz dynamics of dipoles, but involving E_mathrmlocal(mathbfs) as the classical Hamiltonian. Through the renormalization factors c_k, Sunny avoids the large-S assumption, and gives a more accurate result.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Renormalization also applies to the coupling between different sites. In Sunny, couplings will often be expressed as a polynomial of spin operators using set_pair_coupling!, but any such coupling can be decomposed as sum of tensor products of Stevens operators. Without loss of generality, consider a single coupling between two Stevens operators hatmathcalH_mathrmcoupling = hatmathcalO_kq otimes hatmathcalO_kq along a bond connecting sites i and j. Upon constraining to pure dipole states mathbfs_irangle and mathbfs_jrangle, the expected energy takes the form E_mathrmcoupling = c_k c_k mathcalO_kq(mathbfs_i) mathcalO_kq(mathbfs_j), which now involves a product of renormalized Stevens functions. ","category":"page"},{"location":"renormalization.html#Use-:dipole_large_S-mode-to-disable-renormalization","page":"Interaction Strength Renormalization","title":"Use :dipole_large_S mode to disable renormalization","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Although we generally recommend the above renormalization procedure, there are circumstances where it is not desirable. Examples include reproducing a model-system study, or describing a micromagnetic system for which the Stoinfty limit is a good approximation. To simulate dipoles without interaction strength renormalization, construct a System using the mode :dipole_large_S. Symbolic operators in the large-S limit can be constructed by passing Inf to either spin_matrices or stevens_matrices.","category":"page"},{"location":"renormalization.html#Definition-of-Stevens-operators","page":"Interaction Strength Renormalization","title":"Definition of Stevens operators","text":"","category":"section"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"The Stevens operators hatmathcalO_kq are defined as polynomials of angular momentum operators hatS_xyz in some spin-S representation.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Using","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"beginalign*\nX = mathbfhatS cdot mathbfhatS = S (S+1) \nhatS_pm = hatS_x pm i hatS_y \nphi_+ = frac14quad phi_- = frac14 i\nendalign*","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"the relevant Stevens operators are defined as,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"beginalign*\nhatmathcalO_00 =1\n\nhatmathcalO_1pm1 =phi_pm(hatS_+pmhatS_-)+mathrmhc\nhatmathcalO_10 =hatS_z\n\nhatmathcalO_2pm2 =phi_pm(hatS_+^2pmhatS_-^2)+mathrmhc\nhatmathcalO_2pm1 =phi_pm(hatS_+pmhatS_-)hatS_z+mathrmhc\nhatmathcalO_20 =3hatS_z^2-X\n\nhatmathcalO_3pm3 =phi_pm(hatS_+^3pmhatS_-^3)+mathrmhc\nhatmathcalO_3pm2 =phi_pm(hatS_+^2pmhatS_-^2)hatS_z+mathrmhc\nhatmathcalO_3pm1 =phi_pm(hatS_+pmhatS_-)(5hatS_z^2-X-12)+mathrmhc\nhatmathcalO_30 =5hatS_z^3-(3X-1)hatS_z\n\nhatmathcalO_4pm4 =phi_pm(hatS_+^4pmhatS_-^4)+mathrmhc\nhatmathcalO_4pm3 =phi_pm(hatS_+^3pmhatS_-^3)hatS_z+mathrmhc\nhatmathcalO_4pm2 =phi_pm(hatS_+^2pmhatS_-^2)(7hatS_z^2-(X+5))+mathrmhc\nhatmathcalO_4pm1 =phi_pm(hatS_+pmhatS_-)(7hatS_z^3-(3X+1)hatS_z)+mathrmhc\nhatmathcalO_40 =35hatS_z^4-(30X-25)hatS_z^2+(3X^2-6X)\n\nhatmathcalO_5pm5 =phi_pm(hatS_+^5pmhatS_-^5)+mathrmhc\nhatmathcalO_5pm4 =phi_pm(hatS_+^4pmhatS_-^4)hatS_z+mathrmhc\nhatmathcalO_5pm3 =phi_pm(hatS_+^3pmhatS_-^3)(9hatS_z^2-(X+332))+mathrmhc\nhatmathcalO_5pm2 =phi_pm(hatS_+^2pmhatS_-^2)(3hatS_z^3-(X+6)hatS_z)+mathrmhc\nhatmathcalO_5pm1 =phi_pm(hatS_+pmhatS_-)(21hatS_z^4-14XhatS_z^2+(X^2-X+32))+mathrmhc\nhatmathcalO_50 =63hatS_z^5-(70X-105)hatS_z^3+(15X^2-50X+12)hatS_z\n\nhatmathcalO_6pm6 =phi_pm(hatS_+^6pmhatS_-^6)+mathrmhc\nhatmathcalO_6pm5 =phi_pm(hatS_+^5pmhatS_-^5)hatS_z+mathrmhc\nhatmathcalO_6pm4 =phi_pm(hatS_+^4pmhatS_-^4)(11hatS_z^2-X-38)+mathrmhc\nhatmathcalO_6pm3 =phi_pm(hatS_+^3pmhatS_-^3)(11hatS_z^3-(3X+59)hatS_z)+mathrmhc\nhatmathcalO_6pm2 =phi_pm(hatS_+^2pmhatS_-^2)(33hatS_z^4-(18X+123)hatS_z^2+X^2+10X+102)+mathrmhc\nhatmathcalO_6pm1 =phi_pm(hatS_+pmhatS_-)(33hatS_z^5-(30X-15)hatS_z^3+(5X^2-10X+12)hatS_z)+mathrmhc\nhatmathcalO_60 =231hatS_z^6-(315X-735)hatS_z^4+(105X^2-525X+294)hatS_z^2-5X^3+40X^2-60X\nendalign*","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"Computer-generated tables of Stevens operators with k 6 are available from C. Rudowicz and C. Y. Chung, J. Phys.: Condens. Matter 16, 5825 (2004), but these typically do not appear in magnetic simulations.","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"The case k=1 gives the dipole operators,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"(hatmathcalO_11 hatmathcalO_10 hatmathcalO_1-1) = (hatS_x hatS_z hatS_y)","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"The case k=2 gives the quadrupole operators,","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"(hatmathcalO_22 dots hatmathcalO_2-2) = left(hatS_x^2 - hatS_y^2 frachatS_x hatS_z + hatS_z hatS_x2 2hatS_z^2-hatS_x^2-hatS_y^2 frachatS_y hatS_z + hatS_z hatS_y2 hatS_x hatS_y + hatS_y hatS_xright)","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"For each k value, the set of operators hatmathcalO_kq for q = -k dots k form an irreducible representation of the group of rotations O(3). That is, rotation will transform hatmathcalO_kq into a linear combination of hatmathcalO_kq where q varies but k remains fixed. ","category":"page"},{"location":"renormalization.html","page":"Interaction Strength Renormalization","title":"Interaction Strength Renormalization","text":"In taking the large-S limit, each dipole operator is replaced by its expectation value mathbfs = langle hatmathbfS rangle, and only leading-order terms are retained. The operator hatmathcalO_kq becomes a homogeneous polynomial O_kq(mathbfs) of order k in the spin components. One can see these polynomials by constructing stevens_matrices with the argument S = Inf. Due to the normalization constraint, each dipole can be expressed in polar angles, (theta phi). Then the Stevens functions O_kq(mathbfs) correspond to the spherical harmonic functions Y_l^m(theta phi) where l=k and m=q; this correspondence is valid up to k and q-dependent rescaling factors.","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"EditURL = \"../../../../examples/spinw_tutorials/SW15_Ba3NbFe3Si2O14.jl\"","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html#SW15-BaNbFeSiO","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"","category":"section"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"This is a Sunny port of SpinW Tutorial 15, originally authored by Sandor Toth. The goal is to calculate the linear spin wave theory spectrum for Ba₃NbFe₃Si₂O₁₄.","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Load packages","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Build a Crystal for Ba₃NbFe₃Si₂O₁₄ using the crystal structure from Marty et al., Phys. Rev. Lett. 101, 247201 (2008).","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"a = b = 8.539 # (Å)\nc = 5.2414\nlatvecs = lattice_vectors(a, b, c, 90, 90, 120)\ntypes = [\"Fe\",\"Nb\",\"Ba\",\"Si\",\"O\",\"O\",\"O\"]\npositions = [[0.24964,0,0.5],[0,0,0],[0.56598,0,0],[2/3,1/3,0.5220],[2/3,1/3,0.2162],[0.5259,0.7024,0.3536],[0.7840,0.9002,0.7760]]\nlangasite = Crystal(latvecs, positions, 150; types)\ncrystal = subcrystal(langasite, \"Fe\")\nview_crystal(crystal, 7)","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Create a System with a lattice size of (117). The magnetic structure of Ba₃NbFe₃Si₂O₁₄ was determined to have the ordering wavevector 𝐐=(0017) and hence the magnetic unit cell has 7 sites.","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"latsize = (1,1,7)\nS = 5/2\nseed = 5\nsys = System(crystal, latsize, [SpinInfo(1; S, g=2)], :dipole)","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Set exchange interactions as parametrized in Loire et al., Phys. Rev. Lett. 106, 207201 (2011)","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"J₁ = 0.85\nJ₂ = 0.24\nJ₃ = 0.053\nJ₄ = 0.017\nJ₅ = 0.24\nset_exchange!(sys, J₁, Bond(3, 2, [1,1,0]))\nset_exchange!(sys, J₄, Bond(1, 1, [0,0,1]))\nset_exchange!(sys, J₂, Bond(1, 3, [0,0,0]))","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"The final two exchanges define the chirality of the magnetic structure. The crystal chirality, epsilon_T, the chirality of each triangle, ϵ_D and the sense of rotation of the spin helices along c, ϵ_H. The three chiralities are related by ϵ_T=ϵ_D ϵ_H. We now assign J_3 and J_5 according to the crystal chirality.","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"ϵD = -1\nϵH = +1\nϵT = ϵD * ϵH\n\nif ϵT == -1\n set_exchange!(sys, J₃, Bond(2, 3, [-1,-1,1]))\n set_exchange!(sys, J₅, Bond(3, 2, [1,1,1]))\nelseif ϵT == 1\n set_exchange!(sys, J₅, Bond(2, 3, [-1,-1,1]))\n set_exchange!(sys, J₃, Bond(3, 2, [1,1,1]))\nelse\n throw(\"Provide a valid chirality\")\nend","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Whilst Sunny provides tools to optimize the ground state automatically, in this case we already know the model ground state. Set the spiral magnetic order using set_spiral_order_on_sublattice!. It takes an ordering wavevector q, an axis of rotation for the spins axis, and the initial spin S0 for each sublattice.","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"q = [0, 0, 1/7]\naxis = [0,0,1]\nset_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[1, 0, 0])\nset_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[-1/2, -sqrt(3)/2, 0])\nset_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[-1/2, +sqrt(3)/2, 0])\n\nplot_spins(sys; color=[s[1] for s in sys.dipoles])","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Define a path in reciprocal space, 01-1+xi for xi = 0 dots 3.","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"points_rlu = [[0,1,-1],[0,1,-1+1],[0,1,-1+2],[0,1,-1+3]];\ndensity = 100\npath, xticks = reciprocal_space_path(crystal, points_rlu, density);\nnothing #hide","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Calculate broadened intensities","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"swt = SpinWaveTheory(sys)\nγ = 0.15 # width in meV\nbroadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))\nenergies = collect(0:0.01:6) # 0 < ω < 6 (meV).\nis = intensities_broadened(swt, path, energies, broadened_formula);\nnothing #hide","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"Plot","category":"page"},{"location":"examples/spinw/SW15_Ba3NbFe3Si2O14.html","page":"SW15 - Ba₃NbFe₃Si₂O₁₄","title":"SW15 - Ba₃NbFe₃Si₂O₁₄","text":"fig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nheatmap!(ax, 1:size(is,1), energies, is, colorrange=(0,5))\nfig","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"EditURL = \"../../../examples/03_LLD_CoRh2O4.jl\"","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html#.-Landau-Lifshitz-dynamics-of-CoRhO-at-finite-*T*","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"","category":"section"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"using Sunny, GLMakie, Statistics","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html#System-construction","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"System construction","text":"","category":"section"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Construct the system as in the previous CoRh₂O₄ tutorial. After optimization, the system will be in an unfrustrated antiferromagnetic ground state.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"a = 8.5031 # (Å)\nlatvecs = lattice_vectors(a, a, a, 90, 90, 90)\ncryst = Crystal(latvecs, [[0,0,0]], 227, setting=\"1\")\nlatsize = (2, 2, 2)\nS = 3/2\nJ = 0.63 # (meV)\nsys = System(cryst, latsize, [SpinInfo(1; S, g=2)], :dipole; seed=0)\nset_exchange!(sys, J, Bond(1, 3, [0,0,0]))\nrandomize_spins!(sys)\nminimize_energy!(sys)","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Use resize_supercell to build a new system with a lattice of 10×10×10 unit cells. The desired Néel order is retained.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"sys = resize_supercell(sys, (10, 10, 10))\n@assert energy_per_site(sys) ≈ -2J*S^2","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Use the stochastic Landau-Lifshitz dynamics to thermalize system into equilibrium at finite temperature. This is a Langevin equation, which includes damping and noise terms. The strength of the noise term is automatically fixed according to the damping time scale λ and the target temperature, according to a fluctuation-dissipation theorem.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Δt = 0.05/abs(J*S) # Time step\nλ = 0.1 # Dimensionless damping time-scale\nkT = 16 * meV_per_K # 16K, a temperature slightly below ordering\nlangevin = Langevin(Δt; λ, kT);\nnothing #hide","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Because the magnetic order has been initialized correctly, relatively few additional Langevin time-steps are required to reach thermal equilibrium.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"energies = [energy_per_site(sys)]\nfor _ in 1:1000\n step!(sys, langevin)\n push!(energies, energy_per_site(sys))\nend\nplot(energies, color=:blue, figure=(resolution=(600,300),), axis=(xlabel=\"Time steps\", ylabel=\"Energy (meV)\"))","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Thermal fluctuations are apparent in the spin configuration.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"S_ref = sys.dipoles[1,1,1,1]\nplot_spins(sys; color=[s'*S_ref for s in sys.dipoles])","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html#Instantaneous-structure-factor","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"Instantaneous structure factor","text":"","category":"section"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"To visualize the instantaneous (equal-time) structure factor, create an object instant_correlations and use add_sample! to accumulated data for the equilibrated spin configuration.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"sc = instant_correlations(sys)\nadd_sample!(sc, sys) # Accumulate the newly sampled structure factor into `sf`","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Collect 20 additional decorrelated samples. For each sample, about 100 Langevin time-steps is sufficient to collect approximately uncorrelated statistics.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"for _ in 1:20\n for _ in 1:100\n step!(sys, langevin)\n end\n add_sample!(sc, sys)\nend","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Define a slice of momentum space. Wavevectors are specified in reciprocal lattice units (RLU). The notation q1s in -10:0.1:10 indicates that the first q-component ranges from -10 to 10 in intervals of 0.1. That is, q spans over 20 Brillouin zones. To convert to absolute momentum units, each component of q would need to be scaled by a reciprocal lattice vector.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"q1s = -10:0.1:10\nq2s = -10:0.1:10\nqs = [[q1, q2, 0.0] for q1 in q1s, q2 in q2s];\nnothing #hide","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Plot the instantaneous structure factor for the given q-slice. We employ the appropriate FormFactor for Co2⁺. An intensity_formula defines how dynamical correlations correspond to the observable structure factor. The function instant_intensities_interpolated calculates intensities at the target qs by interpolating over the data available at discrete reciprocal-space lattice points.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"formfactors = [FormFactor(\"Co2\")]\ninstant_formula = intensity_formula(sc, :perp; formfactors)\niq = instant_intensities_interpolated(sc, qs, instant_formula);\nnothing #hide","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Plot the resulting intensity data I(𝐪). The color scale is clipped to 50% of the maximum intensity.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"heatmap(q1s, q2s, iq;\n colorrange = (0, maximum(iq)/2),\n axis = (\n xlabel=\"Momentum Transfer Qx (r.l.u)\", xlabelsize=16,\n ylabel=\"Momentum Transfer Qy (r.l.u)\", ylabelsize=16,\n aspect=true,\n )\n)","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html#Dynamical-structure-factor","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"Dynamical structure factor","text":"","category":"section"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"To collect statistics for the dynamical structure factor intensities I(𝐪ω) at finite temperature, use dynamical_correlations. Now, each call to add_sample! will run a classical spin dynamics trajectory. Longer-time trajectories will be required to achieve greater energy resolution, as controlled by nω. Here, we pick a moderate number of energies, nω = 50, which will make the simulation run quickly.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"ωmax = 6.0 # Maximum energy to resolve (meV)\nnω = 50 # Number of energies to resolve\nsc = dynamical_correlations(sys; Δt, nω, ωmax, process_trajectory=:symmetrize)","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Each sample requires running a full dynamical trajectory to measure correlations, so we here restrict to 5 samples.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"for _ in 1:5\n for _ in 1:100\n step!(sys, langevin)\n end\n add_sample!(sc, sys)\nend","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Define points that define a piecewise-linear path through reciprocal space, and a sampling density.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"points = [[3/4, 3/4, 0],\n [ 0, 0, 0],\n [ 0, 1/2, 1/2],\n [1/2, 1, 0],\n [ 0, 1, 0],\n [1/4, 1, 1/4],\n [ 0, 1, 0],\n [ 0, -4, 0]]\ndensity = 50 # (Å)\npath, xticks = reciprocal_space_path(cryst, points, density);\nnothing #hide","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Calculate I(𝐪 ω) intensities along this path with Lorentzian broadening on the scale of 0.1 meV.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"formula = intensity_formula(sc, :perp; formfactors, kT=langevin.kT)\nη = 0.1\niqw = intensities_interpolated(sc, path, formula)\niqwc = broaden_energy(sc, iqw, (ω, ω₀) -> lorentzian(ω-ω₀, η));\nnothing #hide","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Plot the intensity data on a clipped color scale","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"ωs = available_energies(sc)\nheatmap(1:size(iqwc, 1), ωs, iqwc;\n colorrange = (0, maximum(iqwc)/50),\n axis = (;\n xlabel=\"Momentum Transfer (r.l.u)\",\n ylabel=\"Energy Transfer (meV)\",\n xticks,\n xticklabelrotation=π/5,\n aspect = 1.4,\n )\n)","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html#Powder-averaged-intensity","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"Powder averaged intensity","text":"","category":"section"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Define spherical shells in reciprocal space via their radii, in absolute units of 1/Å. For each shell, calculate and average the intensities at 100 𝐪-points, sampled approximately uniformly.","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"radii = 0:0.05:3.5 # (1/Å)\noutput = zeros(Float64, length(radii), length(ωs))\nfor (i, radius) in enumerate(radii)\n pts = reciprocal_space_shell(sc.crystal, radius, 100)\n is = intensities_interpolated(sc, pts, formula)\n is = broaden_energy(sc, is, (ω,ω₀)->lorentzian(ω-ω₀, η))\n output[i, :] = mean(is , dims=1)[1,:]\nend","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"Plot resulting powder-averaged structure factor","category":"page"},{"location":"examples/03_LLD_CoRh2O4.html","page":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","title":"3. Landau-Lifshitz dynamics of CoRh₂O₄ at finite T","text":"heatmap(radii, ωs, output;\n axis = (\n xlabel=\"|Q| (Å⁻¹)\",\n ylabel=\"Energy Transfer (meV)\",\n aspect = 1.4,\n ),\n colorrange = (0, 20.0)\n)","category":"page"},{"location":"versions.html#Version-History","page":"Version History","title":"Version History","text":"","category":"section"},{"location":"versions.html#v0.5.6","page":"Version History","title":"v0.5.6","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Animated spin dynamics is now possible. Call notify on the result of plot_spins to trigger redrawing of the frame. The argument colorfn to plot_spins supports animation of colors.\nGeneral pair couplings are now supported in set_pair_coupling! and set_pair_coupling_at!. :SUN supports interactions of any order, but :dipole mode is limited to bilinear and biquadratic coupling of the spin.\nTo perform a calculation with dipoles in the large-S limit, use the new mode :dipole_large_S when constructing a System.\nDeprecate the option biquad to set_exchange!. Use instead set_pair_coupling!, which generalizes beyond the scalar biquadratic.\nDeprecate spin_operators, stevens_operators, large_S_spin_operators and large_S_stevens_operators. Use instead spin_matrices and stevens_matrices, which require a specific spin-S label. To infer this, one can use spin_label.\nRemove unused option energy_tol in SpinWaveTheory.","category":"page"},{"location":"versions.html#v0.5.5","page":"Version History","title":"v0.5.5","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"reshape_supercell now allows reshaping to multiples of the primitive unit cell, which can speed up certain calculations. This is illustrated in the CoRh₂O₄ powder averaging tutorial.\nresize_supercell now allows all resizings.\nAdded energy_per_site.\nset_spiral_order_on_sublattice! cannot work on reshaped systems.\nVarious bug fixes. In particular, an intensity_formula with :full will now uniformly calculate a 3x3 matrix of complex numbers.","category":"page"},{"location":"versions.html#v0.5.4","page":"Version History","title":"v0.5.4","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Various enhancements to view_crystal. Atoms are now labeled by index, and bonds support interactive inspection (GLMakie only). Font sizes work correctly on Makie v0.20-beta. If using Makie v0.19 on a high-resolution display, pass rescale=1.5 to enlarge font sizes.\nThe function suggest_magnetic_supercell now requires only a list of wavevectors, and will return a 33 matrix that can be programmatically passed to reshape_supercell. The new tolerance parameter tol allows suggest_magnetic_supercell to approximate incommensurate wavevectors with nearby commensurate ones.\nNew functions set_spiral_order! and set_spiral_order_on_sublattice! can be used to initialize a spiral, single-Q order.\nSunny now retains all constant energy shifts that have been introduced by anisotropy operators.\nFix export_vtk functionality.","category":"page"},{"location":"versions.html#v0.5.3","page":"Version History","title":"v0.5.3","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Add large_S_spin_operators and large_S_stevens_operators to support single-ion anisotropies in dipole mode without renormalization. Set large_S=true in set_exchange! to avoid renormalization of biquadratics.\nview_crystal has been rewritten in Makie.\nplot_spins now expects ghost_radius in physical length units.\nSpinWaveTheory will (currently) error if provided a system with enable_dipole_dipole!.","category":"page"},{"location":"versions.html#v0.5.2","page":"Version History","title":"v0.5.2","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Form factors for 5d transition ions.\nDownload links for notebooks and scripts on each doc example\nVarious bug fixes.","category":"page"},{"location":"versions.html#v0.5.1","page":"Version History","title":"v0.5.1","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Fix binning edge cases.\nplot_spins accepts resolution argument.","category":"page"},{"location":"versions.html#v0.5.0","page":"Version History","title":"v0.5.0","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"New features.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Support for Linear Spin Wave Theory in :dipole and :SUN modes. (Thanks Hao Zhang!)","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"New function minimize_energy! to efficiently find an optimal configuration of spin dipoles or SU(N) coherent states.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Major refactors and enhancements to intensity calculations. This new interface allows unification between LSWT and classical spin dynamics calculations. This interface allows: Custom observables as local quantum operators, better support for linebroadening, and automatic binning to facilitate comparison with experimental data. See intensity_formula for documentation. Use load_nxs to load experimental neutron scattering data.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Breaking changes.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Require Julia 1.9.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Replace set_anisotropy! with a new function set_onsite_coupling! (and similarly set_onsite_coupling_at!). The latter expects an explicit matrix representation for the local Hamiltonian. This can be constructed, e.g., as a linear combination of stevens_operators, or as a polynomial of spin_operators. To understand the mapping between these two, the new function print_stevens_expansion acts on an arbitrary local operator.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Remove set_biquadratic!. Instead, use an optional keyword argument biquad to set_exchange!.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename DynamicStructureFactor to dynamical_correlations. Similarly, replace InstantStructureFactor with instant_correlations. The return type has been renamed SampledCorrelations to emphasize that the object may be holding thermodynamic samples, which are collected using add_sample!. Upon construction, the SampledCorrelations object will be empty (no initial sample).","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Remove intensities function. Instead, use one of intensities_interpolated or intensities_binned. These will require an intensity_formula, which defines a calculator (e.g., LSWT).","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename connected_path to reciprocal_space_path, which now returns an xticks object that can be used in plotting. Replace spherical_shell with reciprocal_space_shell that functions similarly.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename polarize_spin! to set_dipole! for consistency with set_coherent!. The behavior of the former function is unchanged: the spin at a given site will still be polarized along the provided direction.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename all_sites to eachsite consistent with Julia convention for iterators.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename reshape_geometry to reshape_supercell, which is the fundamental reshaping function. Rename resize_periodically to resize_supercell.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The constructor SpinInfo now requires a g-factor or tensor as a named argument.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The constructor FormFactor no longer accepts an atom index. Instead, the form factors are associated with site-symmetry classes in order of appearance.","category":"page"},{"location":"versions.html#v0.4.3","page":"Version History","title":"v0.4.3","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Experimental support for linear SpinWaveTheory, implemented in SU(N) mode. This module may evolve rapidly.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Implement renormalization of single-ion anisotropy and biquadratic interactions when in :dipole mode. This makes the model more faithful to the quantum mechanical Hamiltonian, but is also a breaking change.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Various improvements and bugfixes for to_inhomogeneous. Setting inhomogeneous interactions via set_exchange_at! should now infer the correct bond offset direction, or will report an ambiguity error. Ambiguities can be resolved by passing an explicit offset.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The function remove_periodicity! disables periodicity along specified dimensions.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename StaticStructureFactor to InstantStructureFactor.","category":"page"},{"location":"versions.html#v0.4.2","page":"Version History","title":"v0.4.2","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Introduce LocalSampler, a framework for MCMC sampling with local spin updates.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename print_dominant_wavevectors to print_wrapped_intensities to reduce confusion with the physical instantaneous intensities.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The function spherical_shell now takes a radius in physical units of inverse Å.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"New exported functions global_position, magnetic_moment, all_sites.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Remove all uses of Base.deepcopy which resolves crashes.","category":"page"},{"location":"versions.html#v0.4.1","page":"Version History","title":"v0.4.1","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The function to_inhomogeneous creates a system that supports inhomogeneous interactions, which can be set using set_exchange_at!, etc.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"set_biquadratic! replaces set_exchange_with_biquadratic!.","category":"page"},{"location":"versions.html#v0.4.0","page":"Version History","title":"v0.4.0","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"This update includes many breaking changes, and is missing some features of 0.3.0.","category":"page"},{"location":"versions.html#Creating-a-spin-System","page":"Version History","title":"Creating a spin System","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Rename SpinSystem to System. Its constructor now has the form,","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"System(crystal, latsize, infos, mode)","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The parameter infos is now a list of SpinInfo objects. Each defines spin angular momentum S = frac12 1 frac32 , and an optional g-factor or tensor.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The parameter mode is one of :SUN or :dipole.","category":"page"},{"location":"versions.html#Setting-interactions","page":"Version History","title":"Setting interactions","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Interactions are now added mutably to an existing System using the following functions: set_external_field!, set_exchange!, set_onsite_coupling!, enable_dipole_dipole!.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"As a convenience, one can use dmvec(D) to convert a DM vector to a 33 antisymmetric exchange matrix.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Fully general single-ion anisotropy is now possible. The function set_onsite_coupling! expects the single ion anisotropy to be expressed as a polynomial in symbolic spin operators 𝒮, or as a linear combination of symbolic Stevens operators 𝒪. For example, an easy axis anisotropy in the direction n may be written D*(𝒮⋅n)^2.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Stevens operators 𝒪[k,q] admit polynomial expression in spin operators 𝒮[α]. Conversely, a polynomial of spin operators can be expressed as a linear combination of Stevens operators. To see this expansion use print_anisotropy_as_stevens.","category":"page"},{"location":"versions.html#Inhomogeneous-field","page":"Version History","title":"Inhomogeneous field","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"An external field can be applied to a single site with set_external_field_at!. ","category":"page"},{"location":"versions.html#Structure-factor-rewrite","page":"Version History","title":"Structure factor rewrite","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The calculation of structure factors has been completely rewritten. For the new interface see the documentation tutorials.","category":"page"},{"location":"versions.html#Various","page":"Version History","title":"Various","text":"","category":"section"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The \"Sampler\" interface is in flux. Langevin replaces both LangevinHeunP and LangevinSampler. Local spin-flip Monte Carlo sampling methods are temporarily broken.\nrepeat_periodically replaces extend_periodically.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"Additional related functions include resize_periodically and reshape_geometry, the latter being fundamental.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"print_symmetry_table replaces print_bond_table().","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"The new function includes the list of symmetry-allowed single ion anisotropies in addition to exchange interactions.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"When reading CIF files, the field _atom_site_label is now used in place of the field _atom_site_type_symbol.","category":"page"},{"location":"versions.html","page":"Version History","title":"Version History","text":"This is required for correctness. The field _atom_site_label is guaranteed to be present, and is guaranteed to be a distinct label for each symmetry-inequivalent site. Code that explicitly referred to site labels (e.g. in calls to subcrystal) will need to be updated to use the new label.","category":"page"},{"location":"index.html#Overview","page":"Overview","title":"Overview","text":"","category":"section"},{"location":"index.html","page":"Overview","title":"Overview","text":"Sunny is a Julia package for modeling atomic-scale magnetism. It provides powerful tools to study equilibrium and non-equilibrium magnetic phenomena. In particular, it allows estimation of dynamical structure factor intensities, mathcalS(𝐪ω), to support quantitative modeling of experimental scattering data.","category":"page"},{"location":"index.html","page":"Overview","title":"Overview","text":"Features include:","category":"page"},{"location":"index.html","page":"Overview","title":"Overview","text":"Generalized spin dynamics using SU(N) coherent states.\nAbility to specify a crystal from a .cif file or its spacegroup symmetry.\nInteractive visualizations of the 3D crystals and magnetic ordering.\nSymmetry analysis to classify allowed interaction terms, and to propagate them by symmetry.\nSingle-ion anisotropy at arbitrary order, which can be specified using Stevens operators or as a polynomial of spin operators.\nMonte Carlo sampling of spin configurations in thermal equilibrium, and optimization tools.\nMeasurements of dynamical correlations. At low temperature, one can use linear spin wave theory and its multi-boson generalization. This generalizes to finite temperatures using the classical dynamics, which allows for strongly nonlinear effects.\nLong-range dipole-dipole interactions accelerated with the fast Fourier transform (FFT).\nSupport for comparison with experimental data: form factor, dipole factor, temperature-dependent classical-to-quantum factors, intensity binning, etc.","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"EditURL = \"../../../../examples/spinw_tutorials/SW08_Kagome_AFM.jl\"","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html#SW8-Kagome-Antiferromagnet","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"","category":"section"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"This is a Sunny port of SpinW Tutorial 8, originally authored by Bjorn Fak and Sandor Toth. The goal is to calculate the linear spin wave theory spectrum for the sqrt3 times sqrt3 order of a Kagome antiferromagnet.","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Load Packages","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Build a Crystal with Poverline3 space group,","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"a = 1\nlatvecs = lattice_vectors(a, a, 10a, 90, 90, 120)\ncrystal = Crystal(latvecs, [[1/2,0,0]], 147)","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Build a System with antiferrogmanetic nearest neighbor exchange J=1.","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"S = 1\nsys = System(crystal, (3,3,1), [SpinInfo(1; S, g=2)], :dipole)\nJ = 1.0\nset_exchange!(sys, J, Bond(2,3,[0,0,0]))","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Initialize to an energy minimizing magnetic structure, for which nearest-neighbor spins are at 120° angles.","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"q = -[1/3, 1/3, 0]\naxis = [0,0,1]\nset_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[cos(0),sin(0),0])\nset_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[cos(0),sin(0),0])\nset_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[cos(2π/3),sin(2π/3),0])\nplot_spins(sys; dims=2)","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Check energy. Each site participates in 4 bonds with energy JS^2cos(2π3). Factor of 1/2 avoids double counting.","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"@assert energy_per_site(sys) ≈ (4/2)*J*S^2*cos(2π/3)","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Define a path in reciprocal space.","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"points_rlu = [[-1/2, 0, 0], [0, 0, 0], [1/2, 1/2, 0]]\ndensity = 100\npath, xticks = reciprocal_space_path(crystal, points_rlu, density);\nnothing #hide","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Calculate discrete intensities","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"swt = SpinWaveTheory(sys)\nformula = intensity_formula(swt, :perp; kernel=delta_function_kernel)\ndisp, intensity = intensities_bands(swt, path, formula);\nnothing #hide","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"Plot over a restricted color range from [0,1e-2]. Note that the intensities of the flat band at zero-energy are off-scale.","category":"page"},{"location":"examples/spinw/SW08_Kagome_AFM.html","page":"SW8 - Kagome Antiferromagnet","title":"SW8 - Kagome Antiferromagnet","text":"fig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nylims!(ax, -1e-1, 2.3)\nfor i in axes(disp, 2)\n lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange=(0,1e-2))\nend\nfig","category":"page"}]
}
diff --git a/dev/structure-factor.html b/dev/structure-factor.html
index 55979df27..b6e23674b 100644
--- a/dev/structure-factor.html
+++ b/dev/structure-factor.html
@@ -1,9 +1,9 @@
-Structure Factor Calculations · Sunny documentation
The dynamical structure factor is of fundamental importance for characterizing a magnetic system, and facilitates quantitative comparison between theory and experimental scattering data.
Consider, for example, a two-point dynamical spin correlation function, $⟨s^α(𝐱+Δ𝐱, t+Δt) s^β(𝐱, t)⟩$. Here $s^α(𝐱, t)$ represents the time dynamics of a spin dipole component $α$ at position $𝐱$, and brackets represent an average over equilibrium initial conditions and over $(𝐱, t)$. The dynamical structure factor is defined as the Fourier transform of this two-point correlation in both space and time, up to an overall scaling factor. Using the convolution theorem, the result is,
with $V$ the system volume. We will restrict attention to lattice systems with periodic boundaries.
Consider a crystal unit cell defined by three lattice vectors $𝐚_1, 𝐚_2, 𝐚_3$, and linear system sizes $L_1, L_2, L_3$ measured in unit cells. The allowed momentum vectors take on discrete values $𝐪 = \sum_{α=1}^{3} m_α 𝐛_α / L_α$, where $m_α$ are an integers and the reciprocal lattice vectors $𝐛_α$ are defined to satisfy $𝐚_α ⋅ 𝐛_β = 2π δ_{α,β}$. For a Bravais lattice, $𝐪$ will be periodic in the first Brillouin zone, i.e., under any shift $𝐪 → 𝐪 ± 𝐛_α$. More generally, consider a non-Bravais lattice such that each unit cell may contain multiple spins. By partitioning spins $s_j(𝐱,t)$ according to their sublattice index $j$, the relevant momenta $𝐪$ remain discretized as above, but now periodicity in the first Brillouin zone is lost. The structure factor may be written as a phase-average over the displacements between sublattices $𝐫_{j,k}$,
is fundamental. For each sublattice $j$, the data $ŝ_j^α(𝐪, ω)$ can be efficiently obtained by fast Fourier tranformation of a real space configuration $s_j^α(𝐱, t)$. Internally, Sunny will calculate and store the discrete $𝒮̃^{αβ}_{j,k}(𝐪, ω)$ correlation data, and use this to construct $𝒮^{αβ}(𝐪,ω)$ intensities that can be compared with experiment.
Calculating this structure factor involves several steps, with various possible settings. Sunny provides a number of tools to facilitate this calculation and to extract information from the results. These tools are briefly outlined below. Please see the Examples for a "real life" use case. Detailed function information is available in the Library API.
Classical dynamics may be used to estimate structure factor data by analyzing the spin-spin correlations of dynamical trajectories. This is fundamentally a Monte Carlo approach, as the trajectories must be started from an initial spin configuration that is sampled at thermal equilibrium. (Note that it is not possible to estimate a true T=0 dynamical structure factor using this method, but the temperature may be very low.) Samples are accumulated into a SampledCorrelations, from which intensity information may be extracted. The user does not typically build their own SampledCorrelations but instead initializes one by calling either dynamical_correlations or instant_correlations, as described below.
A SampledCorrelations for estimating the dynamical structure factor, $𝒮^{αβ}(𝐪,ω)$, may be created by calling dynamical_correlations. This requires three keyword arguments. These will determine the dynamics used to calculate samples and, consequently, the $ω$ information that will be available.
Δt: Determines the step size used for simulating the dynamics. A smaller number will require proportionally more calculation time. While a smaller Δt will enable the resolution of higher energies, Δt is typically selected to ensure numerical stability rather than to maximize the largest $ω$ value. A safe choice is to use the smaller value of Δt = 0.1/(J* S^2) or Δt = 0.1/(D * S), where S is magnetic moment of the largest local spin (as specified in SpinInfo), J is the parameter governing the largest bilinear interaction (e.g. exchange), and D is the parameter governing the largest single-site term of the Hamiltonian (e.g., anisotropy or Zeeman term).
ωmax: Sets the maximum resolved energy. Note that this is not independent of Δt. If ωmax too large, Sunny will throw an error and ask you to choose a smaller Δt.
nω: Determines the number of energy bins to resolve. A larger number will require more calculation time.
A sample may be added by calling add_sample!(sc, sys). The input sys must be a spin configuration in good thermal equilibrium, e.g., using the continuous Langevin dynamics or using single spin flip trials with LocalSampler. The statistical quality of the $𝒮^{αβ}(𝐪,ω)$ can be improved by repeatedly generating decorrelated spin configurations in sys and calling add_sample! on each configuration.
The outline of typical use case might look like this:
# Make a `SampledCorrelations`
+Structure Factor Calculations · Sunny documentation
The dynamical structure factor is of fundamental importance for characterizing a magnetic system, and facilitates quantitative comparison between theory and experimental scattering data.
Consider, for example, a two-point dynamical spin correlation function, $⟨s^α(𝐱+Δ𝐱, t+Δt) s^β(𝐱, t)⟩$. Here $s^α(𝐱, t)$ represents the time dynamics of a spin dipole component $α$ at position $𝐱$, and brackets represent an average over equilibrium initial conditions and over $(𝐱, t)$. The dynamical structure factor is defined as the Fourier transform of this two-point correlation in both space and time, up to an overall scaling factor. Using the convolution theorem, the result is,
with $V$ the system volume. We will restrict attention to lattice systems with periodic boundaries.
Consider a crystal unit cell defined by three lattice vectors $𝐚_1, 𝐚_2, 𝐚_3$, and linear system sizes $L_1, L_2, L_3$ measured in unit cells. The allowed momentum vectors take on discrete values $𝐪 = \sum_{α=1}^{3} m_α 𝐛_α / L_α$, where $m_α$ are an integers and the reciprocal lattice vectors $𝐛_α$ are defined to satisfy $𝐚_α ⋅ 𝐛_β = 2π δ_{α,β}$. For a Bravais lattice, $𝐪$ will be periodic in the first Brillouin zone, i.e., under any shift $𝐪 → 𝐪 ± 𝐛_α$. More generally, consider a non-Bravais lattice such that each unit cell may contain multiple spins. By partitioning spins $s_j(𝐱,t)$ according to their sublattice index $j$, the relevant momenta $𝐪$ remain discretized as above, but now periodicity in the first Brillouin zone is lost. The structure factor may be written as a phase-average over the displacements between sublattices $𝐫_{j,k}$,
is fundamental. For each sublattice $j$, the data $ŝ_j^α(𝐪, ω)$ can be efficiently obtained by fast Fourier tranformation of a real space configuration $s_j^α(𝐱, t)$. Internally, Sunny will calculate and store the discrete $𝒮̃^{αβ}_{j,k}(𝐪, ω)$ correlation data, and use this to construct $𝒮^{αβ}(𝐪,ω)$ intensities that can be compared with experiment.
Calculating this structure factor involves several steps, with various possible settings. Sunny provides a number of tools to facilitate this calculation and to extract information from the results. These tools are briefly outlined below. Please see the Examples for a "real life" use case. Detailed function information is available in the Library API.
Classical dynamics may be used to estimate structure factor data by analyzing the spin-spin correlations of dynamical trajectories. This is fundamentally a Monte Carlo approach, as the trajectories must be started from an initial spin configuration that is sampled at thermal equilibrium. (Note that it is not possible to estimate a true T=0 dynamical structure factor using this method, but the temperature may be very low.) Samples are accumulated into a SampledCorrelations, from which intensity information may be extracted. The user does not typically build their own SampledCorrelations but instead initializes one by calling either dynamical_correlations or instant_correlations, as described below.
A SampledCorrelations for estimating the dynamical structure factor, $𝒮^{αβ}(𝐪,ω)$, may be created by calling dynamical_correlations. This requires three keyword arguments. These will determine the dynamics used to calculate samples and, consequently, the $ω$ information that will be available.
Δt: Determines the step size used for simulating the dynamics. A smaller number will require proportionally more calculation time. While a smaller Δt will enable the resolution of higher energies, Δt is typically selected to ensure numerical stability rather than to maximize the largest $ω$ value. A safe choice is to use the smaller value of Δt = 0.1/(J* S^2) or Δt = 0.1/(D * S), where S is magnetic moment of the largest local spin (as specified in SpinInfo), J is the parameter governing the largest bilinear interaction (e.g. exchange), and D is the parameter governing the largest single-site term of the Hamiltonian (e.g., anisotropy or Zeeman term).
ωmax: Sets the maximum resolved energy. Note that this is not independent of Δt. If ωmax too large, Sunny will throw an error and ask you to choose a smaller Δt.
nω: Determines the number of energy bins to resolve. A larger number will require more calculation time.
A sample may be added by calling add_sample!(sc, sys). The input sys must be a spin configuration in good thermal equilibrium, e.g., using the continuous Langevin dynamics or using single spin flip trials with LocalSampler. The statistical quality of the $𝒮^{αβ}(𝐪,ω)$ can be improved by repeatedly generating decorrelated spin configurations in sys and calling add_sample! on each configuration.
The outline of typical use case might look like this:
# Make a `SampledCorrelations`
sc = dynamical_correlations(sys; Δt=0.05, ωmax=10.0, nω=100)
# Add samples
for _ in 1:nsamples
decorrelate_system(sys) # Perform some type of Monte Carlo simulation
add_sample!(sc, sys) # Use spins to calculate trajectory and accumulate new sample of 𝒮(𝐪,ω)
-end
The calculation may be configured in a number of ways; see the dynamical_correlations documentation for a list of all keywords.
Sunny provides two methods for calculating instantaneous, or static, structure factors: $𝒮^{αβ}(𝐪)$. The first involves calculating spatial spin-spin correlations at single time slices. The second involves calculating a dynamic structure factor first and integrating out the $ω$ information. The advantage of the latter approach is that it enables application of an $ω$-dependent classical-to-quantum rescaling of structure factor intensities, a method that should be preferred whenever comparing results to experimental data or spin wave calculations. A disadvantage of this approach is that it is computationally more expensive. There are also many cases when it is not straightforward to calculate a meaningful dynamics, as when working with Ising spins. In this section we will discuss how to calculate instantaneous structure factors from static spin configurations. Information about calculating instantaneous data from a dynamical correlations can be found in the following section.
The basic usage for the instantaneous case is very similar to the dynamic case, except one calls instant_correlations instead of dynamical_correlations to configure a SampledCorrelations. Note that there are no required keywords as there is no need to specify any dynamics. instant_correlations will return a SampledCorrelations containing no data. Samples may be added by calling add_sample!(sc, sys), where sc is the SampledCorrelations. When performing a finite-temperature calculation, it is important to ensure that the spin configuration in the sys represents a good equilibrium sample, as in the dynamical case. Note, however, that we recommend calculating instantaneous correlations at finite temperature calculations by using full dynamics (i.e., using dynamical_correlations) and then integrating out the energy axis. An approach to doing this is described in the next section.
The basic function for extracting information from a SampledCorrelations at a particular wave vector, $𝐪$, is intensities_interpolated. It takes a SampledCorrelations, a list of wave vectors, and an intensity_formula. The intensity_formula specifies how to contract and correct correlation data to arrive at a physical intensity. A simple example is formula = intensity_formula(sc, :perp), which will instruct Sunny apply polarization corrections: $\sum_{αβ}(I-q_α q_β) 𝒮^{αβ}(𝐪,ω)$. An intensity at the wave vector $𝐪 = (𝐛_2 + 𝐛_3)/2$ may then be retrieved with intensities_interpolated(sf, [[0.0, 0.5, 0.5]], formula) . intensities_interpolated returns a list of nω elements at each wavevector. The corresponding $ω$ values can be retrieved by calling available_energies on sf.
Since Sunny only calculates the structure factor on a finite lattice when performing classical simulations, it is important to realize that exact information is only available at a discrete set of wave vectors. Specifically, for each axis index $i$, we will get information at $q_i = \frac{n}{L_i}$, where $n$ runs from $(\frac{-L_i}{2}+1)$ to $\frac{L_i}{2}$ and $L_i$ is the linear dimension of the lattice used for the calculation. If you request a wave vector that does not fall into this set, Sunny will automatically round to the nearest $𝐪$ that is available. If intensities_interpolated is given the keyword argument interpolation=:linear, Sunny will use trilinear interpolation to determine a result at the requested wave vector.
To retrieve the intensities at all wave vectors for which there is exact data, first call the function available_wave_vectors to generate a list of qs. This takes an optional keyword argument bzsize, which must be given a tuple of three integers specifying the number of Brillouin zones to calculate, e.g., bzsize=(2,2,2). The resulting list of wave vectors may then be passed to intensities_interpolated.
Alternatively, intensities_binned can be used to place the exact data into histogram bins for comparison with experiment.
The convenience function reciprocal_space_path returns a list of wavevectors sampled along a path that connects specified $𝐪$ points. This list can be used as an input to intensities. Another convenience method, reciprocal_space_shell will generate points on a sphere of a given radius. This is useful for powder averaging.
A number of arguments for intensity_formula are available which modify the calculation of structure factor intensity. It is generally recommended to provide a value of kT corresponding to the temperature of sampled configurations. Given kT, Sunny will include an energy- and temperature-dependent classical-to-quantum rescaling of intensities in the formula.
To retrieve intensity data from a instantaneous structure factor, use instant_intensities_interpolated, which accepts similar arguments to intensities_interpolated. This function may also be used to calculate instantaneous information from a dynamical correlation data, i.e. from a SampledCorrelations created with dynamical_correlations. Note that it is important to supply a value to kT to reap the benefits of this approach over simply calculating a static structure factor at the outset.
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
+end
The calculation may be configured in a number of ways; see the dynamical_correlations documentation for a list of all keywords.
Sunny provides two methods for calculating instantaneous, or static, structure factors: $𝒮^{αβ}(𝐪)$. The first involves calculating spatial spin-spin correlations at single time slices. The second involves calculating a dynamic structure factor first and integrating out the $ω$ information. The advantage of the latter approach is that it enables application of an $ω$-dependent classical-to-quantum rescaling of structure factor intensities, a method that should be preferred whenever comparing results to experimental data or spin wave calculations. A disadvantage of this approach is that it is computationally more expensive. There are also many cases when it is not straightforward to calculate a meaningful dynamics, as when working with Ising spins. In this section we will discuss how to calculate instantaneous structure factors from static spin configurations. Information about calculating instantaneous data from a dynamical correlations can be found in the following section.
The basic usage for the instantaneous case is very similar to the dynamic case, except one calls instant_correlations instead of dynamical_correlations to configure a SampledCorrelations. Note that there are no required keywords as there is no need to specify any dynamics. instant_correlations will return a SampledCorrelations containing no data. Samples may be added by calling add_sample!(sc, sys), where sc is the SampledCorrelations. When performing a finite-temperature calculation, it is important to ensure that the spin configuration in the sys represents a good equilibrium sample, as in the dynamical case. Note, however, that we recommend calculating instantaneous correlations at finite temperature calculations by using full dynamics (i.e., using dynamical_correlations) and then integrating out the energy axis. An approach to doing this is described in the next section.
The basic function for extracting information from a SampledCorrelations at a particular wave vector, $𝐪$, is intensities_interpolated. It takes a SampledCorrelations, a list of wave vectors, and an intensity_formula. The intensity_formula specifies how to contract and correct correlation data to arrive at a physical intensity. A simple example is formula = intensity_formula(sc, :perp), which will instruct Sunny apply polarization corrections: $\sum_{αβ}(I-q_α q_β) 𝒮^{αβ}(𝐪,ω)$. An intensity at the wave vector $𝐪 = (𝐛_2 + 𝐛_3)/2$ may then be retrieved with intensities_interpolated(sf, [[0.0, 0.5, 0.5]], formula) . intensities_interpolated returns a list of nω elements at each wavevector. The corresponding $ω$ values can be retrieved by calling available_energies on sf.
Since Sunny only calculates the structure factor on a finite lattice when performing classical simulations, it is important to realize that exact information is only available at a discrete set of wave vectors. Specifically, for each axis index $i$, we will get information at $q_i = \frac{n}{L_i}$, where $n$ runs from $(\frac{-L_i}{2}+1)$ to $\frac{L_i}{2}$ and $L_i$ is the linear dimension of the lattice used for the calculation. If you request a wave vector that does not fall into this set, Sunny will automatically round to the nearest $𝐪$ that is available. If intensities_interpolated is given the keyword argument interpolation=:linear, Sunny will use trilinear interpolation to determine a result at the requested wave vector.
To retrieve the intensities at all wave vectors for which there is exact data, first call the function available_wave_vectors to generate a list of qs. This takes an optional keyword argument bzsize, which must be given a tuple of three integers specifying the number of Brillouin zones to calculate, e.g., bzsize=(2,2,2). The resulting list of wave vectors may then be passed to intensities_interpolated.
Alternatively, intensities_binned can be used to place the exact data into histogram bins for comparison with experiment.
The convenience function reciprocal_space_path returns a list of wavevectors sampled along a path that connects specified $𝐪$ points. This list can be used as an input to intensities. Another convenience method, reciprocal_space_shell will generate points on a sphere of a given radius. This is useful for powder averaging.
A number of arguments for intensity_formula are available which modify the calculation of structure factor intensity. It is generally recommended to provide a value of kT corresponding to the temperature of sampled configurations. Given kT, Sunny will include an energy- and temperature-dependent classical-to-quantum rescaling of intensities in the formula.
To retrieve intensity data from a instantaneous structure factor, use instant_intensities_interpolated, which accepts similar arguments to intensities_interpolated. This function may also be used to calculate instantaneous information from a dynamical correlation data, i.e. from a SampledCorrelations created with dynamical_correlations. Note that it is important to supply a value to kT to reap the benefits of this approach over simply calculating a static structure factor at the outset.
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
diff --git a/dev/versions.html b/dev/versions.html
index 91b59ecbc..ddaf64102 100644
--- a/dev/versions.html
+++ b/dev/versions.html
@@ -1,2 +1,2 @@
-Version History · Sunny documentation
Animated spin dynamics is now possible. Call notify on the result of plot_spins to trigger redrawing of the frame. The argument colorfn to plot_spins supports animation of colors.
General pair couplings are now supported in set_pair_coupling! and set_pair_coupling_at!. :SUN supports interactions of any order, but :dipole mode is limited to bilinear and biquadratic coupling of the spin.
To perform a calculation with dipoles in the large-$S$ limit, use the new mode :dipole_large_S when constructing a System.
Deprecate the option biquad to set_exchange!. Use instead set_pair_coupling!, which generalizes beyond the scalar biquadratic.
Deprecate spin_operators, stevens_operators, large_S_spin_operators and large_S_stevens_operators. Use instead spin_matrices and stevens_matrices, which require a specific spin-$S$ label. To infer this, one can use spin_label.
reshape_supercell now allows reshaping to multiples of the primitive unit cell, which can speed up certain calculations. This is illustrated in the CoRh₂O₄ powder averaging tutorial.
Various enhancements to view_crystal. Atoms are now labeled by index, and bonds support interactive inspection (GLMakie only). Font sizes work correctly on Makie v0.20-beta. If using Makie v0.19 on a high-resolution display, pass rescale=1.5 to enlarge font sizes.
The function suggest_magnetic_supercell now requires only a list of wavevectors, and will return a $3×3$ matrix that can be programmatically passed to reshape_supercell. The new tolerance parameter tol allows suggest_magnetic_supercell to approximate incommensurate wavevectors with nearby commensurate ones.
Add large_S_spin_operators and large_S_stevens_operators to support single-ion anisotropies in dipole mode without renormalization. Set large_S=true in set_exchange! to avoid renormalization of biquadratics.
Support for Linear Spin Wave Theory in :dipole and :SUN modes. (Thanks Hao Zhang!)
New function minimize_energy! to efficiently find an optimal configuration of spin dipoles or SU(N) coherent states.
Major refactors and enhancements to intensity calculations. This new interface allows unification between LSWT and classical spin dynamics calculations. This interface allows: Custom observables as local quantum operators, better support for linebroadening, and automatic binning to facilitate comparison with experimental data. See intensity_formula for documentation. Use load_nxs to load experimental neutron scattering data.
Breaking changes.
Require Julia 1.9.
Replace set_anisotropy! with a new function set_onsite_coupling! (and similarly set_onsite_coupling_at!). The latter expects an explicit matrix representation for the local Hamiltonian. This can be constructed, e.g., as a linear combination of stevens_operators, or as a polynomial of spin_operators. To understand the mapping between these two, the new function print_stevens_expansion acts on an arbitrary local operator.
Remove set_biquadratic!. Instead, use an optional keyword argument biquad to set_exchange!.
Rename DynamicStructureFactor to dynamical_correlations. Similarly, replace InstantStructureFactor with instant_correlations. The return type has been renamed SampledCorrelations to emphasize that the object may be holding thermodynamic samples, which are collected using add_sample!. Upon construction, the SampledCorrelations object will be empty (no initial sample).
Rename connected_path to reciprocal_space_path, which now returns an xticks object that can be used in plotting. Replace spherical_shell with reciprocal_space_shell that functions similarly.
Rename polarize_spin! to set_dipole! for consistency with set_coherent!. The behavior of the former function is unchanged: the spin at a given site will still be polarized along the provided direction.
Rename all_sites to eachsite consistent with Julia convention for iterators.
Rename reshape_geometry to reshape_supercell, which is the fundamental reshaping function. Rename resize_periodically to resize_supercell.
The constructor SpinInfo now requires a $g$-factor or tensor as a named argument.
The constructor FormFactor no longer accepts an atom index. Instead, the form factors are associated with site-symmetry classes in order of appearance.
Experimental support for linear SpinWaveTheory, implemented in SU(N) mode. This module may evolve rapidly.
Implement renormalization of single-ion anisotropy and biquadratic interactions when in :dipole mode. This makes the model more faithful to the quantum mechanical Hamiltonian, but is also a breaking change.
Various improvements and bugfixes for to_inhomogeneous. Setting inhomogeneous interactions via set_exchange_at! should now infer the correct bond offset direction, or will report an ambiguity error. Ambiguities can be resolved by passing an explicit offset.
The function remove_periodicity! disables periodicity along specified dimensions.
Rename StaticStructureFactor to InstantStructureFactor.
Rename SpinSystem to System. Its constructor now has the form,
System(crystal, latsize, infos, mode)
The parameter infos is now a list of SpinInfo objects. Each defines spin angular momentum $S = \frac{1}{2}, 1, \frac{3}{2}, …$, and an optional $g$-factor or tensor.
As a convenience, one can use dmvec(D) to convert a DM vector to a $3×3$ antisymmetric exchange matrix.
Fully general single-ion anisotropy is now possible. The function set_onsite_coupling! expects the single ion anisotropy to be expressed as a polynomial in symbolic spin operators 𝒮, or as a linear combination of symbolic Stevens operators 𝒪. For example, an easy axis anisotropy in the direction n may be written D*(𝒮⋅n)^2.
Stevens operators 𝒪[k,q] admit polynomial expression in spin operators 𝒮[α]. Conversely, a polynomial of spin operators can be expressed as a linear combination of Stevens operators. To see this expansion use print_anisotropy_as_stevens.
The "Sampler" interface is in flux. Langevin replaces both LangevinHeunP and LangevinSampler. Local spin-flip Monte Carlo sampling methods are temporarily broken.
The new function includes the list of symmetry-allowed single ion anisotropies in addition to exchange interactions.
When reading CIF files, the field _atom_site_label is now used in place of the field _atom_site_type_symbol.
This is required for correctness. The field _atom_site_label is guaranteed to be present, and is guaranteed to be a distinct label for each symmetry-inequivalent site. Code that explicitly referred to site labels (e.g. in calls to subcrystal) will need to be updated to use the new label.
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
Animated spin dynamics is now possible. Call notify on the result of plot_spins to trigger redrawing of the frame. The argument colorfn to plot_spins supports animation of colors.
General pair couplings are now supported in set_pair_coupling! and set_pair_coupling_at!. :SUN supports interactions of any order, but :dipole mode is limited to bilinear and biquadratic coupling of the spin.
To perform a calculation with dipoles in the large-$S$ limit, use the new mode :dipole_large_S when constructing a System.
Deprecate the option biquad to set_exchange!. Use instead set_pair_coupling!, which generalizes beyond the scalar biquadratic.
Deprecate spin_operators, stevens_operators, large_S_spin_operators and large_S_stevens_operators. Use instead spin_matrices and stevens_matrices, which require a specific spin-$S$ label. To infer this, one can use spin_label.
reshape_supercell now allows reshaping to multiples of the primitive unit cell, which can speed up certain calculations. This is illustrated in the CoRh₂O₄ powder averaging tutorial.
Various enhancements to view_crystal. Atoms are now labeled by index, and bonds support interactive inspection (GLMakie only). Font sizes work correctly on Makie v0.20-beta. If using Makie v0.19 on a high-resolution display, pass rescale=1.5 to enlarge font sizes.
The function suggest_magnetic_supercell now requires only a list of wavevectors, and will return a $3×3$ matrix that can be programmatically passed to reshape_supercell. The new tolerance parameter tol allows suggest_magnetic_supercell to approximate incommensurate wavevectors with nearby commensurate ones.
Add large_S_spin_operators and large_S_stevens_operators to support single-ion anisotropies in dipole mode without renormalization. Set large_S=true in set_exchange! to avoid renormalization of biquadratics.
Support for Linear Spin Wave Theory in :dipole and :SUN modes. (Thanks Hao Zhang!)
New function minimize_energy! to efficiently find an optimal configuration of spin dipoles or SU(N) coherent states.
Major refactors and enhancements to intensity calculations. This new interface allows unification between LSWT and classical spin dynamics calculations. This interface allows: Custom observables as local quantum operators, better support for linebroadening, and automatic binning to facilitate comparison with experimental data. See intensity_formula for documentation. Use load_nxs to load experimental neutron scattering data.
Breaking changes.
Require Julia 1.9.
Replace set_anisotropy! with a new function set_onsite_coupling! (and similarly set_onsite_coupling_at!). The latter expects an explicit matrix representation for the local Hamiltonian. This can be constructed, e.g., as a linear combination of stevens_operators, or as a polynomial of spin_operators. To understand the mapping between these two, the new function print_stevens_expansion acts on an arbitrary local operator.
Remove set_biquadratic!. Instead, use an optional keyword argument biquad to set_exchange!.
Rename DynamicStructureFactor to dynamical_correlations. Similarly, replace InstantStructureFactor with instant_correlations. The return type has been renamed SampledCorrelations to emphasize that the object may be holding thermodynamic samples, which are collected using add_sample!. Upon construction, the SampledCorrelations object will be empty (no initial sample).
Rename connected_path to reciprocal_space_path, which now returns an xticks object that can be used in plotting. Replace spherical_shell with reciprocal_space_shell that functions similarly.
Rename polarize_spin! to set_dipole! for consistency with set_coherent!. The behavior of the former function is unchanged: the spin at a given site will still be polarized along the provided direction.
Rename all_sites to eachsite consistent with Julia convention for iterators.
Rename reshape_geometry to reshape_supercell, which is the fundamental reshaping function. Rename resize_periodically to resize_supercell.
The constructor SpinInfo now requires a $g$-factor or tensor as a named argument.
The constructor FormFactor no longer accepts an atom index. Instead, the form factors are associated with site-symmetry classes in order of appearance.
Experimental support for linear SpinWaveTheory, implemented in SU(N) mode. This module may evolve rapidly.
Implement renormalization of single-ion anisotropy and biquadratic interactions when in :dipole mode. This makes the model more faithful to the quantum mechanical Hamiltonian, but is also a breaking change.
Various improvements and bugfixes for to_inhomogeneous. Setting inhomogeneous interactions via set_exchange_at! should now infer the correct bond offset direction, or will report an ambiguity error. Ambiguities can be resolved by passing an explicit offset.
The function remove_periodicity! disables periodicity along specified dimensions.
Rename StaticStructureFactor to InstantStructureFactor.
Rename SpinSystem to System. Its constructor now has the form,
System(crystal, latsize, infos, mode)
The parameter infos is now a list of SpinInfo objects. Each defines spin angular momentum $S = \frac{1}{2}, 1, \frac{3}{2}, …$, and an optional $g$-factor or tensor.
As a convenience, one can use dmvec(D) to convert a DM vector to a $3×3$ antisymmetric exchange matrix.
Fully general single-ion anisotropy is now possible. The function set_onsite_coupling! expects the single ion anisotropy to be expressed as a polynomial in symbolic spin operators 𝒮, or as a linear combination of symbolic Stevens operators 𝒪. For example, an easy axis anisotropy in the direction n may be written D*(𝒮⋅n)^2.
Stevens operators 𝒪[k,q] admit polynomial expression in spin operators 𝒮[α]. Conversely, a polynomial of spin operators can be expressed as a linear combination of Stevens operators. To see this expansion use print_anisotropy_as_stevens.
The "Sampler" interface is in flux. Langevin replaces both LangevinHeunP and LangevinSampler. Local spin-flip Monte Carlo sampling methods are temporarily broken.
The new function includes the list of symmetry-allowed single ion anisotropies in addition to exchange interactions.
When reading CIF files, the field _atom_site_label is now used in place of the field _atom_site_type_symbol.
This is required for correctness. The field _atom_site_label is guaranteed to be present, and is guaranteed to be a distinct label for each symmetry-inequivalent site. Code that explicitly referred to site labels (e.g. in calls to subcrystal) will need to be updated to use the new label.
Settings
This document was generated with Documenter.jl version 1.1.2 on Wednesday 1 November 2023. Using Julia version 1.9.3.
The 4D correlation data produced by Sunny is too high-dimensional to visualize directly. This page describes how to export 3D slices of correlation data from Sunny to the Visual ToolKit (VTK) format, which is compatible with the ParaView visualization software. ParaView supports volumetric rendering:
First, generate some correlation data in Sunny. We will use a 2D lattice, since the correlation data $S(Q_x,Q_y,\omega)$ is 3D and can be exported in its entirety. The following code sets up the system, thermalizes it, and records the correlation data in a SampledCorrelations called dsf.
using Sunny
+ParaView Rendering · Sunny documentation
The 4D correlation data produced by Sunny is too high-dimensional to visualize directly. This page describes how to export 3D slices of correlation data from Sunny to the Visual ToolKit (VTK) format, which is compatible with the ParaView visualization software. ParaView supports volumetric rendering:
First, generate some correlation data in Sunny. We will use a 2D lattice, since the correlation data $S(Q_x,Q_y,\omega)$ is 3D and can be exported in its entirety. The following code sets up the system, thermalizes it, and records the correlation data in a SampledCorrelations called dsf.
using Sunny
# Single layer 12x12 periodic square lattice
latsize = (12,12,1)
@@ -61,4 +61,4 @@
signal = sum(signal; dims = 4)
# Export to ParaView
-export_vtk("experiment_data_as_vtk", params, signal)
Settings
This document was generated with Documenter.jl version 1.1.2 on Tuesday 31 October 2023. Using Julia version 1.9.3.
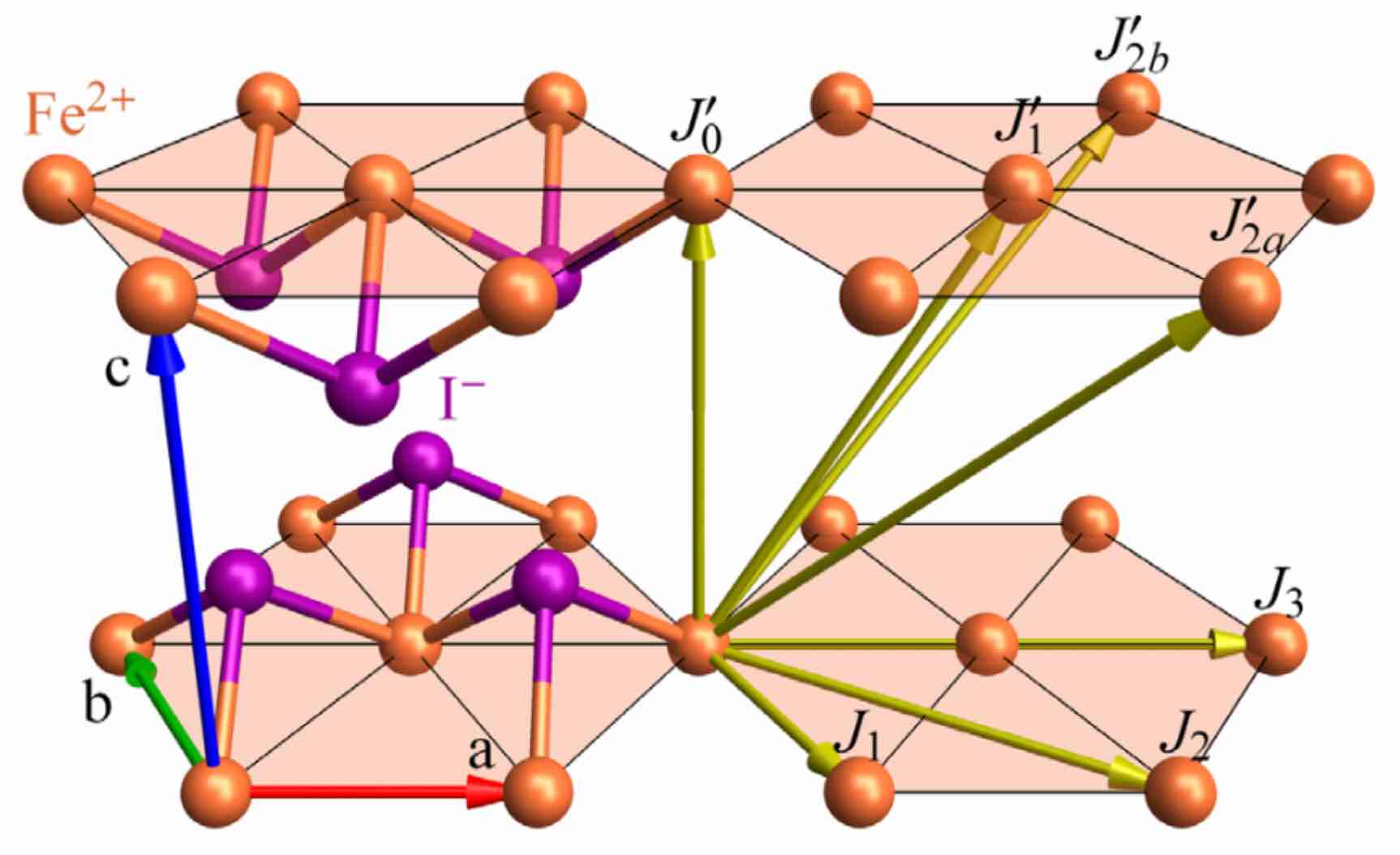





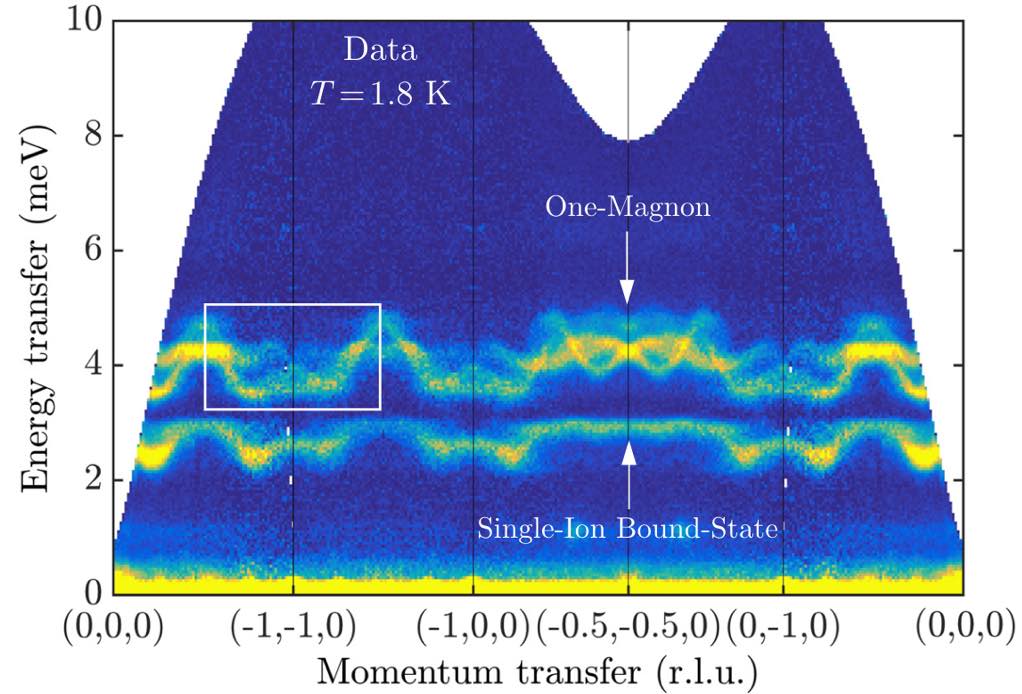



















































 ","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The Fe atoms are arranged in stacked triangular layers. The effective spin Hamiltonian takes the form,","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"mathcalH=sum_(ij) 𝐒_i J_ij 𝐒_j - Dsum_i left(S_i^zright)^2","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"where the set of exchange matrices J_ij between bonded sites (ij) includes competing ferromagnetic and antiferromagnetic interactions. This model also includes a strong easy axis anisotropy, D 0.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"We will formulate this Hamiltonian in Sunny and then calculate its dynamic structure factor.","category":"page"},{"location":"examples/fei2_tutorial.html#Get-Julia-and-Sunny","page":"Case Study: FeI₂","title":"Get Julia and Sunny","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Sunny is implemented in Julia. This is a relatively new programming language that allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users may wish to take a look at our Getting Started with Julia guide. Sunny requires Julia 1.9 or later.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"From the Julia prompt, load Sunny. For plotting, one can choose either GLMakie (a pop-up window) or WGLMakie (inline plots for a Jupyter notebook or VSCode).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"If these packages are not yet installed, Julia should offer to install them using its built-in package management system. If old versions are installed, you may need to update them to run this tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html#Crystals","page":"Case Study: FeI₂","title":"Crystals","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A Crystal describes the crystallographic unit cell and will usually be loaded from a .cif file. Here, we instead build a crystal by listing all atoms and their types.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"a = b = 4.05012 # Lattice constants for triangular lattice\nc = 6.75214 # Spacing in the z-direction\n\nlatvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that\n # define the conventional unit cell\npositions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions\n # of lattice vectors\ntypes = [\"Fe\", \"I\", \"I\"]\nFeI2 = Crystal(latvecs, positions; types)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Observe that Sunny inferred the space group, 'P -3 m 1' (164) and labeled the atoms according to their point group symmetries.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Only the Fe atoms are magnetic, so we discard the I ions using subcrystal.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"cryst = subcrystal(FeI2, \"Fe\")","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Observe that cryst retains the spacegroup symmetry of the full FeI₂ crystal. This information will be used, for example, to propagate exchange interactions between symmetry-equivalent bonds.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In a running Julia environment, the crystal can be viewed interactively using view_crystal.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"view_crystal(cryst, 8.0)","category":"page"},{"location":"examples/fei2_tutorial.html#Symmetry-analysis","page":"Case Study: FeI₂","title":"Symmetry analysis","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The command print_symmetry_table provides a list of all the symmetry-allowed interactions up to a cutoff distance.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"print_symmetry_table(cryst, 8.0)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The allowed g-tensor is expressed as a 3×3 matrix in the free coefficients A, B, ... The allowed single-ion anisotropy is expressed as a linear combination of Stevens operators. The latter correspond to polynomials of the spin operators, as we will describe below.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The allowed exchange interactions are given as a 3×3 matrix for representative bonds. The notation Bond(i, j, n) indicates a bond between atom indices i and j, with cell offset n. In the general case, it will be necessary to associate atom indices with their positions in the unit cell; these can be viewed with display(cryst). Note that the order of the pair (i j) is significant if the exchange tensor contains antisymmetric Dzyaloshinskii–Moriya (DM) interactions.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In the case of FeI₂, Bond(1, 1, [1,0,0]) is one of the 6 nearest-neighbor Fe-Fe bonds on a triangular lattice layer, and Bond(1, 1, [0,0,1]) is an Fe-Fe bond between layers.","category":"page"},{"location":"examples/fei2_tutorial.html#Building-a-spin-System","page":"Case Study: FeI₂","title":"Building a spin System","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In constructing a spin System, we must provide several additional details about the spins.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"sys = System(cryst, (4, 4, 4), [SpinInfo(1, S=1, g=2)], :SUN, seed=2)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"This system includes 444 unit cells, i.e. 64 Fe atoms, each with spin S=1 and a g-factor of 2. Quantum mechanically, spin S=1 involves a superposition of 2S+1=3 distinct angular momentum states. In :SUN mode, this superposition will be modeled explicitly using the formalism of SU(3) coherent states, which captures both dipolar and quadrupolar fluctuations. For the more traditional dipole dynamics, use :dipole mode instead.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Next we will use set_exchange! to assign interaction to bonds. Sunny will automatically propagate each interaction to all symmetry-equivalent bonds in the unit cell. The FeI₂ interactions below follow Bai et al.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"J1pm = -0.236\nJ1pmpm = -0.161\nJ1zpm = -0.261\nJ2pm = 0.026\nJ3pm = 0.166\nJ′0pm = 0.037\nJ′1pm = 0.013\nJ′2apm = 0.068\n\nJ1zz = -0.236\nJ2zz = 0.113\nJ3zz = 0.211\nJ′0zz = -0.036\nJ′1zz = 0.051\nJ′2azz = 0.073\n\nJ1xx = J1pm + J1pmpm\nJ1yy = J1pm - J1pmpm\nJ1yz = J1zpm\n\nset_exchange!(sys, [J1xx 0.0 0.0;\n 0.0 J1yy J1yz;\n 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))\nset_exchange!(sys, [J2pm 0.0 0.0;\n 0.0 J2pm 0.0;\n 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))\nset_exchange!(sys, [J3pm 0.0 0.0;\n 0.0 J3pm 0.0;\n 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))\nset_exchange!(sys, [J′0pm 0.0 0.0;\n 0.0 J′0pm 0.0;\n 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))\nset_exchange!(sys, [J′1pm 0.0 0.0;\n 0.0 J′1pm 0.0;\n 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))\nset_exchange!(sys, [J′2apm 0.0 0.0;\n 0.0 J′2apm 0.0;\n 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function set_onsite_coupling! assigns a single-ion anisotropy. The argument can be constructed using spin_matrices or stevens_matrices. Here we use Julia's anonymous function syntax to assign an easy-axis anisotropy along the direction hatz.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"D = 2.165\nset_onsite_coupling!(sys, S -> -D*S[3]^2, 1)","category":"page"},{"location":"examples/fei2_tutorial.html#Calculating-structure-factor-intensities","page":"Case Study: FeI₂","title":"Calculating structure factor intensities","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In the remainder of this tutorial, we will examine Sunny's tools for calculating the dynamical structure factor using a multi-boson generalization of linear spin wave theory (LSWT). This theory describes non-interacting quasi-particle excitations that hybridize dipolar and quadrupolar modes.","category":"page"},{"location":"examples/fei2_tutorial.html#Finding-the-ground-state","page":"Case Study: FeI₂","title":"Finding the ground state","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Begin with a random configuration and use minimize_energy! to find a configuration of the SU(3) coherent states (i.e. spin dipoles and quadrupoles) that locally minimizes energy.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"randomize_spins!(sys)\nminimize_energy!(sys)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A positive number above indicates that the procedure has converged to a local energy minimum. The configuration, however, may still have defects. This can be checked by visualizing the spins, colored according to their z-components.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"plot_spins(sys; color=[s[3] for s in sys.dipoles])","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A different understanding of the magnetic ordering can be obtained by moving to Fourier space. The 'instant' structure factor 𝒮(𝐪) is an experimental observable. To investigate 𝒮(𝐪) as true 3D data, Sunny provides instant_correlations and related functions. Here, however, we will use print_wrapped_intensities, which gives average intensities for the individual Bravais sublattices (in effect, all wavevectors are wrapped to the first Brillouin zone).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"print_wrapped_intensities(sys)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The result will likely be approximately consistent with the known zero-field energy-minimizing magnetic structure of FeI₂, which is single-Q (two-up, two-down antiferromagnetic order). Mathematically, spontaneous symmetry breaking should select one of Q = 0 -14 14, 14 0 14, or -141414, associated with the three-fold rotational symmetry of the crystal spacegroup. In nature, however, one will frequently encounter competing \"domains\" associated with the three possible orientations of the ground state.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"If the desired ground state is already known, as with FeI₂, it could be entered by hand using set_dipole!. Alternatively, in the case of FeI₂, we could repeatedly employ the above randomization and minimization procedure until a defect-free configuration is found. Some systems will have more complicated ground states, which can be much more challenging to find. For this, Sunny provides experimental support for powerful simulated annealing via parallel tempering, but that is outside the scope of this tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Here, let's break the three-fold symmetry of FeI₂ by hand. Given one or more desired Q modes, Sunny can suggest a magnetic supercell with appropriate periodicity. Let's arbitrarily select one of the three possible ordering wavevectors, Q = 0 -14 14. Sunny suggests a corresponding magnetic supercell in units of the crystal lattice vectors.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"suggest_magnetic_supercell([[0, -1/4, 1/4]])","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The system returned by reshape_supercell is smaller, and is sheared relative to the original system. This makes it much easier to find the global energy minimum.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"sys_min = reshape_supercell(sys, [1 0 0; 0 2 1; 0 -2 1])\nrandomize_spins!(sys_min)\nminimize_energy!(sys_min);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Plot the system again, now including \"ghost\" spins out to 12Å","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"plot_spins(sys_min; color=[s[3] for s in sys_min.dipoles], ghost_radius=12)","category":"page"},{"location":"examples/fei2_tutorial.html#Linear-spin-wave-theory","page":"Case Study: FeI₂","title":"Linear spin wave theory","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Now that we have found the ground state for a magnetic supercell, we can immediately proceed to perform zero-temperature calculations using linear spin wave theory. We begin by instantiating a SpinWaveTheory type using the supercell.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"swt = SpinWaveTheory(sys_min)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Select a sequence of wavevectors that will define a piecewise linear interpolation in reciprocal lattice units (RLU).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"q_points = [[0,0,0], [1,0,0], [0,1,0], [1/2,0,0], [0,1,0], [0,0,0]];\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function reciprocal_space_path will linearly sample a path between the provided q-points with a given density. The xticks return value provides labels for use in plotting.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"density = 50\npath, xticks = reciprocal_space_path(cryst, q_points, density);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The dispersion function defines the quasiparticle excitation energies ω_i(𝐪) for each point 𝐪 along the reciprocal space path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp = dispersion(swt, path);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In addition to the band energies ω_i(𝐪), Sunny can calculate the inelastic neutron scattering intensity I_i(𝐪) for each band i according to an intensity_formula. We choose to apply a polarization correction (1 - 𝐪𝐪) by setting the mode argument to :perp. Selecting delta_function_kernel specifies that we want the energy and intensity of each band individually.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function intensities_bands uses linear spin wave theory to calculate both the dispersion and intensity data for the provided path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp, intensity = intensities_bands(swt, path, formula);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"These can be plotted in GLMakie.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"fig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nylims!(ax, 0.0, 7.5)\nxlims!(ax, 1, size(disp, 1))\ncolorrange = extrema(intensity)\nfor i in axes(disp, 2)\n lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange)\nend\nfig","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"To make comparisons with inelastic neutron scattering (INS) data, it is helpful to employ an empirical broadening kernel, e.g., a lorentzian.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"γ = 0.15 # width in meV\nbroadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The intensities_broadened function requires an energy range in addition to the 𝐪-space path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"energies = collect(0:0.01:10) # 0 < ω < 10 (meV).\nis1 = intensities_broadened(swt, path, energies, broadened_formula);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A real FeI₂ sample will exhibit competing magnetic domains associated with spontaneous symmetry breaking of the 6-fold rotational symmetry of the triangular lattice. Note that the wavevectors 𝐪 and -𝐪 are equivalent in the structure factor, which leaves three distinct domain orientations, which are related by 120° rotations about the z-axis. Rather than rotating the spin configuration directly, on can rotate the 𝐪-space path. Below, we use rotation_in_rlu to average the intensities over all three possible orientations.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"R = rotation_in_rlu(cryst, [0, 0, 1], 2π/3)\nis2 = intensities_broadened(swt, [R*q for q in path], energies, broadened_formula)\nis3 = intensities_broadened(swt, [R*R*q for q in path], energies, broadened_formula)\nis_averaged = (is1 + is2 + is3) / 3\n\nfig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nheatmap!(ax, 1:size(is_averaged, 1), energies, is_averaged)\nfig","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"This result can be directly compared to experimental neutron scattering data from Bai et al.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"
","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The Fe atoms are arranged in stacked triangular layers. The effective spin Hamiltonian takes the form,","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"mathcalH=sum_(ij) 𝐒_i J_ij 𝐒_j - Dsum_i left(S_i^zright)^2","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"where the set of exchange matrices J_ij between bonded sites (ij) includes competing ferromagnetic and antiferromagnetic interactions. This model also includes a strong easy axis anisotropy, D 0.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"We will formulate this Hamiltonian in Sunny and then calculate its dynamic structure factor.","category":"page"},{"location":"examples/fei2_tutorial.html#Get-Julia-and-Sunny","page":"Case Study: FeI₂","title":"Get Julia and Sunny","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Sunny is implemented in Julia. This is a relatively new programming language that allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users may wish to take a look at our Getting Started with Julia guide. Sunny requires Julia 1.9 or later.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"From the Julia prompt, load Sunny. For plotting, one can choose either GLMakie (a pop-up window) or WGLMakie (inline plots for a Jupyter notebook or VSCode).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"If these packages are not yet installed, Julia should offer to install them using its built-in package management system. If old versions are installed, you may need to update them to run this tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html#Crystals","page":"Case Study: FeI₂","title":"Crystals","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A Crystal describes the crystallographic unit cell and will usually be loaded from a .cif file. Here, we instead build a crystal by listing all atoms and their types.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"a = b = 4.05012 # Lattice constants for triangular lattice\nc = 6.75214 # Spacing in the z-direction\n\nlatvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that\n # define the conventional unit cell\npositions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions\n # of lattice vectors\ntypes = [\"Fe\", \"I\", \"I\"]\nFeI2 = Crystal(latvecs, positions; types)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Observe that Sunny inferred the space group, 'P -3 m 1' (164) and labeled the atoms according to their point group symmetries.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Only the Fe atoms are magnetic, so we discard the I ions using subcrystal.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"cryst = subcrystal(FeI2, \"Fe\")","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Observe that cryst retains the spacegroup symmetry of the full FeI₂ crystal. This information will be used, for example, to propagate exchange interactions between symmetry-equivalent bonds.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In a running Julia environment, the crystal can be viewed interactively using view_crystal.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"view_crystal(cryst, 8.0)","category":"page"},{"location":"examples/fei2_tutorial.html#Symmetry-analysis","page":"Case Study: FeI₂","title":"Symmetry analysis","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The command print_symmetry_table provides a list of all the symmetry-allowed interactions up to a cutoff distance.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"print_symmetry_table(cryst, 8.0)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The allowed g-tensor is expressed as a 3×3 matrix in the free coefficients A, B, ... The allowed single-ion anisotropy is expressed as a linear combination of Stevens operators. The latter correspond to polynomials of the spin operators, as we will describe below.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The allowed exchange interactions are given as a 3×3 matrix for representative bonds. The notation Bond(i, j, n) indicates a bond between atom indices i and j, with cell offset n. In the general case, it will be necessary to associate atom indices with their positions in the unit cell; these can be viewed with display(cryst). Note that the order of the pair (i j) is significant if the exchange tensor contains antisymmetric Dzyaloshinskii–Moriya (DM) interactions.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In the case of FeI₂, Bond(1, 1, [1,0,0]) is one of the 6 nearest-neighbor Fe-Fe bonds on a triangular lattice layer, and Bond(1, 1, [0,0,1]) is an Fe-Fe bond between layers.","category":"page"},{"location":"examples/fei2_tutorial.html#Building-a-spin-System","page":"Case Study: FeI₂","title":"Building a spin System","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In constructing a spin System, we must provide several additional details about the spins.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"sys = System(cryst, (4, 4, 4), [SpinInfo(1, S=1, g=2)], :SUN, seed=2)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"This system includes 444 unit cells, i.e. 64 Fe atoms, each with spin S=1 and a g-factor of 2. Quantum mechanically, spin S=1 involves a superposition of 2S+1=3 distinct angular momentum states. In :SUN mode, this superposition will be modeled explicitly using the formalism of SU(3) coherent states, which captures both dipolar and quadrupolar fluctuations. For the more traditional dipole dynamics, use :dipole mode instead.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Next we will use set_exchange! to assign interaction to bonds. Sunny will automatically propagate each interaction to all symmetry-equivalent bonds in the unit cell. The FeI₂ interactions below follow Bai et al.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"J1pm = -0.236\nJ1pmpm = -0.161\nJ1zpm = -0.261\nJ2pm = 0.026\nJ3pm = 0.166\nJ′0pm = 0.037\nJ′1pm = 0.013\nJ′2apm = 0.068\n\nJ1zz = -0.236\nJ2zz = 0.113\nJ3zz = 0.211\nJ′0zz = -0.036\nJ′1zz = 0.051\nJ′2azz = 0.073\n\nJ1xx = J1pm + J1pmpm\nJ1yy = J1pm - J1pmpm\nJ1yz = J1zpm\n\nset_exchange!(sys, [J1xx 0.0 0.0;\n 0.0 J1yy J1yz;\n 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))\nset_exchange!(sys, [J2pm 0.0 0.0;\n 0.0 J2pm 0.0;\n 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))\nset_exchange!(sys, [J3pm 0.0 0.0;\n 0.0 J3pm 0.0;\n 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))\nset_exchange!(sys, [J′0pm 0.0 0.0;\n 0.0 J′0pm 0.0;\n 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))\nset_exchange!(sys, [J′1pm 0.0 0.0;\n 0.0 J′1pm 0.0;\n 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))\nset_exchange!(sys, [J′2apm 0.0 0.0;\n 0.0 J′2apm 0.0;\n 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function set_onsite_coupling! assigns a single-ion anisotropy. The argument can be constructed using spin_matrices or stevens_matrices. Here we use Julia's anonymous function syntax to assign an easy-axis anisotropy along the direction hatz.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"D = 2.165\nset_onsite_coupling!(sys, S -> -D*S[3]^2, 1)","category":"page"},{"location":"examples/fei2_tutorial.html#Calculating-structure-factor-intensities","page":"Case Study: FeI₂","title":"Calculating structure factor intensities","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In the remainder of this tutorial, we will examine Sunny's tools for calculating the dynamical structure factor using a multi-boson generalization of linear spin wave theory (LSWT). This theory describes non-interacting quasi-particle excitations that hybridize dipolar and quadrupolar modes.","category":"page"},{"location":"examples/fei2_tutorial.html#Finding-the-ground-state","page":"Case Study: FeI₂","title":"Finding the ground state","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Begin with a random configuration and use minimize_energy! to find a configuration of the SU(3) coherent states (i.e. spin dipoles and quadrupoles) that locally minimizes energy.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"randomize_spins!(sys)\nminimize_energy!(sys)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A positive number above indicates that the procedure has converged to a local energy minimum. The configuration, however, may still have defects. This can be checked by visualizing the spins, colored according to their z-components.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"plot_spins(sys; color=[s[3] for s in sys.dipoles])","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A different understanding of the magnetic ordering can be obtained by moving to Fourier space. The 'instant' structure factor 𝒮(𝐪) is an experimental observable. To investigate 𝒮(𝐪) as true 3D data, Sunny provides instant_correlations and related functions. Here, however, we will use print_wrapped_intensities, which gives average intensities for the individual Bravais sublattices (in effect, all wavevectors are wrapped to the first Brillouin zone).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"print_wrapped_intensities(sys)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The result will likely be approximately consistent with the known zero-field energy-minimizing magnetic structure of FeI₂, which is single-Q (two-up, two-down antiferromagnetic order). Mathematically, spontaneous symmetry breaking should select one of Q = 0 -14 14, 14 0 14, or -141414, associated with the three-fold rotational symmetry of the crystal spacegroup. In nature, however, one will frequently encounter competing \"domains\" associated with the three possible orientations of the ground state.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"If the desired ground state is already known, as with FeI₂, it could be entered by hand using set_dipole!. Alternatively, in the case of FeI₂, we could repeatedly employ the above randomization and minimization procedure until a defect-free configuration is found. Some systems will have more complicated ground states, which can be much more challenging to find. For this, Sunny provides experimental support for powerful simulated annealing via parallel tempering, but that is outside the scope of this tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Here, let's break the three-fold symmetry of FeI₂ by hand. Given one or more desired Q modes, Sunny can suggest a magnetic supercell with appropriate periodicity. Let's arbitrarily select one of the three possible ordering wavevectors, Q = 0 -14 14. Sunny suggests a corresponding magnetic supercell in units of the crystal lattice vectors.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"suggest_magnetic_supercell([[0, -1/4, 1/4]])","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The system returned by reshape_supercell is smaller, and is sheared relative to the original system. This makes it much easier to find the global energy minimum.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"sys_min = reshape_supercell(sys, [1 0 0; 0 2 1; 0 -2 1])\nrandomize_spins!(sys_min)\nminimize_energy!(sys_min);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Plot the system again, now including \"ghost\" spins out to 12Å","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"plot_spins(sys_min; color=[s[3] for s in sys_min.dipoles], ghost_radius=12)","category":"page"},{"location":"examples/fei2_tutorial.html#Linear-spin-wave-theory","page":"Case Study: FeI₂","title":"Linear spin wave theory","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Now that we have found the ground state for a magnetic supercell, we can immediately proceed to perform zero-temperature calculations using linear spin wave theory. We begin by instantiating a SpinWaveTheory type using the supercell.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"swt = SpinWaveTheory(sys_min)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Select a sequence of wavevectors that will define a piecewise linear interpolation in reciprocal lattice units (RLU).","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"q_points = [[0,0,0], [1,0,0], [0,1,0], [1/2,0,0], [0,1,0], [0,0,0]];\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function reciprocal_space_path will linearly sample a path between the provided q-points with a given density. The xticks return value provides labels for use in plotting.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"density = 50\npath, xticks = reciprocal_space_path(cryst, q_points, density);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The dispersion function defines the quasiparticle excitation energies ω_i(𝐪) for each point 𝐪 along the reciprocal space path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp = dispersion(swt, path);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"In addition to the band energies ω_i(𝐪), Sunny can calculate the inelastic neutron scattering intensity I_i(𝐪) for each band i according to an intensity_formula. We choose to apply a polarization correction (1 - 𝐪𝐪) by setting the mode argument to :perp. Selecting delta_function_kernel specifies that we want the energy and intensity of each band individually.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The function intensities_bands uses linear spin wave theory to calculate both the dispersion and intensity data for the provided path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp, intensity = intensities_bands(swt, path, formula);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"These can be plotted in GLMakie.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"fig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nylims!(ax, 0.0, 7.5)\nxlims!(ax, 1, size(disp, 1))\ncolorrange = extrema(intensity)\nfor i in axes(disp, 2)\n lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange)\nend\nfig","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"To make comparisons with inelastic neutron scattering (INS) data, it is helpful to employ an empirical broadening kernel, e.g., a lorentzian.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"γ = 0.15 # width in meV\nbroadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The intensities_broadened function requires an energy range in addition to the 𝐪-space path.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"energies = collect(0:0.01:10) # 0 < ω < 10 (meV).\nis1 = intensities_broadened(swt, path, energies, broadened_formula);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"A real FeI₂ sample will exhibit competing magnetic domains associated with spontaneous symmetry breaking of the 6-fold rotational symmetry of the triangular lattice. Note that the wavevectors 𝐪 and -𝐪 are equivalent in the structure factor, which leaves three distinct domain orientations, which are related by 120° rotations about the z-axis. Rather than rotating the spin configuration directly, on can rotate the 𝐪-space path. Below, we use rotation_in_rlu to average the intensities over all three possible orientations.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"R = rotation_in_rlu(cryst, [0, 0, 1], 2π/3)\nis2 = intensities_broadened(swt, [R*q for q in path], energies, broadened_formula)\nis3 = intensities_broadened(swt, [R*R*q for q in path], energies, broadened_formula)\nis_averaged = (is1 + is2 + is3) / 3\n\nfig = Figure()\nax = Axis(fig[1,1]; xlabel=\"Momentum (r.l.u.)\", ylabel=\"Energy (meV)\", xticks, xticklabelrotation=π/6)\nheatmap!(ax, 1:size(is_averaged, 1), energies, is_averaged)\nfig","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"This result can be directly compared to experimental neutron scattering data from Bai et al.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":" ","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"(The publication figure accidentally used a non-standard coordinate system to label the wave vectors.)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"To get this agreement, the use of SU(3) coherent states is essential. In other words, we needed a theory of multi-flavored bosons. The lower band has large quadrupolar character, and arises from the strong easy-axis anisotropy of FeI₂. By setting mode = :SUN, the calculation captures this coupled dipole-quadrupole dynamics.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"An interesting exercise is to repeat the same study, but using mode = :dipole instead of :SUN. That alternative choice would constrain the coherent state dynamics to the space of dipoles only.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The full dynamical spin structure factor (DSSF) can be retrieved as a 33 matrix with the dssf function, for a given path of 𝐪-vectors.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp, is = dssf(swt, path);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The first output disp is identical to that obtained from dispersion. The second output is contains a list of 33 matrix of intensities. For example, is[q,n][2,3] yields the (yz) component of the structure factor intensity for nth mode at the qth wavevector in the path.","category":"page"},{"location":"examples/fei2_tutorial.html#What's-next?","page":"Case Study: FeI₂","title":"What's next?","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The multi-boson linear spin wave theory, applied above, can be understood as the quantization of a certain generalization of the Landau-Lifshitz spin dynamics. Rather than dipoles, this dynamics takes places on the space of SU(N) coherent states.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The full SU(N) coherent state dynamics, with appropriate quantum correction factors, can be useful to model finite temperature scattering data. In particular, it captures certain anharmonic effects due to thermal fluctuations. This is the subject of our FeI₂ at Finite Temperature tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The classical dynamics is also a good starting point to study non-equilibrium phenomena. Empirical noise and damping terms can be used to model coupling to a thermal bath. This yields a Langevin dynamics of SU(N) coherent states. Our CP² Skyrmion Quench tutorial shows how this dynamics gives rise to the formation of novel topological defects in a temperature quench.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Relative to LSWT calculations, it can take much more time to estimate mathcalS(𝐪ω) intensities using classical dynamics simulation. See the SunnyTutorials notebooks for examples of \"production-scale\" simulations.","category":"page"},{"location":"structure-factor.html#Structure-Factor-Calculations","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"","category":"section"},{"location":"structure-factor.html#Overview","page":"Structure Factor Calculations","title":"Overview","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The dynamical structure factor is of fundamental importance for characterizing a magnetic system, and facilitates quantitative comparison between theory and experimental scattering data.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Consider, for example, a two-point dynamical spin correlation function, s^α(𝐱+Δ𝐱 t+Δt) s^β(𝐱 t). Here s^α(𝐱 t) represents the time dynamics of a spin dipole component α at position 𝐱, and brackets represent an average over equilibrium initial conditions and over (𝐱 t). The dynamical structure factor is defined as the Fourier transform of this two-point correlation in both space and time, up to an overall scaling factor. Using the convolution theorem, the result is,","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ(𝐪 ω) = frac1V s^α(𝐪 ω)^ast s^β(𝐪 ω) ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"with V the system volume. We will restrict attention to lattice systems with periodic boundaries.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Consider a crystal unit cell defined by three lattice vectors 𝐚_1 𝐚_2 𝐚_3, and linear system sizes L_1 L_2 L_3 measured in unit cells. The allowed momentum vectors take on discrete values 𝐪 = sum_α=1^3 m_α 𝐛_α L_α, where m_α are an integers and the reciprocal lattice vectors 𝐛_α are defined to satisfy 𝐚_α 𝐛_β = 2π δ_αβ. For a Bravais lattice, 𝐪 will be periodic in the first Brillouin zone, i.e., under any shift 𝐪 𝐪 𝐛_α. More generally, consider a non-Bravais lattice such that each unit cell may contain multiple spins. By partitioning spins s_j(𝐱t) according to their sublattice index j, the relevant momenta 𝐪 remain discretized as above, but now periodicity in the first Brillouin zone is lost. The structure factor may be written as a phase-average over the displacements between sublattices 𝐫_jk,","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ(𝐪 ω) = _jk e^i 𝐫_jk 𝐪 𝒮^αβ_jk(𝐪 ω) ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"From a theoretical perspective, the quantity","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ_jk(𝐪 ω) = frac1V s_j^α(𝐪 ω)^ast s_k^β(𝐪 ω)","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"is fundamental. For each sublattice j, the data s_j^α(𝐪 ω) can be efficiently obtained by fast Fourier tranformation of a real space configuration s_j^α(𝐱 t). Internally, Sunny will calculate and store the discrete 𝒮^αβ_jk(𝐪 ω) correlation data, and use this to construct 𝒮^αβ(𝐪ω) intensities that can be compared with experiment.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Calculating this structure factor involves several steps, with various possible settings. Sunny provides a number of tools to facilitate this calculation and to extract information from the results. These tools are briefly outlined below. Please see the Examples for a \"real life\" use case. Detailed function information is available in the Library API.","category":"page"},{"location":"structure-factor.html#Estimating-stucture-factors-with-classical-dynamics","page":"Structure Factor Calculations","title":"Estimating stucture factors with classical dynamics","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Classical dynamics may be used to estimate structure factor data by analyzing the spin-spin correlations of dynamical trajectories. This is fundamentally a Monte Carlo approach, as the trajectories must be started from an initial spin configuration that is sampled at thermal equilibrium. (Note that it is not possible to estimate a true T=0 dynamical structure factor using this method, but the temperature may be very low.) Samples are accumulated into a SampledCorrelations, from which intensity information may be extracted. The user does not typically build their own SampledCorrelations but instead initializes one by calling either dynamical_correlations or instant_correlations, as described below.","category":"page"},{"location":"structure-factor.html#Estimating-a-dynamical-structure-factor:-𝒮(𝐪,ω)","page":"Structure Factor Calculations","title":"Estimating a dynamical structure factor: 𝒮(𝐪ω)","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A SampledCorrelations for estimating the dynamical structure factor, 𝒮^αβ(𝐪ω), may be created by calling dynamical_correlations. This requires three keyword arguments. These will determine the dynamics used to calculate samples and, consequently, the ω information that will be available. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Δt: Determines the step size used for simulating the dynamics. A smaller number will require proportionally more calculation time. While a smaller Δt will enable the resolution of higher energies, Δt is typically selected to ensure numerical stability rather than to maximize the largest ω value. A safe choice is to use the smaller value of Δt = 0.1/(J* S^2) or Δt = 0.1/(D * S), where S is magnetic moment of the largest local spin (as specified in SpinInfo), J is the parameter governing the largest bilinear interaction (e.g. exchange), and D is the parameter governing the largest single-site term of the Hamiltonian (e.g., anisotropy or Zeeman term).\nωmax: Sets the maximum resolved energy. Note that this is not independent of Δt. If ωmax too large, Sunny will throw an error and ask you to choose a smaller Δt. \nnω: Determines the number of energy bins to resolve. A larger number will require more calculation time.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A sample may be added by calling add_sample!(sc, sys). The input sys must be a spin configuration in good thermal equilibrium, e.g., using the continuous Langevin dynamics or using single spin flip trials with LocalSampler. The statistical quality of the 𝒮^αβ(𝐪ω) can be improved by repeatedly generating decorrelated spin configurations in sys and calling add_sample! on each configuration.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The outline of typical use case might look like this:","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"# Make a `SampledCorrelations`\nsc = dynamical_correlations(sys; Δt=0.05, ωmax=10.0, nω=100) \n\n# Add samples\nfor _ in 1:nsamples\n decorrelate_system(sys) # Perform some type of Monte Carlo simulation\n add_sample!(sc, sys) # Use spins to calculate trajectory and accumulate new sample of 𝒮(𝐪,ω)\nend","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The calculation may be configured in a number of ways; see the dynamical_correlations documentation for a list of all keywords.","category":"page"},{"location":"structure-factor.html#Estimating-an-instantaneous-(\"static\")-structure-factor:-𝒮(𝐪)","page":"Structure Factor Calculations","title":"Estimating an instantaneous (\"static\") structure factor: 𝒮(𝐪)","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Sunny provides two methods for calculating instantaneous, or static, structure factors: 𝒮^αβ(𝐪). The first involves calculating spatial spin-spin correlations at single time slices. The second involves calculating a dynamic structure factor first and integrating out the ω information. The advantage of the latter approach is that it enables application of an ω-dependent classical-to-quantum rescaling of structure factor intensities, a method that should be preferred whenever comparing results to experimental data or spin wave calculations. A disadvantage of this approach is that it is computationally more expensive. There are also many cases when it is not straightforward to calculate a meaningful dynamics, as when working with Ising spins. In this section we will discuss how to calculate instantaneous structure factors from static spin configurations. Information about calculating instantaneous data from a dynamical correlations can be found in the following section.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The basic usage for the instantaneous case is very similar to the dynamic case, except one calls instant_correlations instead of dynamical_correlations to configure a SampledCorrelations. Note that there are no required keywords as there is no need to specify any dynamics. instant_correlations will return a SampledCorrelations containing no data. Samples may be added by calling add_sample!(sc, sys), where sc is the SampledCorrelations. When performing a finite-temperature calculation, it is important to ensure that the spin configuration in the sys represents a good equilibrium sample, as in the dynamical case. Note, however, that we recommend calculating instantaneous correlations at finite temperature calculations by using full dynamics (i.e., using dynamical_correlations) and then integrating out the energy axis. An approach to doing this is described in the next section.","category":"page"},{"location":"structure-factor.html#Extracting-information-from-sampled-correlation-data","page":"Structure Factor Calculations","title":"Extracting information from sampled correlation data","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The basic function for extracting information from a SampledCorrelations at a particular wave vector, 𝐪, is intensities_interpolated. It takes a SampledCorrelations, a list of wave vectors, and an intensity_formula. The intensity_formula specifies how to contract and correct correlation data to arrive at a physical intensity. A simple example is formula = intensity_formula(sc, :perp), which will instruct Sunny apply polarization corrections: sum_αβ(I-q_α q_β) 𝒮^αβ(𝐪ω). An intensity at the wave vector 𝐪 = (𝐛_2 + 𝐛_3)2 may then be retrieved with intensities_interpolated(sf, [[0.0, 0.5, 0.5]], formula) . intensities_interpolated returns a list of nω elements at each wavevector. The corresponding ω values can be retrieved by calling available_energies on sf.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Since Sunny only calculates the structure factor on a finite lattice when performing classical simulations, it is important to realize that exact information is only available at a discrete set of wave vectors. Specifically, for each axis index i, we will get information at q_i = fracnL_i, where n runs from (frac-L_i2+1) to fracL_i2 and L_i is the linear dimension of the lattice used for the calculation. If you request a wave vector that does not fall into this set, Sunny will automatically round to the nearest 𝐪 that is available. If intensities_interpolated is given the keyword argument interpolation=:linear, Sunny will use trilinear interpolation to determine a result at the requested wave vector. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"To retrieve the intensities at all wave vectors for which there is exact data, first call the function available_wave_vectors to generate a list of qs. This takes an optional keyword argument bzsize, which must be given a tuple of three integers specifying the number of Brillouin zones to calculate, e.g., bzsize=(2,2,2). The resulting list of wave vectors may then be passed to intensities_interpolated.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Alternatively, intensities_binned can be used to place the exact data into histogram bins for comparison with experiment.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The convenience function reciprocal_space_path returns a list of wavevectors sampled along a path that connects specified 𝐪 points. This list can be used as an input to intensities. Another convenience method, reciprocal_space_shell will generate points on a sphere of a given radius. This is useful for powder averaging. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A number of arguments for intensity_formula are available which modify the calculation of structure factor intensity. It is generally recommended to provide a value of kT corresponding to the temperature of sampled configurations. Given kT, Sunny will include an energy- and temperature-dependent classical-to-quantum rescaling of intensities in the formula.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"To retrieve intensity data from a instantaneous structure factor, use instant_intensities_interpolated, which accepts similar arguments to intensities_interpolated. This function may also be used to calculate instantaneous information from a dynamical correlation data, i.e. from a SampledCorrelations created with dynamical_correlations. Note that it is important to supply a value to kT to reap the benefits of this approach over simply calculating a static structure factor at the outset. ","category":"page"},{"location":"writevtk.html#ParaView-Rendering","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The 4D correlation data produced by Sunny is too high-dimensional to visualize directly. This page describes how to export 3D slices of correlation data from Sunny to the Visual ToolKit (VTK) format, which is compatible with the ParaView visualization software. ParaView supports volumetric rendering:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"(The publication figure accidentally used a non-standard coordinate system to label the wave vectors.)","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"To get this agreement, the use of SU(3) coherent states is essential. In other words, we needed a theory of multi-flavored bosons. The lower band has large quadrupolar character, and arises from the strong easy-axis anisotropy of FeI₂. By setting mode = :SUN, the calculation captures this coupled dipole-quadrupole dynamics.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"An interesting exercise is to repeat the same study, but using mode = :dipole instead of :SUN. That alternative choice would constrain the coherent state dynamics to the space of dipoles only.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The full dynamical spin structure factor (DSSF) can be retrieved as a 33 matrix with the dssf function, for a given path of 𝐪-vectors.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"disp, is = dssf(swt, path);\nnothing #hide","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The first output disp is identical to that obtained from dispersion. The second output is contains a list of 33 matrix of intensities. For example, is[q,n][2,3] yields the (yz) component of the structure factor intensity for nth mode at the qth wavevector in the path.","category":"page"},{"location":"examples/fei2_tutorial.html#What's-next?","page":"Case Study: FeI₂","title":"What's next?","text":"","category":"section"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The multi-boson linear spin wave theory, applied above, can be understood as the quantization of a certain generalization of the Landau-Lifshitz spin dynamics. Rather than dipoles, this dynamics takes places on the space of SU(N) coherent states.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The full SU(N) coherent state dynamics, with appropriate quantum correction factors, can be useful to model finite temperature scattering data. In particular, it captures certain anharmonic effects due to thermal fluctuations. This is the subject of our FeI₂ at Finite Temperature tutorial.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"The classical dynamics is also a good starting point to study non-equilibrium phenomena. Empirical noise and damping terms can be used to model coupling to a thermal bath. This yields a Langevin dynamics of SU(N) coherent states. Our CP² Skyrmion Quench tutorial shows how this dynamics gives rise to the formation of novel topological defects in a temperature quench.","category":"page"},{"location":"examples/fei2_tutorial.html","page":"Case Study: FeI₂","title":"Case Study: FeI₂","text":"Relative to LSWT calculations, it can take much more time to estimate mathcalS(𝐪ω) intensities using classical dynamics simulation. See the SunnyTutorials notebooks for examples of \"production-scale\" simulations.","category":"page"},{"location":"structure-factor.html#Structure-Factor-Calculations","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"","category":"section"},{"location":"structure-factor.html#Overview","page":"Structure Factor Calculations","title":"Overview","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The dynamical structure factor is of fundamental importance for characterizing a magnetic system, and facilitates quantitative comparison between theory and experimental scattering data.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Consider, for example, a two-point dynamical spin correlation function, s^α(𝐱+Δ𝐱 t+Δt) s^β(𝐱 t). Here s^α(𝐱 t) represents the time dynamics of a spin dipole component α at position 𝐱, and brackets represent an average over equilibrium initial conditions and over (𝐱 t). The dynamical structure factor is defined as the Fourier transform of this two-point correlation in both space and time, up to an overall scaling factor. Using the convolution theorem, the result is,","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ(𝐪 ω) = frac1V s^α(𝐪 ω)^ast s^β(𝐪 ω) ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"with V the system volume. We will restrict attention to lattice systems with periodic boundaries.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Consider a crystal unit cell defined by three lattice vectors 𝐚_1 𝐚_2 𝐚_3, and linear system sizes L_1 L_2 L_3 measured in unit cells. The allowed momentum vectors take on discrete values 𝐪 = sum_α=1^3 m_α 𝐛_α L_α, where m_α are an integers and the reciprocal lattice vectors 𝐛_α are defined to satisfy 𝐚_α 𝐛_β = 2π δ_αβ. For a Bravais lattice, 𝐪 will be periodic in the first Brillouin zone, i.e., under any shift 𝐪 𝐪 𝐛_α. More generally, consider a non-Bravais lattice such that each unit cell may contain multiple spins. By partitioning spins s_j(𝐱t) according to their sublattice index j, the relevant momenta 𝐪 remain discretized as above, but now periodicity in the first Brillouin zone is lost. The structure factor may be written as a phase-average over the displacements between sublattices 𝐫_jk,","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ(𝐪 ω) = _jk e^i 𝐫_jk 𝐪 𝒮^αβ_jk(𝐪 ω) ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"From a theoretical perspective, the quantity","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"𝒮^αβ_jk(𝐪 ω) = frac1V s_j^α(𝐪 ω)^ast s_k^β(𝐪 ω)","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"is fundamental. For each sublattice j, the data s_j^α(𝐪 ω) can be efficiently obtained by fast Fourier tranformation of a real space configuration s_j^α(𝐱 t). Internally, Sunny will calculate and store the discrete 𝒮^αβ_jk(𝐪 ω) correlation data, and use this to construct 𝒮^αβ(𝐪ω) intensities that can be compared with experiment.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Calculating this structure factor involves several steps, with various possible settings. Sunny provides a number of tools to facilitate this calculation and to extract information from the results. These tools are briefly outlined below. Please see the Examples for a \"real life\" use case. Detailed function information is available in the Library API.","category":"page"},{"location":"structure-factor.html#Estimating-stucture-factors-with-classical-dynamics","page":"Structure Factor Calculations","title":"Estimating stucture factors with classical dynamics","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Classical dynamics may be used to estimate structure factor data by analyzing the spin-spin correlations of dynamical trajectories. This is fundamentally a Monte Carlo approach, as the trajectories must be started from an initial spin configuration that is sampled at thermal equilibrium. (Note that it is not possible to estimate a true T=0 dynamical structure factor using this method, but the temperature may be very low.) Samples are accumulated into a SampledCorrelations, from which intensity information may be extracted. The user does not typically build their own SampledCorrelations but instead initializes one by calling either dynamical_correlations or instant_correlations, as described below.","category":"page"},{"location":"structure-factor.html#Estimating-a-dynamical-structure-factor:-𝒮(𝐪,ω)","page":"Structure Factor Calculations","title":"Estimating a dynamical structure factor: 𝒮(𝐪ω)","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A SampledCorrelations for estimating the dynamical structure factor, 𝒮^αβ(𝐪ω), may be created by calling dynamical_correlations. This requires three keyword arguments. These will determine the dynamics used to calculate samples and, consequently, the ω information that will be available. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Δt: Determines the step size used for simulating the dynamics. A smaller number will require proportionally more calculation time. While a smaller Δt will enable the resolution of higher energies, Δt is typically selected to ensure numerical stability rather than to maximize the largest ω value. A safe choice is to use the smaller value of Δt = 0.1/(J* S^2) or Δt = 0.1/(D * S), where S is magnetic moment of the largest local spin (as specified in SpinInfo), J is the parameter governing the largest bilinear interaction (e.g. exchange), and D is the parameter governing the largest single-site term of the Hamiltonian (e.g., anisotropy or Zeeman term).\nωmax: Sets the maximum resolved energy. Note that this is not independent of Δt. If ωmax too large, Sunny will throw an error and ask you to choose a smaller Δt. \nnω: Determines the number of energy bins to resolve. A larger number will require more calculation time.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A sample may be added by calling add_sample!(sc, sys). The input sys must be a spin configuration in good thermal equilibrium, e.g., using the continuous Langevin dynamics or using single spin flip trials with LocalSampler. The statistical quality of the 𝒮^αβ(𝐪ω) can be improved by repeatedly generating decorrelated spin configurations in sys and calling add_sample! on each configuration.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The outline of typical use case might look like this:","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"# Make a `SampledCorrelations`\nsc = dynamical_correlations(sys; Δt=0.05, ωmax=10.0, nω=100) \n\n# Add samples\nfor _ in 1:nsamples\n decorrelate_system(sys) # Perform some type of Monte Carlo simulation\n add_sample!(sc, sys) # Use spins to calculate trajectory and accumulate new sample of 𝒮(𝐪,ω)\nend","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The calculation may be configured in a number of ways; see the dynamical_correlations documentation for a list of all keywords.","category":"page"},{"location":"structure-factor.html#Estimating-an-instantaneous-(\"static\")-structure-factor:-𝒮(𝐪)","page":"Structure Factor Calculations","title":"Estimating an instantaneous (\"static\") structure factor: 𝒮(𝐪)","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Sunny provides two methods for calculating instantaneous, or static, structure factors: 𝒮^αβ(𝐪). The first involves calculating spatial spin-spin correlations at single time slices. The second involves calculating a dynamic structure factor first and integrating out the ω information. The advantage of the latter approach is that it enables application of an ω-dependent classical-to-quantum rescaling of structure factor intensities, a method that should be preferred whenever comparing results to experimental data or spin wave calculations. A disadvantage of this approach is that it is computationally more expensive. There are also many cases when it is not straightforward to calculate a meaningful dynamics, as when working with Ising spins. In this section we will discuss how to calculate instantaneous structure factors from static spin configurations. Information about calculating instantaneous data from a dynamical correlations can be found in the following section.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The basic usage for the instantaneous case is very similar to the dynamic case, except one calls instant_correlations instead of dynamical_correlations to configure a SampledCorrelations. Note that there are no required keywords as there is no need to specify any dynamics. instant_correlations will return a SampledCorrelations containing no data. Samples may be added by calling add_sample!(sc, sys), where sc is the SampledCorrelations. When performing a finite-temperature calculation, it is important to ensure that the spin configuration in the sys represents a good equilibrium sample, as in the dynamical case. Note, however, that we recommend calculating instantaneous correlations at finite temperature calculations by using full dynamics (i.e., using dynamical_correlations) and then integrating out the energy axis. An approach to doing this is described in the next section.","category":"page"},{"location":"structure-factor.html#Extracting-information-from-sampled-correlation-data","page":"Structure Factor Calculations","title":"Extracting information from sampled correlation data","text":"","category":"section"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The basic function for extracting information from a SampledCorrelations at a particular wave vector, 𝐪, is intensities_interpolated. It takes a SampledCorrelations, a list of wave vectors, and an intensity_formula. The intensity_formula specifies how to contract and correct correlation data to arrive at a physical intensity. A simple example is formula = intensity_formula(sc, :perp), which will instruct Sunny apply polarization corrections: sum_αβ(I-q_α q_β) 𝒮^αβ(𝐪ω). An intensity at the wave vector 𝐪 = (𝐛_2 + 𝐛_3)2 may then be retrieved with intensities_interpolated(sf, [[0.0, 0.5, 0.5]], formula) . intensities_interpolated returns a list of nω elements at each wavevector. The corresponding ω values can be retrieved by calling available_energies on sf.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Since Sunny only calculates the structure factor on a finite lattice when performing classical simulations, it is important to realize that exact information is only available at a discrete set of wave vectors. Specifically, for each axis index i, we will get information at q_i = fracnL_i, where n runs from (frac-L_i2+1) to fracL_i2 and L_i is the linear dimension of the lattice used for the calculation. If you request a wave vector that does not fall into this set, Sunny will automatically round to the nearest 𝐪 that is available. If intensities_interpolated is given the keyword argument interpolation=:linear, Sunny will use trilinear interpolation to determine a result at the requested wave vector. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"To retrieve the intensities at all wave vectors for which there is exact data, first call the function available_wave_vectors to generate a list of qs. This takes an optional keyword argument bzsize, which must be given a tuple of three integers specifying the number of Brillouin zones to calculate, e.g., bzsize=(2,2,2). The resulting list of wave vectors may then be passed to intensities_interpolated.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"Alternatively, intensities_binned can be used to place the exact data into histogram bins for comparison with experiment.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"The convenience function reciprocal_space_path returns a list of wavevectors sampled along a path that connects specified 𝐪 points. This list can be used as an input to intensities. Another convenience method, reciprocal_space_shell will generate points on a sphere of a given radius. This is useful for powder averaging. ","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"A number of arguments for intensity_formula are available which modify the calculation of structure factor intensity. It is generally recommended to provide a value of kT corresponding to the temperature of sampled configurations. Given kT, Sunny will include an energy- and temperature-dependent classical-to-quantum rescaling of intensities in the formula.","category":"page"},{"location":"structure-factor.html","page":"Structure Factor Calculations","title":"Structure Factor Calculations","text":"To retrieve intensity data from a instantaneous structure factor, use instant_intensities_interpolated, which accepts similar arguments to intensities_interpolated. This function may also be used to calculate instantaneous information from a dynamical correlation data, i.e. from a SampledCorrelations created with dynamical_correlations. Note that it is important to supply a value to kT to reap the benefits of this approach over simply calculating a static structure factor at the outset. ","category":"page"},{"location":"writevtk.html#ParaView-Rendering","page":"ParaView Rendering","title":"ParaView Rendering","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The 4D correlation data produced by Sunny is too high-dimensional to visualize directly. This page describes how to export 3D slices of correlation data from Sunny to the Visual ToolKit (VTK) format, which is compatible with the ParaView visualization software. ParaView supports volumetric rendering:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":" ","category":"page"},{"location":"writevtk.html#Simulation-data","page":"ParaView Rendering","title":"Simulation data","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"First, generate some correlation data in Sunny. We will use a 2D lattice, since the correlation data S(Q_xQ_yomega) is 3D and can be exported in its entirety. The following code sets up the system, thermalizes it, and records the correlation data in a SampledCorrelations called dsf.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"using Sunny\n\n# Single layer 12x12 periodic square lattice\nlatsize = (12,12,1)\n\nlatvecs = lattice_vectors(8.,8.,12.,90,100,90)\npositions = [[0,0,0]]\ntypes = [\"Cu\"]\nformfactors = [FormFactor(\"Cu2\")]\nxtal = Crystal(latvecs, positions; types)\n\nsys = System(xtal, latsize, [SpinInfo(1, S=1/2, g=2)], :SUN; seed=1)\n\nJ = 10.\nset_exchange!(sys, J, Bond(1,1,[1,0,0]))\nset_exchange!(sys, J, Bond(1,1,[0,1,0]))\n\nΔt = 0.01\nkT = 0.5\nlangevin = Langevin(Δt; λ=0.5, kT)\nrandomize_spins!(sys)\nfor i in 1:10_000 # Long enough to reach equilibrium\n step!(sys, langevin)\nend \n\nωmax=10.\n\ndsf = dynamical_correlations(sys\n ;Δt=Δt\n ,nω=48\n ,ωmax=ωmax\n ,process_trajectory=:symmetrize)\n\nnsamples = 10\nfor _ in 1:nsamples\n for _ in 1:1000 \n step!(sys, langevin)\n end\n add_sample!(dsf, sys)\nend","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The default histogram BinningParameters are already integrated over the z direction because the system is 2D:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"unit_resolution_binning_parameters(dsf)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 12 bins from -0.042 to +0.958 along [+1.27 dx] (Δ = 0.065)\n⊡ 12 bins from -0.042 to +0.958 along [+1.27 dy] (Δ = 0.065)\n∫ Integrated from +0.000 to +0.000 along [-0.33 dx +1.88 dz] (Δ = 0.524)\n⊡ 48 bins from -0.107 to +10.134 along [+1.00 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The histogram is very oblong; it's approximately 1x1x10. To make it a nicer shape, we will rescale the energy axis to be be fractions of ωmax:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"params = unit_resolution_binning_parameters(dsf)\nscale_factor = ωmax\nparams.binend[4] /= scale_factor\nparams.binstart[4] /= scale_factor\nparams.binwidth[4] /= scale_factor\nparams.covectors[4,:] ./= scale_factor","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Doing this changes the last axis of the histogram to fit in [0,1]:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 49 bins from -0.011 to +1.013 along [+0.10 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that our histogram is a cube, we compute the intensity in the histogram bins using the usual intensities_binned:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"formula = intensity_formula(dsf,:trace)\nsignal, counts = intensities_binned(dsf, params; formula)\nintensity = signal ./ counts","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that we have our intensity data and the binning parameters, we can export to VTK format using export_vtk and move to ParaView for the visualization.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"# Importing WriteVTK enables Sunny's export-to-VTK functions\nimport WriteVTK\n\n# [1,2,4] specifies that the (x,y,z) axes in ParaView are (Qx,Qy,ω)\nexport_vtk(\"square_lattice\", params, intensity; dims_kept = [1,2,4])\n# Writes a file square_lattice.vti in the current directory","category":"page"},{"location":"writevtk.html#Loading-in-ParaView","page":"ParaView Rendering","title":"Loading in ParaView","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In ParaView, use File > Open to open square_lattice.vti. This will add the file to the Pipeline Browser with a closed eye icon, indicating that the data is ready to be loaded.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In the Properties panel, both bin_centers and data will be selected for import by default. Uncheck bin_centers because we don't need that information for the visualization. Click the green Apply button to load the data.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"By default, only the outline of the data is shown in the 3D viewport. Since we adjusted the energy axis, the outline is a 1x1x1 cube. Optionally enable the axes grid under \"View\", and customize using the adjacent edit button.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"writevtk.html#Simulation-data","page":"ParaView Rendering","title":"Simulation data","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"First, generate some correlation data in Sunny. We will use a 2D lattice, since the correlation data S(Q_xQ_yomega) is 3D and can be exported in its entirety. The following code sets up the system, thermalizes it, and records the correlation data in a SampledCorrelations called dsf.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"using Sunny\n\n# Single layer 12x12 periodic square lattice\nlatsize = (12,12,1)\n\nlatvecs = lattice_vectors(8.,8.,12.,90,100,90)\npositions = [[0,0,0]]\ntypes = [\"Cu\"]\nformfactors = [FormFactor(\"Cu2\")]\nxtal = Crystal(latvecs, positions; types)\n\nsys = System(xtal, latsize, [SpinInfo(1, S=1/2, g=2)], :SUN; seed=1)\n\nJ = 10.\nset_exchange!(sys, J, Bond(1,1,[1,0,0]))\nset_exchange!(sys, J, Bond(1,1,[0,1,0]))\n\nΔt = 0.01\nkT = 0.5\nlangevin = Langevin(Δt; λ=0.5, kT)\nrandomize_spins!(sys)\nfor i in 1:10_000 # Long enough to reach equilibrium\n step!(sys, langevin)\nend \n\nωmax=10.\n\ndsf = dynamical_correlations(sys\n ;Δt=Δt\n ,nω=48\n ,ωmax=ωmax\n ,process_trajectory=:symmetrize)\n\nnsamples = 10\nfor _ in 1:nsamples\n for _ in 1:1000 \n step!(sys, langevin)\n end\n add_sample!(dsf, sys)\nend","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The default histogram BinningParameters are already integrated over the z direction because the system is 2D:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"unit_resolution_binning_parameters(dsf)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 12 bins from -0.042 to +0.958 along [+1.27 dx] (Δ = 0.065)\n⊡ 12 bins from -0.042 to +0.958 along [+1.27 dy] (Δ = 0.065)\n∫ Integrated from +0.000 to +0.000 along [-0.33 dx +1.88 dz] (Δ = 0.524)\n⊡ 48 bins from -0.107 to +10.134 along [+1.00 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The histogram is very oblong; it's approximately 1x1x10. To make it a nicer shape, we will rescale the energy axis to be be fractions of ωmax:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"params = unit_resolution_binning_parameters(dsf)\nscale_factor = ωmax\nparams.binend[4] /= scale_factor\nparams.binstart[4] /= scale_factor\nparams.binwidth[4] /= scale_factor\nparams.covectors[4,:] ./= scale_factor","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Doing this changes the last axis of the histogram to fit in [0,1]:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 49 bins from -0.011 to +1.013 along [+0.10 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that our histogram is a cube, we compute the intensity in the histogram bins using the usual intensities_binned:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"formula = intensity_formula(dsf,:trace)\nsignal, counts = intensities_binned(dsf, params; formula)\nintensity = signal ./ counts","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that we have our intensity data and the binning parameters, we can export to VTK format using export_vtk and move to ParaView for the visualization.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"# Importing WriteVTK enables Sunny's export-to-VTK functions\nimport WriteVTK\n\n# [1,2,4] specifies that the (x,y,z) axes in ParaView are (Qx,Qy,ω)\nexport_vtk(\"square_lattice\", params, intensity; dims_kept = [1,2,4])\n# Writes a file square_lattice.vti in the current directory","category":"page"},{"location":"writevtk.html#Loading-in-ParaView","page":"ParaView Rendering","title":"Loading in ParaView","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In ParaView, use File > Open to open square_lattice.vti. This will add the file to the Pipeline Browser with a closed eye icon, indicating that the data is ready to be loaded.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In the Properties panel, both bin_centers and data will be selected for import by default. Uncheck bin_centers because we don't need that information for the visualization. Click the green Apply button to load the data.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"By default, only the outline of the data is shown in the 3D viewport. Since we adjusted the energy axis, the outline is a 1x1x1 cube. Optionally enable the axes grid under \"View\", and customize using the adjacent edit button.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":" ","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"To enable the volumetric render:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Select \"Volume\" from the \"Representation\" drop-down menu under \"Display\".\nThe \"Coloring\" drop-down should automatically select data because it's the only data loaded.\nOpen the Color Map Editor to adjust the opacity of the fog, which may be too faint to see by default.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"To enable the volumetric render:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Select \"Volume\" from the \"Representation\" drop-down menu under \"Display\".\nThe \"Coloring\" drop-down should automatically select data because it's the only data loaded.\nOpen the Color Map Editor to adjust the opacity of the fog, which may be too faint to see by default.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":" ","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Depending on your computer and your dataset size, the volumetric rendering may be slow, but our dataset is relatively small, so the render should be fast.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"If nothing shows up at first, don't despair. Often, there are Bragg-like peaks in the correlation data which outshine everything else. To see this, enable Display Data Histogram in the Color Map Editor panel. To zoom in on the lower-intensity data, click and drag the right side handle of the opacity transfer function box to the middle a few times.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Depending on your computer and your dataset size, the volumetric rendering may be slow, but our dataset is relatively small, so the render should be fast.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"If nothing shows up at first, don't despair. Often, there are Bragg-like peaks in the correlation data which outshine everything else. To see this, enable Display Data Histogram in the Color Map Editor panel. To zoom in on the lower-intensity data, click and drag the right side handle of the opacity transfer function box to the middle a few times.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":" ","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"After suitable color mapping, the dispersion curve should become visible:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"After suitable color mapping, the dispersion curve should become visible:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":" ","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"EditURL = \"../../../examples/out_of_equilibrium.jl\"","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/out_of_equilibrium.html#CP-Skyrmion-Quench","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"","category":"section"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"This example demonstrates Sunny's ability to simulate the out-of-equilibrium dynamics of generalized spin systems. We will implement the model Hamiltonian of Zhang et al., Nature Communications 14, 3626 (2023), which supports a novel type of topological defect, a CP² skyrmion, that involves both the dipolar and quadrupolar parts of a quantum spin.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Beginning from an initial high-temperature state, a disordered gas of CP² skyrmions can be formed by rapidly quenching to low temperature. To model the coupled dynamics of dipoles and quadrupoles, Sunny uses a recently developed generalization of the Landau-Lifshitz spin dynamics, Dahlbom et al., Phys. Rev. B 106, 235154 (2022).","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The Hamiltonian we will implement,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"mathcalH = sum_langle ij rangle J_ij( hatS_i^x hatS_j^x + hatS_i^y hatS_j^y + DeltahatS_i^z hatS_j^z) - hsum_ihatS_i^z + Dsum_i(hatS_i^z)^2","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"contains competing ferromagnetic nearest-neightbor and antiferromagnetic next-nearest-neighbor exchange terms on a triangular lattice. Both exchanges exhibit anisotropy on the z-term. Additionally, there is an external magnetic field, h, and easy-plane single-ion anisotropy, D 0. We begin by implementing the Crystal.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"lat_vecs = lattice_vectors(1.0, 1.0, 2.0, 90, 90, 120)\nbasis_vecs = [[0,0,0]]\ncryst = Crystal(lat_vecs, basis_vecs)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The crystal is then used to create a spin System. All parameters in this model system are dimensionless, so we select \"theory\" units and set the g-factor to one.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"L = 40\ndims = (L, L, 1)\nsys = System(cryst, dims, [SpinInfo(1, S=1, g=1)], :SUN; seed=101, units=Units.theory)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"We proceed to implement each term of the Hamiltonian, selecting our parameters so that the system occupies a region of the phase diagram that supports skyrmions. The exchange interactions are set as follows.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"J1 = -1 # Nearest-neighbor ferromagnetic\nJ2 = (2.0/(1+√5)) # Tune competing exchange to set skyrmion scale length\nΔ = 2.6 # Exchange anisotropy\n\nex1 = J1 * [1.0 0.0 0.0;\n 0.0 1.0 0.0;\n 0.0 0.0 Δ]\nex2 = J2 * [1.0 0.0 0.0;\n 0.0 1.0 0.0;\n 0.0 0.0 Δ]\nset_exchange!(sys, ex1, Bond(1, 1, [1, 0, 0]))\nset_exchange!(sys, ex2, Bond(1, 1, [1, 2, 0]))","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Next we add the external field,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"h = 15.5\nfield = set_external_field!(sys, [0.0 0.0 h])","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"and finally an easy-plane single-ion anisotropy,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"D = 19.0\nset_onsite_coupling!(sys, S -> D*S[3]^2, 1)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Initialize system to an infinite temperature (fully randomized) initial condition.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"randomize_spins!(sys)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"We are now ready to simulate the quenching process using a generalized Langevin spin dynamics. If we were working with spin dipoles only, then Langevin dynamics would be the usual Landau-Lifshitz spin dynamics, augmented with damping and noise terms. In the present study, we are instead working with quantum spin-1 (an (N=3)-level system that includes both dipoles and quadrupoles). Here, Langevin captures the coupled dipole-quadrupole dynamics using the formalism of SU(N) coherent states.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Selecting kT = 0 in the Langevin dynamics will effective disable the noise term. Then the parameter λ effectively determines the damping time-scale.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Δt = 0.2/D # Integration time step (inverse meV). Typically this will be\n # inversely proportional to the largest energy scale in the\n # system. We can use a fairly large time-step here because\n # accuracy isn't critical.\nkT = 0 # Target equilibrium temperature (meV)\nλ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)\nintegrator = Langevin(Δt; kT, λ)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Finally we run the dynamics. We will record the state of the system at three different times during the quenching process by copying the coherents field of the System.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"τs = [4., 16, 256] # Times to record snapshots\nframes = [] # Empty array to store snapshots\nfor i in eachindex(τs)\n dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate\n numsteps = round(Int, dur/Δt)\n for _ in 1:numsteps # Perform the integration\n step!(sys, integrator)\n end\n push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration\nend","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"To visualize the state of the system contained in each snapshot, we will calculate and plot the skyrmion density on each plaquette of our lattice. The function plot_triangular_plaquettes is not part of the core Sunny package, but rather something you could define yourself. We are using the definition in plotting2d.jl from the Sunny examples/extra directory.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"include(pkgdir(Sunny, \"examples\", \"extra\", \"plotting2d.jl\"))\n\nfunction sun_berry_curvature(z₁, z₂, z₃)\n z₁, z₂, z₃ = normalize.((z₁, z₂, z₃))\n n₁ = z₁ ⋅ z₂\n n₂ = z₂ ⋅ z₃\n n₃ = z₃ ⋅ z₁\n return angle(n₁ * n₂ * n₃)\nend\n\nplot_triangular_plaquettes(sun_berry_curvature, frames; resolution=(1800,600),\n offset_spacing=10, texts = [\"\\tt = \"*string(τ) for τ in τs], text_offset = (0.0, 6.0)\n)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The times are given in hbarJ_1. The white background corresponds to a quantum paramagnetic state, where the local spin exhibits a strong quadrupole moment and little or no dipole moment. Observe that the process has generated a number of well-formed skyrmions of both positive (red) and negative (blue) charge in addition to a number of other metastable spin configurations. A full-sized version of this figure is available in Dahlbom et al..","category":"page"},{"location":"library.html#Library-API","page":"Library API","title":"Library API","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"This page describes the public types and functions exported by Sunny. This documentation can be also be accessed using the Julia help system (enter ? at the Julia command prompt).","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"Sunny.plot_spins\nSunny.view_crystal\nSunny.export_vtk","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"Modules = [Sunny]\nPrivate = false","category":"page"},{"location":"library.html#Sunny.Site","page":"Library API","title":"Sunny.Site","text":"(cell1, cell2, cell3, i) :: Site\n\nFour indices identifying a single site in a System. The first three indices select the lattice cell and the last selects the sublattice (i.e., the atom within the unit cell).\n\nThis object can be used to index dipoles and coherents fields of a System. A Site is also required to specify inhomogeneous interactions via functions such as set_external_field_at! or set_exchange_at!.\n\nNote that the definition of a cell may change when a system is reshaped. In this case, it is convenient to construct the Site using position_to_site, which always takes a position in fractional coordinates of the original lattice vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Units","page":"Library API","title":"Sunny.Units","text":"Units.meV\nUnits.theory\n\nThe unit system is implicitly determined by the definition of two physical constants: the vacuum permeability μ₀ and the Bohr magneton μ_B. Temperatures are effectively measured in units of energy (k_B = 1) and time is effectively measured in units of inverse energy (ħ = 1). The default unit system, Units.meV, employs (meV, Å, tesla). Select alternatively Units.theory for a units system defined so that μ₀ = μ_B = 1.\n\nSee also meV_per_K\n\n\n\n\n\n","category":"constant"},{"location":"library.html#Sunny.meV_per_K","page":"Library API","title":"Sunny.meV_per_K","text":"meV_per_K = 0.086173332621451774\n\nA physical constant. Useful for converting kelvin into the default energy units, meV.\n\n\n\n\n\n","category":"constant"},{"location":"library.html#Sunny.BinningParameters","page":"Library API","title":"Sunny.BinningParameters","text":"BinningParameters(binstart,binend,binwidth;covectors = I(4))\nBinningParameters(binstart,binend;numbins,covectors = I(4))\n\nDescribes a 4D parallelepided histogram in a format compatible with experimental Inelasitic Neutron Scattering data. See generate_mantid_script_from_binning_parameters to convert BinningParameters to a format understandable by the Mantid software, or load_nxs to load BinningParameters from a Mantid .nxs file.\n\nThe coordinates of the histogram axes are specified by multiplication of (q,ω) with each row of the covectors matrix, with q given in [R.L.U.]. Since the default covectors matrix is the identity matrix, the default axes are (qx,qy,qz,ω) in absolute units.\n\nThe convention for the binning scheme is that:\n\nThe left edge of the first bin starts at binstart\nThe bin width is binwidth\nThe last bin contains binend\nThere are no \"partial bins;\" the last bin may contain values greater than binend. C.f. count_bins.\n\nA value can be binned by computing its bin index:\n\ncoords = covectors * value\nbin_ix = 1 .+ floor.(Int64,(coords .- binstart) ./ binwidth)\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Bond","page":"Library API","title":"Sunny.Bond","text":"Bond(i, j, n)\n\nRepresents a bond between atom indices i and j. n is a vector of three integers specifying unit cell displacement in terms of lattice vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Crystal","page":"Library API","title":"Sunny.Crystal","text":"An object describing a crystallographic unit cell and its space group symmetry. Constructors are as follows:\n\nCrystal(filename; symprec=1e-5)\n\nReads the crystal from a .cif file located at the path filename. The optional parameter symprec controls the precision tolerance for spacegroup symmetries.\n\nCrystal(latvecs, positions; types=nothing, symprec=1e-5)\n\nConstructs a crystal from the complete list of atom positions positions, with coordinates (between 0 and 1) in units of lattice vectors latvecs. Spacegroup symmetry information is automatically inferred. The optional parameter types is a list of strings, one for each atom, and can be used to break symmetry-equivalence between atoms.\n\nCrystal(latvecs, positions, spacegroup_number; types=nothing, setting=nothing, symprec=1e-5)\n\nBuilds a crystal by applying symmetry operators for a given international spacegroup number. For certain spacegroups, there are multiple possible unit cell settings; in this case, a warning message will be printed, and a list of crystals will be returned, one for every possible setting. Alternatively, the optional setting string will disambiguate between unit cell conventions.\n\nCurrently, crystals built using only the spacegroup number will be missing some symmetry information. It is generally preferred to build a crystal from a .cif file or from the full specification of the unit cell.\n\nExamples\n\n# Read a Crystal from a .cif file\nCrystal(\"filename.cif\")\n\n# Build an FCC crystal using the primitive unit cell. The spacegroup number\n# 225 is inferred.\nlatvecs = [1 1 0;\n 1 0 1;\n 0 1 1] / 2\npositions = [[0, 0, 0]]\nCrystal(latvecs, positions)\n\n# Build a CsCl crystal (two cubic sublattices). By providing distinct type\n# strings, the spacegroup number 221 is inferred.\nlatvecs = lattice_vectors(1, 1, 1, 90, 90, 90)\npositions = [[0,0,0], [0.5,0.5,0.5]]\ntypes = [\"Na\", \"Cl\"]\ncryst = Crystal(latvecs, positions; types)\n\n# Build a diamond cubic crystal from its spacegroup number 227. This\n# spacegroup has two possible settings (\"1\" or \"2\"), which determine an\n# overall unit cell translation.\nlatvecs = lattice_vectors(1, 1, 1, 90, 90, 90)\npositions = [[1, 1, 1] / 4]\ncryst = Crystal(latvecs, positions, 227; setting=\"1\")\n\nSee also lattice_vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.FormFactor-Tuple{String}","page":"Library API","title":"Sunny.FormFactor","text":"FormFactor(ion::String; g_lande=2)\n\nThe magnetic form factor for a given magnetic ion and charge state. When passed to an intensity_formula, determines a 𝐪-dependent scaling of the structure factor.\n\nThe parameter ion must be one of the following strings:\n\nAm2, Am3, Am4, Am5, Am6, Am7, Au1, Au2, Au3, Au4, Au5, Ce2, Co0, Co1, Co2, Co3,\nCo4, Cr0, Cr1, Cr2, Cr3, Cr4, Cu0, Cu1, Cu2, Cu3, Cu4, Dy2, Dy3, Er2, Er3, Eu2,\nEu3, Fe0, Fe1, Fe2, Fe3, Fe4, Gd2, Gd3, Hf2, Hf3, Ho2, Ho3, Ir0a, Ir0b, Ir0c,\nIr1a, Ir1b, Ir2, Ir3, Ir4, Ir5, Ir6, Mn0, Mn1, Mn2, Mn3, Mn4, Mo0, Mo1, Nb0,\nNb1, Nd2, Nd3, Ni0, Ni1, Ni2, Ni3, Ni4, Np3, Np4, Np5, Np6, Os0a, Os0b, Os0c,\nOs1a, Os1b, Os2, Os3, Os4, Os5, Os6, Os7, Pd0, Pd1, Pr3, Pt1, Pt2, Pt3, Pt4,\nPt5, Pt6, Pu3, Pu4, Pu5, Pu6, Re0a, Re0b, Re0c, Re1a, Re1b, Re2, Re3, Re4, Re5,\nRe6, Rh0, Rh1, Ru0, Ru1, Sc0, Sc1, Sc2, Sm2, Sm3, Ta2, Ta3, Ta4, Tb2, Tb3, Tc0,\nTc1, Ti0, Ti1, Ti2, Ti3, Tm2, Tm3, U3, U4, U5, V0, V1, V2, V3, V4, W0a, W0b,\nW0c, W1a, W1b, W2c, W3, W4, W5, Y0, Yb2, Yb3, Zr0, Zr1\n\nThe trailing number denotes ionization state. For example, \"Fe0\" denotes a neutral iron atom, while \"Fe2\" denotes Fe²⁺. If multiple electronic configurations are possible, they will be distinguished by a trailing letter (a, b, ...). Omitting this letter will print an informative error,\n\nFormFactor(\"Ir0\")\n\nERROR: Disambiguate form factor according to electronic configuration:\n \"Ir0a\" -- 6s⁰5d⁹\n \"Ir0b\" -- 6s¹5d⁸\n \"Ir0c\" -- 6s²5d⁷\n\nThe form factor is approximated as\n\nF(s) = j_0(s) + frac2-gg j_2(s) s^2,\n\ninvolving the Landé g-factor. The j_l(s) are radial integrals associated with the lth Bessel function of the magnetic dipole, where s = k4π, and k is the magnitude of momentum transfer. \n\nThe radial integrals have been calculated using Hartree-Fock for transition metals, or Dirac-Fock for the rare earths and actinide series [1–3]. Sunny uses approximate fits as a sum of Gaussians,\n\nj_0(s) = A e^-as^2 + B e^-bs^2 + C e^-cs^2 + D e^-ds^2 + E \nj_l(s) = (A e^-as^2 + B e^-bs^2 + C e^-cs^2 + D e^-ds^2 + E) s^2\n\nReferences:\n\nhttps://www.ill.eu/sites/ccsl/ffacts/ffachtml.html\nJ. Brown, The Neutron Data Booklet, 2nd ed., Sec. 2.5 Magnetic Form Factors (2003)\nK. Kobayashi, T. Nagao, M. Ito, Acta Cryst. A, 67 pp 473–480 (2011)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.ImplicitMidpoint","page":"Library API","title":"Sunny.ImplicitMidpoint","text":"ImplicitMidpoint(Δt::Float64; atol=1e-12) where N\n\nEnergy-conserving spin dynamics. One call to the step! function will advance a System by Δt units of time.\n\nUses the spherical midpoint integration scheme for dipole systems and the Schrödinger midpoint integration scheme for SU(N) spin systems. Both integration schemes are symplectic, and therefore avoid energy drift over long periods of simulation time.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Langevin","page":"Library API","title":"Sunny.Langevin","text":"Langevin(Δt::Float64; λ::Float64, kT::Float64)\n\nSpin dynamics with coupling to a Langevin thermostat, which includes damping and noise terms. One call to the step! function will advance a System by Δt units of time.\n\nAssuming ergodicity, the Langevin dynamics will sample from thermal equilibrium for the target temperature kT. The empirical parameter λ determines the strength of the coupling to the thermal bath. In other words, 1/λ is the decorrelation time-scale. If λ = 0, then the spin dynamics coincides with ImplicitMidpoint.\n\nAn alternative approach to sampling is LocalSampler, which may be preferred when the allowed spin values become effective discrete (e.g. Ising spins).\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.LocalSampler","page":"Library API","title":"Sunny.LocalSampler","text":"LocalSampler(; kT, nsweeps=1.0, propose=propose_uniform)\n\nMonte Carlo simulation involving Metropolis updates to individual spins. One call to the step! function will perform nsweeps of MCMC sampling for a provided System. The default value of 1.0 means that step! performs, on average, one trial update per spin.\n\nAssuming ergodicity, the LocalSampler will sample from thermal equilibrium for the target temperature kT. \n\nThe trial spin updates are sampled using the propose function. Built-in options include propose_uniform, propose_flip, and propose_delta. Multiple proposals can be mixed with the macro @mix_proposals.\n\nThe returned object stores fields ΔE and Δs, which represent the cumulative change to the net energy and dipole, respectively.\n\nAn alternative approach to sampling is Langevin, which may be preferred for simulating continuous spins, especially in the presence of long-range dipole-dipole interactions (cf. enable_dipole_dipole!).\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SampledCorrelations","page":"Library API","title":"Sunny.SampledCorrelations","text":"SampledCorrelations\n\nBasic data type for storing sampled correlation data. A SampleCorrelations is initialized by calling either dynamical_correlations or instant_correlations.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SpinInfo","page":"Library API","title":"Sunny.SpinInfo","text":"SpinInfo(atom::Int; S, g=2)\n\nCharacterizes the spin at a given atom index within the crystal unit cell. S is an integer multiple of 1/2 and gives the spin angular momentum in units of ħ. g is the g-factor or tensor, such that an angular momentum dipole s produces a magnetic moment g s in units of the Bohr magneton.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SpinWaveTheory","page":"Library API","title":"Sunny.SpinWaveTheory","text":"SpinWaveTheory(sys, energy_ϵ::Float64=1e-8)\n\nConstructs an object to perform linear spin wave theory. Use it with dispersion and dssf functions.\n\nThe optional parameter energy_ϵ adds a small positive shift to the diagonal of the dynamical matrix D to avoid numerical issues with zero-energy quasi-particle modes.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.System-Tuple{Crystal, Tuple{Int64, Int64, Int64}, Vector{SpinInfo}, Symbol}","page":"Library API","title":"Sunny.System","text":"System(crystal::Crystal, latsize, infos, mode; units=Units.meV, seed::Int)\n\nConstruct a System of spins for a given Crystal symmetry. The latsize parameter determines the number of unit cells in each lattice vector direction. The infos parameter is a list of SpinInfo objects, which determine the magnitude S and g-tensor of each spin.\n\nThe two primary options for mode are :SUN and :dipole. In the former, each spin-S degree of freedom is described as an SU(N) coherent state, i.e. a quantum superposition of N = 2S + 1 levels. This formalism can be useful to capture multipolar spin fluctuations or local entanglement effects. \n\nMode :dipole projects the SU(N) dynamics onto the restricted space of pure dipoles. In practice this means that Sunny will simulate Landau-Lifshitz dynamics, but single-ion anisotropy and biquadratic exchange interactions will be renormalized to improve accuracy. To disable this renormalization, use the mode :dipole_large_S which applies the S classical limit. For details, see the documentation page: Interaction Strength Renormalization.\n\nThe default units system of (meV, Å, tesla) can be overridden by with the units parameter; see Units. \n\nAn optional seed may be provided to achieve reproducible random number generation.\n\nAll spins are initially polarized in the z-direction.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.add_sample!-Tuple{SampledCorrelations, System}","page":"Library API","title":"Sunny.add_sample!","text":"add_sample!(sc::SampledCorrelations, sys::System)\n\nadd_trajectory uses the spin configuration contained in the System to generate a correlation data and accumulate it into sc. For static structure factors, this involves analyzing the spin-spin correlations of the spin configuration provided. For a dynamic structure factor, a trajectory is calculated using the given spin configuration as an initial condition. The spin-spin correlations are then calculating in time and accumulated into sc. \n\nThis function will change the state of sys when calculating dynamical structure factor data. To preserve the initial state of sys, it must be saved separately prior to calling add_sample!. Alternatively, the initial spin configuration may be copied into a new System and this new System can be passed to add_sample!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.available_energies-Tuple{SampledCorrelations}","page":"Library API","title":"Sunny.available_energies","text":"available_energies(sc::SampledCorrelations; negative_energies=false)\n\nReturn the ω values for the energy index of a SampledCorrelations. By default, only returns values for non-negative energies, which corresponds to the default output of intensities. Set negative_energies to true to retrieve all ω values.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.available_wave_vectors-Tuple{SampledCorrelations}","page":"Library API","title":"Sunny.available_wave_vectors","text":"available_wave_vectors(sc::SampledCorrelations; bzsize=(1,1,1))\n\nReturns all wave vectors for which sc contains exact values. bsize specifies the number of Brillouin zones to be included.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.axes_bincenters-Tuple{Any, Any, Any}","page":"Library API","title":"Sunny.axes_bincenters","text":"axes_bincenters(params::BinningParameters)\n\nReturns tick marks which label the bins of the histogram described by BinningParameters by their bin centers.\n\nThe following alternative syntax can be used to compute bin centers for a single axis:\n\naxes_bincenters(binstart,binend,binwidth)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.broaden_energy-Tuple{SampledCorrelations, Any, Function}","page":"Library API","title":"Sunny.broaden_energy","text":"broaden_energy(sc::SampledCorrelations, vals, kernel::Function; negative_energies=false)\n\nPerforms a real-space convolution along the energy axis of an array of intensities. Assumes the format of the intensities array corresponds to what would be returned by intensities_interpolated. kernel must be a function that takes two numbers: kernel(ω, ω₀), where ω is a frequency, and ω₀ is the center frequency of the kernel. Sunny provides lorentzian for the most common use case:\n\nnewvals = broaden_energy(sc, vals, (ω, ω₀) -> lorentzian(ω-ω₀, 0.2))\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.count_bins-Tuple{Any, Any, Any}","page":"Library API","title":"Sunny.count_bins","text":"count_bins(binstart,binend,binwidth)\n\nReturns the number of bins in the binning scheme implied by binstart, binend, and binwidth. To count the bins in a BinningParameters, use params.numbins.\n\nThis function defines how partial bins are handled, so it should be used preferentially over computing the number of bins manually.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dispersion-Tuple{SpinWaveTheory, Any}","page":"Library API","title":"Sunny.dispersion","text":"dispersion(swt::SpinWaveTheory, qs)\n\nComputes the spin excitation energy dispersion relations given a SpinWaveTheory and an array of wave vectors qs. Each element q of qs must be a 3-vector in units of reciprocal lattice units. I.e., qᵢ is given in 2πaᵢ with aᵢ the lattice constant of the original chemical lattice.\n\nThe first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of the array is an energy.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dmvec-Tuple{Any}","page":"Library API","title":"Sunny.dmvec","text":"dmvec(D)\n\nAntisymmetric matrix representation of the Dzyaloshinskii-Moriya pseudo-vector,\n\n [ 0 D[3] -D[2]\n -D[3] 0 D[1]\n D[2] -D[1] 0 ]\n\nUseful in the context of set_exchange!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dssf-Tuple{SpinWaveTheory, Any}","page":"Library API","title":"Sunny.dssf","text":"dssf(swt::SpinWaveTheory, qs)\n\nGiven a SpinWaveTheory object, computes the dynamical spin structure factor,\n\n 𝒮^αβ(𝐤 ω) = 1(2πN)dt _𝐫 expi(ωt - 𝐤𝐫) S^α(𝐫 t)S^β(0 0)\n\nusing the result from linear spin-wave theory,\n\n 𝒮^αβ(𝐤 ω) = _n A_n^αβ(𝐤)^2 δω-ω_n(𝐤)\n\nqs is an array of wave vectors of arbitrary dimension. Each element q of qs must be a 3-vector in reciprocal lattice units (RLU), i.e., in the basis of reciprocal lattice vectors.\n\nThe first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of this array is a tensor (3×3 matrix) corresponding to the indices α and β.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dynamical_correlations-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.dynamical_correlations","text":"dynamical_correlations(sys::System; Δt, nω, ωmax, \n process_trajectory=:none, observables=nothing, correlations=nothing)\n\nCreates an empty SampledCorrelations object for calculating and storing dynamical structure factor intensities 𝒮(𝐪ω). Call add_sample! to accumulate data for the given configuration of a spin system. Internally, this will run a dynamical trajectory and measure time correlations. The 𝒮(𝐪ω) data can be retrieved by calling intensities_interpolated. Alternatively, instant_intensities_interpolated will integrate out ω to obtain 𝒮(𝐪), optionally applying classical-to-quantum correction factors.\n\nThree keywords are required to specify the dynamics used for the trajectory calculation.\n\nΔt: The time step used for calculating the trajectory from which dynamic spin-spin correlations are calculated. The trajectories are calculated with an ImplicitMidpoint integrator.\nωmax: The maximum energy, ω, that will be resolved.\nnω: The number of energy bins to calculated between 0 and ωmax.\n\nAdditional keyword options are the following:\n\nobservables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.\ncorrelations: Specify which correlation functions are calculated, i.e. which matrix elements αβ of 𝒮^αβ(qω) are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.eachsite-Tuple{System}","page":"Library API","title":"Sunny.eachsite","text":"eachsite(sys::System)\n\nAn iterator over all Sites in the system. \n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.enable_dipole_dipole!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.enable_dipole_dipole!","text":"enable_dipole_dipole!(sys::System)\n\nEnables long-range dipole-dipole interactions,\n\n -(μ_04π) _ij (3 (𝐌_j𝐫_ij)(𝐌_i𝐫_ij) - 𝐌_i𝐌_j) 𝐫_ij^3\n\nwhere the sum is over all pairs of spins (singly counted), including periodic images, regularized using the Ewald summation convention. The magnetic moments are 𝐌_i = μ_B g 𝐒_i where g is the g-factor or g-tensor, and 𝐒_i is the spin angular momentum dipole in units of ħ. The Bohr magneton μ_B and vacuum permeability μ_0 are physical constants, with numerical values determined by the unit system.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.energy-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.energy","text":"energy(sys::System)\n\nThe total system energy. See also energy_per_site.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.energy_per_site-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.energy_per_site","text":"energy_per_site(sys::System)\n\nThe total system energy divided by the number of sites.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.generate_mantid_script_from_binning_parameters-Tuple{Any}","page":"Library API","title":"Sunny.generate_mantid_script_from_binning_parameters","text":"generate_mantid_script_from_binning_parameters(params::BinningParameters)\n\nGenerate a Mantid script which bins data according to the given BinningParameters.\n\nwarning: Units\nTake care to ensure the units are correct (R.L.U. or absolute). You may want to call Sunny.bin_rlu_as_absolute_units! or Sunny.bin_absolute_units_as_rlu! first.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.global_position-Tuple{System, Any}","page":"Library API","title":"Sunny.global_position","text":"global_position(sys::System, site::Site)\n\nPosition of a Site in global coordinates.\n\nTo precompute a full list of positions, one can use eachsite as below:\n\npos = [global_position(sys, site) for site in eachsite(sys)]\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.instant_correlations-Tuple{System}","page":"Library API","title":"Sunny.instant_correlations","text":"instant_correlations(sys::System; process_trajectory=:none, observables=nothing, correlations=nothing)\n\nCreates an empty SampledCorrelations object for calculating and storing instantaneous structure factor intensities 𝒮(𝐪). Call add_sample! to accumulate data for the given configuration of a spin system. Call instant_intensities_interpolated to retrieve averaged 𝒮(𝐪) data.\n\nImportant note: When dealing with continuous (non-Ising) spins, consider creating using dynamical_correlations instead of instant_correlations. The former will provide full 𝒮(𝐪ω) data, from which 𝒮(𝐪) can be obtained by integrating out ω. During this integration step, Sunny can incorporate temperature- and ω-dependent classical-to-quantum correction factors to produce more accurate 𝒮(𝐪) estimates. See instant_intensities_interpolated for more information.\n\nThe following optional keywords are available:\n\nobservables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.\ncorrelations: Specify which correlation functions are calculated, i.e. which matrix elements αβ of 𝒮^αβ(qω) are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.instant_intensities_interpolated-Tuple{SampledCorrelations, Any, Any}","page":"Library API","title":"Sunny.instant_intensities_interpolated","text":"instant_intensities_interpolated(sc::SampledCorrelations, qs; kwargs...)\n\nReturn 𝒮(𝐪) intensities at wave vectors qs. The functionality is very similar to intensities_interpolated, except the returned array has dimensions identical to qs. If called on a SampledCorrelations with dynamical information, i.e., 𝒮(𝐪ω), the ω information is integrated out.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.integrate_axes!-Tuple{BinningParameters}","page":"Library API","title":"Sunny.integrate_axes!","text":"integrate_axes!(params::BinningParameters; axes)\n\nIntegrate over one or more axes of the histogram by setting the number of bins in that axis to 1. Examples:\n\nintegrate_axes!(params; axes = [2,3])\nintegrate_axes!(params; axes = 2)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.integrated_lorentzian-Tuple{Float64}","page":"Library API","title":"Sunny.integrated_lorentzian","text":"integrated_lorentzian(η)\n\nReturns x mapsto atan(xη)π for use with intensities_binned.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_bands-Tuple{SpinWaveTheory, Any, Sunny.SpinWaveIntensityFormula}","page":"Library API","title":"Sunny.intensities_bands","text":"dispersion, intensities = intensities_bands(swt::SpinWaveTheory, ks, formula::SpinWaveIntensityFormula)\n\nComputes the scattering intensities at each energy band for each momentum transfer k in ks, according to Linear Spin Wave Theory and the given intensity formula. The formula must have a delta-function kernel, e.g.:\n\nformula = intensity_formula(swt, :perp, formula; kernel = delta_function_kernel)\n\nor else the bands will be broadened, and their intensity can not be computed.\n\nThe outputs will be arrays with indices identical to ks, with the last index giving the band index. dispersions reports the energy of each band, while intensities reports the scattering intensity.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_binned-Tuple{SampledCorrelations, BinningParameters, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.intensities_binned","text":"intensity, counts = intensities_binned(sc::SampledCorrelations, params::BinningParameters, formula; integrated_kernel)\n\nGiven correlation data contained in a SampledCorrelations and BinningParameters describing the shape of a histogram, compute the intensity and normalization for each histogram bin using a given intensity_formula.\n\nThe BinningParameters are expected to accept (q,ω) in R.L.U. for the (possibly reshaped) crystal associated with sc.\n\nThis is an alternative to intensities_interpolated which bins the scattering intensities into a histogram instead of interpolating between them at specified qs values. See unit_resolution_binning_parameters for a reasonable default choice of BinningParameters which roughly emulates intensities_interpolated with interpolation = :round.\n\nIf a function integrated_kernel(Δω) is passed, it will be used as the CDF of a kernel function for energy broadening. For example, integrated_kernel = Δω -> atan(Δω/η)/pi (c.f. integrated_lorentzian implements Lorentzian broadening with parameter η. Energy-dependent energy broadening can be achieved by providing an integrated_kernel(ω,Δω) whose first argument is the energy transfer ω.\n\nCurrently, energy broadening is only supported if the BinningParameters are such that the first three axes are purely spatial and the last (energy) axis is [0,0,0,1].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_broadened-Tuple{SpinWaveTheory, Any, Any, Any}","page":"Library API","title":"Sunny.intensities_broadened","text":"intensities_broadened(swt::SpinWaveTheory, ks, ωvals, formula)\n\nComputes the scattering intensities at each (Q,ω) according to Linear Spin Wave Theory and the given intensity formula. The required formula must have a non-delta-function kernel, e.g.:\n\nformula = intensity_formula(swt, :perp; kernel = lorentzian(0.05))\n\nor else the intensity at ωvals which are not exactly on the dispersion curve can not be calculated.\n\nThe intensity is computed at each wave vector in ks and each energy in ωvals. The output will be an array with indices identical to ks, with the last index matching ωvals.\n\nNote that ks is an array of wave vectors of arbitrary dimension. Each element k of ks must be a 3-wavevector in absolute units.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_interpolated-Tuple{SampledCorrelations, Any, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.intensities_interpolated","text":"intensities_interpolated(sc::SampledCorrelations, qs, formula:ClassicalIntensityFormula; interpolation=nothing, negative_energies=false)\n\nThe basic function for retrieving 𝒮(𝐪ω) information from a SampledCorrelations. Maps an array of wave vectors qs to an array of structure factor intensities, including an additional energy index. The values of ω associated with the energy index can be retrieved by calling available_energies. The three coordinates of each wave vector are measured in reciprocal lattice units, i.e., multiples of the reciprocal lattice vectors.\n\ninterpolation: Since 𝒮(𝐪 ω) is calculated on a finite lattice, data is only available at discrete wave vectors. By default, Sunny will round a requested q to the nearest available wave vector. Linear interpolation can be applied by setting interpolation=:linear.\nnegative_energies: If set to true, Sunny will return the periodic extension of the energy axis. Most users will not want this.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SampledCorrelations, AbstractVector{Int64}}","page":"Library API","title":"Sunny.intensity_formula","text":"formula = intensity_formula(sc::SampledCorrelations)\n\nEstablish a formula for computing the intensity of the discrete scattering modes (q,ω) using the correlation data 𝒮^αβ(qω) stored in the SampledCorrelations. The formula returned from intensity_formula can be passed to intensities_interpolated or intensities_binned.\n\nintensity_formula(sc,...; kT = Inf, formfactors = ...)\n\nThere are keyword arguments providing temperature and form factor corrections:\n\nkT: If a temperature is provided, the intensities will be rescaled by a temperature- and ω-dependent classical-to-quantum factor. kT should be specified when making comparisons with spin wave calculations or experimental data. If kT is not specified, infinite temperature (no correction) is assumed.\nformfactors: To apply form factor corrections, provide this keyword with a list of FormFactors, one for each symmetry-distinct site in the crystal. The order of FormFactors must correspond to the order of site symmetry classes, e.g., as they appear when printed in display(crystal).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SampledCorrelations, Any}","page":"Library API","title":"Sunny.intensity_formula","text":"A custom intensity formula can be specifed by providing a function intensity = f(q,ω,correlations) and specifying which correlations it requires:\n\nintensity_formula(f,sc::SampledCorrelations, required_correlations; kwargs...)\n\nThe function is intended to be specified using do notation. For example, this custom formula sums the off-diagonal correlations:\n\nrequired = [(:Sx,:Sy),(:Sy,:Sz),(:Sx,:Sz)]\nintensity_formula(sc,required,return_type = ComplexF64) do k, ω, off_diagonal_correlations\n sum(off_diagonal_correlations)\nend\n\nIf your custom formula returns a type other than Float64, use the return_type keyword argument to flag this.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SpinWaveTheory, AbstractVector{Int64}}","page":"Library API","title":"Sunny.intensity_formula","text":"formula = intensity_formula(swt::SpinWaveTheory; kernel = ...)\n\nEstablish a formula for computing the scattering intensity by diagonalizing the hamiltonian H(q) using Linear Spin Wave Theory.\n\nIf kernel = delta_function_kernel, then the resulting formula can be used with intensities_bands.\n\nIf kernel is an energy broadening kernel function, then the resulting formula can be used with intensities_broadened. Energy broadening kernel functions can either be a function of Δω only, e.g.:\n\nkernel = Δω -> ...\n\nor a function of both the energy transfer ω and of Δω, e.g.:\n\nkernel = (ω,Δω) -> ...\n\nThe integral of a properly normalized kernel function over all Δω is one.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{SpinWaveTheory, Symbol}","page":"Library API","title":"Sunny.intensity_formula","text":"intensity_formula([swt or sc], contraction_mode::Symbol)\n\nSunny has several built-in formulas that can be selected by setting contraction_mode to one of these values:\n\n:trace (default), which yields operatornametr 𝒮(qω) = _α 𝒮^αα(qω)\n:perp, which contracts 𝒮^αβ(qω) with the dipole factor δ_αβ - q_αq_β, returning the unpolarized intensity.\n:full, which will return all elements 𝒮^αβ(𝐪ω) without contraction.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lattice_params-Tuple{StaticArraysCore.SMatrix{3, 3, Float64, 9}}","page":"Library API","title":"Sunny.lattice_params","text":"lattice_params(latvecs::Mat3)\n\nCompute the lattice parameters (a b c α β γ) for the three lattice vectors provided as columns of latvecs. The inverse mapping is lattice_vectors.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lattice_vectors-NTuple{6, Any}","page":"Library API","title":"Sunny.lattice_vectors","text":"lattice_vectors(a, b, c, α, β, γ)\n\nReturn the lattice vectors, as columns of the 33 output matrix, that correspond to the conventional unit cell defined by the lattice constants (a b c) and the angles (α β γ) in degrees. The inverse mapping is lattice_params.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.load_nxs-Tuple{Any}","page":"Library API","title":"Sunny.load_nxs","text":"params, signal = load_nxs(filename)\n\nGiven the name of a Mantid-exported MDHistoWorkspace file, load the BinningParameters and the signal from that file.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lorentzian-Tuple{Any, Any}","page":"Library API","title":"Sunny.lorentzian","text":"lorentzian(x, η)\n\nReturns η(π(x^2 + η^2)).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.magnetic_moment-Tuple{System, Any}","page":"Library API","title":"Sunny.magnetic_moment","text":"magnetic_moment(sys::System, site::Site)\n\nGet the magnetic moment for a Site. This is the spin dipole multiplied by the Bohr magneton and the local g-tensor.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.merge_correlations-Union{Tuple{Array{SampledCorrelations{N}, 1}}, Tuple{N}} where N","page":"Library API","title":"Sunny.merge_correlations","text":"merge_correlations(scs::Vector{SampledCorrelations)\n\nAccumulate a list of SampledCorrelations into a single, summary SampledCorrelations. Useful for reducing the results of parallel computations.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.minimize_energy!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.minimize_energy!","text":"minimize_energy!(sys::System{N}; maxiters=100, subiters=20,\n method=Optim.ConjugateGradient(), kwargs...) where N\n\nOptimizes the spin configuration in sys to minimize energy. A total of maxiters iterations will be attempted, with restarts after every subiters iterations. The remaining kwargs will be forwarded to the optimize method of the Optim.jl package.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.polarize_spins!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.polarize_spins!","text":"polarize_spins!(sys::System, dir)\n\nPolarize all spins in the system along the direction dir.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.position_to_site-Tuple{System, Any}","page":"Library API","title":"Sunny.position_to_site","text":"position_to_site(sys::System, r)\n\nConverts a position r to four indices of a Site. The coordinates of r are given in units of the lattice vectors for the original crystal. This function can be useful for working with systems that have been reshaped using reshape_supercell.\n\nExample\n\n# Find the `site` at the center of a unit cell which is displaced by four\n# multiples of the first lattice vector\nsite = position_to_site(sys, [4.5, 0.5, 0.5])\n\n# Print the dipole at this site\nprintln(sys.dipoles[site])\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.powder_average_binned-Tuple{SampledCorrelations, Any, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.powder_average_binned","text":"powder_average_binned(sc::SampledCorrelations, radial_binning_parameters; formula\n ω_binning_parameters, integrated_kernel = nothing, bzsize = nothing)\n\nThis function emulates the experimental situation of \"powder averaging,\" where only the magnitude (and not the direction) of the momentum transfer is resolvable. The intensities are binned similarly to intensities_binned, but the histogram x-axis is |k| in absolute units, which is a nonlinear function of kx,ky,kz. The y-axis is energy.\n\nRadial binning parameters are specified as tuples (start,end,bin_width), e.g. radial_binning_parameters = (0,6π,6π/55).\n\nEnergy broadening is supported in the same way as intensities_binned, and this function accepts the same kind of intensity_formula.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.primitive_cell_shape-Tuple{Crystal}","page":"Library API","title":"Sunny.primitive_cell_shape","text":"primitive_cell_shape(cryst::Crystal)\n\nReturns the shape of the primitive cell as a 3×3 matrix, in fractional coordinates of the conventional lattice vectors. May be useful for constructing inputs to reshape_supercell.\n\nExamples\n\n# Valid if `cryst` has not been reshaped\n@assert cryst.prim_latvecs ≈ cryst.latvecs * primitive_cell_shape(cryst)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_bond-Tuple{Crystal, Bond}","page":"Library API","title":"Sunny.print_bond","text":"print_bond(cryst::Crystal, bond::Bond; b_ref::Bond)\n\nPrints symmetry information for bond bond. A symmetry-equivalent reference bond b_ref can optionally be provided to fix the meaning of the coefficients A, B, ...\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_site-Tuple{Any, Any}","page":"Library API","title":"Sunny.print_site","text":"print_site(cryst, i; R=I)\n\nPrint symmetry information for the site i, including allowed g-tensor and allowed anisotropy operator. An optional rotation matrix R can be provided to define the reference frame for expression of the anisotropy.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_stevens_expansion-Tuple{AbstractMatrix}","page":"Library API","title":"Sunny.print_stevens_expansion","text":"function print_stevens_expansion(op)\n\nPrints a local Hermitian operator as a linear combination of Stevens operators. The operator op may be a finite-dimensional matrix or an abstract spin polynomial in the large-S limit.\n\nExamples\n\nS = spin_matrices(2)\nprint_stevens_expansion(S[1]^4 + S[2]^4 + S[3]^4)\n# Prints: (1/20)𝒪₄₀ + (1/4)𝒪₄₄ + 102/5\n\nS = spin_matrices(Inf)\nprint_stevens_expansion(S[1]^4 + S[2]^4 + S[3]^4)\n# Prints: (1/20)𝒪₄₀ + (1/4)𝒪₄₄ + (3/5)𝒮⁴\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_suggested_frame-Tuple{Crystal, Int64}","page":"Library API","title":"Sunny.print_suggested_frame","text":"print_suggested_frame(cryst, i; digits=4)\n\nPrint a suggested reference frame, as a rotation matrix R, that can be used as input to print_site(). The purpose is to simplify the description of allowed anisotropies.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_symmetry_table-Tuple{Crystal, Any}","page":"Library API","title":"Sunny.print_symmetry_table","text":"print_symmetry_table(cryst::Crystal, max_dist)\n\nPrint symmetry information for all equivalence classes of sites and bonds, up to a maximum bond distance of max_dist. Equivalent to calling print_bond(cryst, b) for every bond b in reference_bonds(cryst, max_dist), where Bond(i, i, [0,0,0]) refers to a single site i.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_wrapped_intensities-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.print_wrapped_intensities","text":"print_wrapped_intensities(sys::System; nmax=10)\n\nFor Bravais lattices: Prints up to nmax wavevectors according to their instantaneous (static) structure factor intensities, listed in descending order. For non-Bravais lattices: Performs the same analysis for each spin sublattice independently; the output weights are naïvely averaged over sublattices, without incorporating phase shift information. This procedure therefore wraps all wavevectors into the first Brillouin zone. Each wavevector coordinate is given between -12 and 12 in reciprocal lattice units (RLU). The output from this function will typically be used as input to suggest_magnetic_supercell.\n\nBecause this function does not incorporate phase information in its averaging over sublattices, the printed weights are not directly comparable with experiment. For that purpose, use instant_correlations instead.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_delta-Tuple{Any}","page":"Library API","title":"Sunny.propose_delta","text":"propose_delta(magnitude)\n\nGenerate a proposal function that adds a Gaussian perturbation to the existing spin state. In :dipole mode, the procedure is to first introduce a random three-vector perturbation 𝐬 = 𝐬 + 𝐬 ξ and then return the properly normalized spin 𝐬 (𝐬𝐬). Each component of the random vector ξ is Gaussian distributed with a standard deviation of magnitude; the latter is dimensionless and typically smaller than one. \n\nIn :SUN mode, the procedure is analogous, but now involving Gaussian perturbations to each of the N complex components of an SU(N) coherent state.\n\nIn the limit of very large magnitude, this function coincides with propose_uniform.\n\nFor use with LocalSampler.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_flip-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.propose_flip","text":"propose_flip\n\nFunction to propose pure spin flip updates in the context of a LocalSampler. Dipoles are flipped as 𝐬 -𝐬. SU(N) coherent states are flipped using the time-reversal operator.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_uniform","page":"Library API","title":"Sunny.propose_uniform","text":"propose_uniform\n\nFunction to propose a uniformly random spin update in the context of a LocalSampler. In :dipole mode, the result is a random three-vector with appropriate normalization. In :SUN mode, the result is a random SU(N) coherent state with appropriate normalization.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.randomize_spins!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.randomize_spins!","text":"randomize_spins!(sys::System)\n\nRandomizes all spins under appropriate the uniform distribution.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_path-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.reciprocal_space_path","text":"reciprocal_space_path(cryst::Crystal, qs, density)\n\nReturns a pair (path, xticks). The path return value is a list of wavevectors that samples linearly between the provided wavevectors qs. The xticks return value can be used to label the special 𝐪 values on the x-axis of a plot.\n\nSpecial note about units: the wavevectors qs must be provided in reciprocal lattice units (RLU) for the given crystal, but the sampling density must be specified in the global frame. Specifically, the density is given as number of sample points per unit of radian inverse length, where the unit of length is the same as that used to specify the lattice vectors of the Crystal. The path will therefore include more samples between q-points that are further apart in absolute Fourier distance.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_path_bins-Tuple{Any, Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.reciprocal_space_path_bins","text":"reciprocal_space_path_bins(sc,qs,density,args...;kwargs...)\n\nTakes a list of wave vectors, qs in R.L.U., and builds a series of histogram BinningParameters whose first axis traces a path through the provided points. The second and third axes are integrated over according to the args and kwargs, which are passed through to slice_2D_binning_parameters.\n\nAlso returned is a list of marker indices corresponding to the input points, and a list of ranges giving the indices of each histogram x-axis within a concatenated histogram. The density parameter is given in samples per reciprocal lattice unit (R.L.U.).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_shell-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.reciprocal_space_shell","text":"reciprocal_space_shell(cryst::Crystal, radius, n)\n\nSample n points on the reciprocal space sphere with a given radius (units of inverse length).\n\nExamples\n\n# Sample wavevectors on the sphere at fixed density\nreciprocal_space_shell(cryst, r, 4π*r^2*density)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reference_bonds-Tuple{Crystal, Float64}","page":"Library API","title":"Sunny.reference_bonds","text":"reference_bonds(cryst::Crystal, max_dist)\n\nReturns a full list of bonds, one for each symmetry equivalence class, up to distance max_dist. The reference bond b for each equivalence class is selected according to a scoring system that prioritizes simplification of the elements in basis_for_symmetry_allowed_couplings(cryst, b).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.remove_periodicity!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.remove_periodicity!","text":"remove_periodicity!(sys::System, dims)\n\nRemove periodic interactions along the dimensions where dims is true. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nExample\n\n# Remove periodic boundaries along the 1st and 3rd dimensions\nremove_periodicity!(sys::System, (true, false, true))\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.repeat_periodically-Union{Tuple{N}, Tuple{System{N}, Tuple{Int64, Int64, Int64}}} where N","page":"Library API","title":"Sunny.repeat_periodically","text":"repeat_periodically(sys::System{N}, counts::NTuple{3,Int}) where N\n\nCreates a System identical to sys but repeated a given number of times in each dimension, specified by the tuple counts.\n\nSee also reshape_supercell.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reshape_supercell-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.reshape_supercell","text":"reshape_supercell(sys::System, shape)\n\nMaps an existing System to a new one that has the shape and periodicity of a requested supercell. The columns of the 33 integer matrix shape represent the supercell lattice vectors measured in units of the original crystal lattice vectors.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.resize_supercell-Union{Tuple{N}, Tuple{System{N}, Tuple{Int64, Int64, Int64}}} where N","page":"Library API","title":"Sunny.resize_supercell","text":"resize_supercell(sys::System{N}, latsize::NTuple{3,Int}) where N\n\nCreates a System with a given number of conventional unit cells in each lattice vector direction. Interactions and other settings will be inherited from sys.\n\nConvenience function for:\n\nreshape_supercell(sys, [latsize[1] 0 0; 0 latsize[2] 0; 0 0 latsize[3]])\n\nSee also reshape_supercell.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.rotate_operator-Tuple{LinearAlgebra.Hermitian{ComplexF64, Matrix{ComplexF64}}, Any}","page":"Library API","title":"Sunny.rotate_operator","text":"rotate_operator(A, R)\n\nRotates the local quantum operator A according to the 33 rotation matrix R.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.rotation_in_rlu-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.rotation_in_rlu","text":"rotation_in_rlu(cryst::Crystal, axis, angle)\n\nReturns a 33 matrix that rotates wavevectors in reciprocal lattice units (RLU). The axis vector is a real-space direction in absolute units (but arbitrary magnitude), and the angle is in radians.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_coherent!-Union{Tuple{N}, Tuple{System{N}, Any, Any}} where N","page":"Library API","title":"Sunny.set_coherent!","text":"set_coherent!(sys::System, Z, site::Site)\n\nSet a coherent spin state at a Site using the N complex amplitudes in Z.\n\nFor a standard SpinInfo, these amplitudes will be interpreted in the eigenbasis of 𝒮ᶻ. That is, Z[1] represents the amplitude for the basis state fully polarized along the z-direction, and subsequent components represent states with decreasing angular momentum along this axis (m = S S-1 -S).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_dipole!-Union{Tuple{N}, Tuple{System{N}, Any, Any}} where N","page":"Library API","title":"Sunny.set_dipole!","text":"set_dipole!(sys::System, dir, site::Site)\n\nPolarize the spin at a Site along the direction dir.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_exchange!-Union{Tuple{N}, Tuple{System{N}, Any, Bond}} where N","page":"Library API","title":"Sunny.set_exchange!","text":"set_exchange!(sys::System, J, bond::Bond)\n\nSets a 3×3 spin-exchange matrix J along bond, yielding a pairwise interaction energy 𝐒_iJ 𝐒_j. This interaction will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells.\n\nThe parameter J may be scalar or matrix-valued. As a convenience, dmvec(D) can be used to construct the antisymmetric part of the exchange, where D is the Dzyaloshinskii-Moriya pseudo-vector. The resulting interaction will be 𝐃(𝐒_i𝐒_j).\n\nFor more general interactions, such as biquadratic, use set_pair_coupling! instead.\n\nExamples\n\n# An explicit exchange matrix\nJ1 = [2 3 0;\n -3 2 0;\n 0 0 2]\nset_exchange!(sys, J1, bond)\n\n# An equivalent Heisenberg + DM exchange \nJ2 = 2*I + dmvec([0,0,3])\nset_exchange!(sys, J2, bond)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_exchange_at!-Union{Tuple{N}, Tuple{System{N}, Any, Union{NTuple{4, Int64}, CartesianIndex{4}}, Union{NTuple{4, Int64}, CartesianIndex{4}}}} where N","page":"Library API","title":"Sunny.set_exchange_at!","text":"set_exchange_at!(sys::System, J, site1::Site, site2::Site; offset=nothing)\n\nSets an exchange interaction `𝐒_i⋅J 𝐒_j along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nIf the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.\n\nFor more general interactions, such as biquadratic, use set_pair_coupling_at! instead.\n\nSee also set_exchange!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_external_field!-Tuple{System, Any}","page":"Library API","title":"Sunny.set_external_field!","text":"set_external_field!(sys::System, B::Vec3)\n\nSets the external field B that couples to all spins.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_external_field_at!-Tuple{System, Any, Any}","page":"Library API","title":"Sunny.set_external_field_at!","text":"set_external_field_at!(sys::System, B::Vec3, site::Site)\n\nSets a Zeeman coupling between a field B and a single spin. Site includes a unit cell and a sublattice index.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_onsite_coupling!-Tuple{System, Any, Int64}","page":"Library API","title":"Sunny.set_onsite_coupling!","text":"set_onsite_coupling!(sys::System, op, i::Int)\n\nSet the single-ion anisotropy for the ith atom of every unit cell, as well as all symmetry-equivalent atoms. The operator op may be provided as an abstract function of the local spin operators, as a polynomial of spin_matrices, or as a linear combination of stevens_matrices.\n\nExamples\n\n# An easy axis anisotropy in the z-direction\nset_onsite_coupling!(sys, S -> -D*S[3]^3, i)\n\n# The unique quartic single-ion anisotropy for a site with cubic point group\n# symmetry\nset_onsite_coupling!(sys, S -> 20*(S[1]^4 + S[2]^4 + S[3]^4), i)\n\n# An equivalent expression of this quartic anisotropy, up to a constant shift\nO = stevens_matrices(spin_label(sys, i))\nset_onsite_coupling!(sys, O[4,0] + 5*O[4,4], i)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_onsite_coupling_at!-Tuple{System, Any, Union{NTuple{4, Int64}, CartesianIndex{4}}}","page":"Library API","title":"Sunny.set_onsite_coupling_at!","text":"set_onsite_coupling_at!(sys::System, op, site::Site)\n\nSets the single-ion anisotropy operator op for a single Site, ignoring crystal symmetry. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nSee also set_onsite_coupling!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_pair_coupling!-Union{Tuple{N}, Tuple{System{N}, AbstractMatrix, Any}} where N","page":"Library API","title":"Sunny.set_pair_coupling!","text":"set_pair_coupling!(sys::System, op, bond)\n\nSets an arbitrary coupling op along bond. This coupling will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites.\n\nExamples\n\n# Bilinear+biquadratic exchange involving 3×3 matrices J1 and J2\nset_pair_coupling!(sys, (Si, Sj) -> Si'*J1*Sj + (Si'*J2*Sj)^2, bond)\n\n# Equivalent expression using an appropriate fixed matrix representation\nS = spin_matrices(1/2)\nSi, Sj = to_product_space(S, S)\nset_pair_coupling!(sys, Si'*J1*Sj + (Si'*J2*Sj)^2, bond)\n\nSee also spin_matrices, to_product_space.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_pair_coupling_at!-Union{Tuple{N}, Tuple{System{N}, AbstractMatrix, Union{NTuple{4, Int64}, CartesianIndex{4}}, Union{NTuple{4, Int64}, CartesianIndex{4}}}} where N","page":"Library API","title":"Sunny.set_pair_coupling_at!","text":"set_pair_coupling_at!(sys::System, op, bond)\n\nSets an arbitrary coupling along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites. The documentation for set_pair_coupling! provides examples constructing op.\n\nThe system must support inhomogeneous interactions via to_inhomogeneous.\n\nIf the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_spiral_order!-Tuple{Any}","page":"Library API","title":"Sunny.set_spiral_order!","text":"set_spiral_order!(sys; q, axis, S0)\n\nInitializes the system with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0 at the real-space origin. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.\n\nExample\n\n# Spiral order for a wavevector propagating in the direction of the first\n# reciprocal lattice vector (i.e., orthogonal to the lattice vectors ``𝐚_2``\n# and ``𝐚_3``), repeating with a period of 10 lattice constants, and spiraling\n# about the ``ẑ``-axis. The spin at the origin will point in the direction\n# ``𝐒_0 = ŷ + ẑ``. Here, ``(x̂, ŷ, ẑ)`` are the axes of Cartesian coordinate\n# system in the global frame.\nset_spiral_order!(sys; q=[1/10, 0, 0], axis=[0, 0, 1], S0=[0, 1, 1])\n\nSee also set_spiral_order_on_sublattice!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_spiral_order_on_sublattice!-Tuple{Any, Any}","page":"Library API","title":"Sunny.set_spiral_order_on_sublattice!","text":"set_spiral_order_on_sublattice!(sys, i; q, axis, S0)\n\nInitializes sublattice i with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0. The phase is selected such that the spin at sys.dipole[1,1,1,i] will point in the direction of S0. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.\n\nThis function is not available for systems with reshaped unit cells.\n\nSee also set_spiral_order!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_vacancy_at!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.set_vacancy_at!","text":"set_vacancy_at!(sys::System, site::Site)\n\nMake a single site nonmagnetic. Site includes a unit cell and a sublattice index.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.slice_2D_binning_parameters-Tuple{Vector{Float64}, Any, Any, Int64, Any}","page":"Library API","title":"Sunny.slice_2D_binning_parameters","text":"slice_2D_binning_parameter(sc::SampledCorrelations, cut_from_q, cut_to_q, cut_bins::Int64, cut_width::Float64; plane_normal = [0,0,1],cut_height = cutwidth)\n\nCreates BinningParameters which make a cut along one dimension of Q-space.\n\nThe x-axis of the resulting histogram consists of cut_bins-many bins ranging from cut_from_q to cut_to_q. The width of the bins in the transverse direciton is controlled by cut_width and cut_height.\n\nThe binning in the transverse directions is defined in the following way, which sets their normalization and orthogonality properties:\n\ncut_covector = normalize(cut_to_q - cut_from_q)\ntransverse_covector = normalize(plane_normal × cut_covector)\ncotransverse_covector = normalize(transverse_covector × cut_covector)\n\nIn other words, the axes are orthonormal with respect to the Euclidean metric.\n\nIf the cut is too narrow, there will be very few scattering vectors per bin, or the number per bin will vary substantially along the cut. If the output appears under-resolved, try increasing cut_width.\n\nThe four axes of the resulting histogram are:\n\nAlong the cut\nFist transverse Q direction\nSecond transverse Q direction\nEnergy\n\nThis function can be used without reference to a SampledCorrelations using this alternate syntax to manually specify the bin centers for the energy axis:\n\nslice_2D_binning_parameter(ω_bincenters, cut_from, cut_to,...)\n\nwhere ω_bincenters specifies the energy axis, and both cut_from and cut_to are arbitrary covectors, in any units.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.spin_label-Tuple{System, Int64}","page":"Library API","title":"Sunny.spin_label","text":"spin_label(sys::System, i::Int)\n\nIf atom i carries a single spin-S moment, then returns the half-integer label S. Otherwise, throws an error.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.spin_matrices-Tuple{Any}","page":"Library API","title":"Sunny.spin_matrices","text":"spin_matrices(S)\n\nReturns a triple of NN spin matrices, where N = 2S+1. These are the generators of SU(2) in the spin-S representation.\n\nIf S == Inf, then the return values are abstract symbols denoting infinite-dimensional matrices that commute. These can be useful for repeating historical studies, or modeling micromagnetic systems. A technical discussion appears in the Sunny documentation page: Interaction Strength Renormalization.\n\nExample\n\nS = spin_matrices(3/2)\n@assert S'*S ≈ (3/2)*(3/2+1)*I\n@assert S[1]*S[2] - S[2]*S[1] ≈ im*S[3]\n\nS = spin_matrices(Inf)\n@assert S[1]*S[2] - S[2]*S[1] == 0\n\nSee also print_stevens_expansion.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.step!","page":"Library API","title":"Sunny.step!","text":"step!(sys::System, dynamics)\n\nAdvance the spin configuration one dynamical time-step. The dynamics object may be a continuous spin dynamics, such as Langevin or ImplicitMidpoint, or it may be a discrete Monte Carlo sampling scheme such as LocalSampler.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.stevens_matrices-Tuple{Any}","page":"Library API","title":"Sunny.stevens_matrices","text":"stevens_matrices(S)\n\nReturns a generator of Stevens operators in the spin-S representation. The return value O can be indexed as O[k,q], where 0 k 6 labels an irrep of SO(3) and -k q k. This will produce an NN matrix where ``N = 2S\n\n1``. Linear combinations of Stevens operators can be used as a \"physical\n\nbasis\" for decomposing local observables. To see this decomposition, use print_stevens_expansion.\n\nIf S == Inf, then symbolic operators will be returned. In this infinite dimensional limit, the Stevens operators become homogeneous polynomials of commuting spin operators.\n\nExample\n\nO = stevens_matrices(2)\nS = spin_matrices(2)\n\nA = (1/20)O[4,0] + (1/4)O[4,4] + (102/5)I\nB = S[1]^4 + S[2]^4 + S[3]^4\n@assert A ≈ B\n\nSee also spin_matrices and Interaction Strength Renormalization.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.subcrystal-Union{Tuple{N}, Tuple{Crystal, Vararg{String, N}}} where N","page":"Library API","title":"Sunny.subcrystal","text":"subcrystal(cryst, types) :: Crystal\n\nFilters sublattices of a Crystal by atom types, keeping the space group unchanged.\n\nsubcrystal(cryst, classes) :: Crystal\n\nFilters sublattices of Crystal by equivalence classes, keeping the space group unchanged.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.suggest_magnetic_supercell-Tuple{Any}","page":"Library API","title":"Sunny.suggest_magnetic_supercell","text":"suggest_magnetic_supercell(qs; tol=1e-12, maxsize=100)\n\nSuggests a magnetic supercell, in units of the crystal lattice vectors, that is consistent with periodicity of the wavevectors qs in RLU. If the wavevectors are incommensurate (with respect to the maximum supercell size maxsize), one can select a larger error tolerance tol to find a supercell that is almost commensurate.\n\nPrints a 33 matrix of integers that is suitable for use in reshape_supercell.\n\nExamples\n\n# A magnetic supercell for a single-Q structure. Will print\nq1 = [0, -1/4, 1/4]\nsuggest_magnetic_supercell([q1]) # [1 0 0; 0 2 1; 0 -2 1]\n\n# A larger magnetic supercell for a double-Q structure\nq2 = [1/4, 0, 1/4]\nsuggest_magnetic_supercell([q1, q2]) # [1 2 2; -1 2 -2; -1 2 2]\n\n# If given incommensurate wavevectors, find an approximate supercell that\n# is exactly commensurate for nearby wavevectors.\nsuggest_magnetic_supercell([[0, 0, 1/√5], [0, 0, 1/√7]]; tol=1e-2)\n\n# This prints [1 0 0; 0 1 0; 0 0 16], which becomes commensurate under the\n# approximations `1/√5 ≈ 7/16` and `1/√7 ≈ 3/8`.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.symmetry_equivalent_bonds-Tuple{System, Bond}","page":"Library API","title":"Sunny.symmetry_equivalent_bonds","text":"symmetry_equivalent_bonds(sys::System, bond::Bond)\n\nGiven a Bond for the original (unreshaped) crystal, return all symmetry equivalent bonds in the System. Each returned bond is represented as a pair of Sites, which may be used as input to set_exchange_at!. Reverse bonds are not included (no double counting).\n\nExample\n\nfor (site1, site2, offset) in symmetry_equivalent_bonds(sys, bond)\n @assert site1 < site2\n set_exchange_at!(sys, J, site1, site2; offset)\nend\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.to_inhomogeneous-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.to_inhomogeneous","text":"to_inhomogeneous(sys::System)\n\nReturns a copy of the system that allows for inhomogeneous interactions, which can be set using set_onsite_coupling_at!, set_exchange_at!, and set_vacancy_at!.\n\nInhomogeneous systems do not support symmetry-propagation of interactions or system reshaping.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.to_product_space-Tuple{Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.to_product_space","text":"to_product_space(A, B, Cs...)\n\nGiven lists of operators acting on local Hilbert spaces individually, return the corresponding operators that act on the tensor product space. In typical usage, the inputs will represent local physical observables and the outputs will be used to define quantum couplings.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.unit_resolution_binning_parameters-Tuple{Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.unit_resolution_binning_parameters","text":"unit_resolution_binning_parameters(sc::SampledCorrelations)\n\nCreate BinningParameters which place one histogram bin centered at each possible (q,ω) scattering vector of the crystal. This is the finest possible binning without creating bins with zero scattering vectors in them.\n\nThis function can be used without reference to a SampledCorrelations using an alternate syntax to manually specify the bin centers for the energy axis and the lattice size:\n\nunit_resolution_binning_parameters(ω_bincenters,latsize,[reciprocal lattice vectors])\n\nThe last argument may be a 3x3 matrix specifying the reciprocal lattice vectors, or a Crystal.\n\nLastly, binning parameters for a single axis may be specifed by their bin centers:\n\n(binstart,binend,binwidth) = unit_resolution_binning_parameters(bincenters::Vector{Float64})\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.@mix_proposals-Tuple","page":"Library API","title":"Sunny.@mix_proposals","text":"@mix_proposals weight1 propose1 weight2 propose2 ...\n\nMacro to generate a proposal function that randomly selects among the provided functions according to the provided probability weights. For use with LocalSampler.\n\nExample\n\n# A proposal function that proposes a spin flip 40% of the time, and a\n# Gaussian perturbation 60% of the time.\n@mix_proposals 0.4 propose_flip 0.6 propose_delta(0.2)\n\n\n\n\n\n","category":"macro"},{"location":"library.html#Optional-Makie-extensions","page":"Library API","title":"Optional Makie extensions","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"The following will be enabled through a package extension if either GLMakie or WGLMakie is loaded.","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"plot_spins\nview_crystal","category":"page"},{"location":"library.html#Sunny.plot_spins","page":"Library API","title":"Sunny.plot_spins","text":"plot_spins(sys::System; arrowscale=1.0, color=:red, colorfn=nothing,\n colormap=:viridis, colorrange=nothing, show_cell=true, orthographic=false,\n ghost_radius=0, dims=3\n\nPlot the spin configuration defined by sys. Optional parameters are:\n\narrowscale: Scale all arrows by dimensionless factor.\ncolor: Arrow colors. May be symbolic or numeric. If scalar, will be shared among all sites.\ncolorfn: Function that dynamically maps from a site index to a numeric color value. Useful for animations.\ncolormap, colorrange: Used to populate colors from numbers following Makie conventions.\nshow_cell: Show original crystallographic unit cell.\northographic: Use camera with orthographic projection.\nghost_radius: Show translucent periodic images up to a given distance (length units).\ndims: Spatial dimensions of system (1, 2, or 3).\n\nCalling notify on the return value will animate the figure.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.view_crystal","page":"Library API","title":"Sunny.view_crystal","text":"view_crystal(crystal::Crystal, max_dist::Real; show_axis=true, orthographic=false)\n\nAn interactive crystal viewer, with bonds up to max_dist.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Optional-WriteVTK-extensions","page":"Library API","title":"Optional WriteVTK extensions","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"The following will be enabled through a package extension if WriteVTK is loaded.","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"export_vtk","category":"page"},{"location":"library.html#Sunny.export_vtk","page":"Library API","title":"Sunny.export_vtk","text":"export_vtk(filename,params::BinningParameters,data)\n\nExport a VTK-compatible file to filename (do not include file extension when specifying the file name) which contains the data as VTK Cell Data on a grid parameterized by params.\n\nAt least one axis of the BinningParameters must be integrated over, since VTK does not support 4D data. See integrate_axes!.\n\n\n\n\n\n","category":"function"},{"location":"examples/contributed/MgCr2O4-tutorial.html#MgCr2O4-at-Finite-Temperature","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Author: Martin Mourigal
","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"EditURL = \"../../../examples/out_of_equilibrium.jl\"","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Download this example as Jupyter notebook or Julia script.","category":"page"},{"location":"examples/out_of_equilibrium.html#CP-Skyrmion-Quench","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"","category":"section"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"This example demonstrates Sunny's ability to simulate the out-of-equilibrium dynamics of generalized spin systems. We will implement the model Hamiltonian of Zhang et al., Nature Communications 14, 3626 (2023), which supports a novel type of topological defect, a CP² skyrmion, that involves both the dipolar and quadrupolar parts of a quantum spin.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Beginning from an initial high-temperature state, a disordered gas of CP² skyrmions can be formed by rapidly quenching to low temperature. To model the coupled dynamics of dipoles and quadrupoles, Sunny uses a recently developed generalization of the Landau-Lifshitz spin dynamics, Dahlbom et al., Phys. Rev. B 106, 235154 (2022).","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"using Sunny, GLMakie","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The Hamiltonian we will implement,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"mathcalH = sum_langle ij rangle J_ij( hatS_i^x hatS_j^x + hatS_i^y hatS_j^y + DeltahatS_i^z hatS_j^z) - hsum_ihatS_i^z + Dsum_i(hatS_i^z)^2","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"contains competing ferromagnetic nearest-neightbor and antiferromagnetic next-nearest-neighbor exchange terms on a triangular lattice. Both exchanges exhibit anisotropy on the z-term. Additionally, there is an external magnetic field, h, and easy-plane single-ion anisotropy, D 0. We begin by implementing the Crystal.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"lat_vecs = lattice_vectors(1.0, 1.0, 2.0, 90, 90, 120)\nbasis_vecs = [[0,0,0]]\ncryst = Crystal(lat_vecs, basis_vecs)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The crystal is then used to create a spin System. All parameters in this model system are dimensionless, so we select \"theory\" units and set the g-factor to one.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"L = 40\ndims = (L, L, 1)\nsys = System(cryst, dims, [SpinInfo(1, S=1, g=1)], :SUN; seed=101, units=Units.theory)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"We proceed to implement each term of the Hamiltonian, selecting our parameters so that the system occupies a region of the phase diagram that supports skyrmions. The exchange interactions are set as follows.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"J1 = -1 # Nearest-neighbor ferromagnetic\nJ2 = (2.0/(1+√5)) # Tune competing exchange to set skyrmion scale length\nΔ = 2.6 # Exchange anisotropy\n\nex1 = J1 * [1.0 0.0 0.0;\n 0.0 1.0 0.0;\n 0.0 0.0 Δ]\nex2 = J2 * [1.0 0.0 0.0;\n 0.0 1.0 0.0;\n 0.0 0.0 Δ]\nset_exchange!(sys, ex1, Bond(1, 1, [1, 0, 0]))\nset_exchange!(sys, ex2, Bond(1, 1, [1, 2, 0]))","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Next we add the external field,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"h = 15.5\nfield = set_external_field!(sys, [0.0 0.0 h])","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"and finally an easy-plane single-ion anisotropy,","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"D = 19.0\nset_onsite_coupling!(sys, S -> D*S[3]^2, 1)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Initialize system to an infinite temperature (fully randomized) initial condition.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"randomize_spins!(sys)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"We are now ready to simulate the quenching process using a generalized Langevin spin dynamics. If we were working with spin dipoles only, then Langevin dynamics would be the usual Landau-Lifshitz spin dynamics, augmented with damping and noise terms. In the present study, we are instead working with quantum spin-1 (an (N=3)-level system that includes both dipoles and quadrupoles). Here, Langevin captures the coupled dipole-quadrupole dynamics using the formalism of SU(N) coherent states.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Selecting kT = 0 in the Langevin dynamics will effective disable the noise term. Then the parameter λ effectively determines the damping time-scale.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Δt = 0.2/D # Integration time step (inverse meV). Typically this will be\n # inversely proportional to the largest energy scale in the\n # system. We can use a fairly large time-step here because\n # accuracy isn't critical.\nkT = 0 # Target equilibrium temperature (meV)\nλ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)\nintegrator = Langevin(Δt; kT, λ)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"Finally we run the dynamics. We will record the state of the system at three different times during the quenching process by copying the coherents field of the System.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"τs = [4., 16, 256] # Times to record snapshots\nframes = [] # Empty array to store snapshots\nfor i in eachindex(τs)\n dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate\n numsteps = round(Int, dur/Δt)\n for _ in 1:numsteps # Perform the integration\n step!(sys, integrator)\n end\n push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration\nend","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"To visualize the state of the system contained in each snapshot, we will calculate and plot the skyrmion density on each plaquette of our lattice. The function plot_triangular_plaquettes is not part of the core Sunny package, but rather something you could define yourself. We are using the definition in plotting2d.jl from the Sunny examples/extra directory.","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"include(pkgdir(Sunny, \"examples\", \"extra\", \"plotting2d.jl\"))\n\nfunction sun_berry_curvature(z₁, z₂, z₃)\n z₁, z₂, z₃ = normalize.((z₁, z₂, z₃))\n n₁ = z₁ ⋅ z₂\n n₂ = z₂ ⋅ z₃\n n₃ = z₃ ⋅ z₁\n return angle(n₁ * n₂ * n₃)\nend\n\nplot_triangular_plaquettes(sun_berry_curvature, frames; resolution=(1800,600),\n offset_spacing=10, texts = [\"\\tt = \"*string(τ) for τ in τs], text_offset = (0.0, 6.0)\n)","category":"page"},{"location":"examples/out_of_equilibrium.html","page":"CP² Skyrmion Quench","title":"CP² Skyrmion Quench","text":"The times are given in hbarJ_1. The white background corresponds to a quantum paramagnetic state, where the local spin exhibits a strong quadrupole moment and little or no dipole moment. Observe that the process has generated a number of well-formed skyrmions of both positive (red) and negative (blue) charge in addition to a number of other metastable spin configurations. A full-sized version of this figure is available in Dahlbom et al..","category":"page"},{"location":"library.html#Library-API","page":"Library API","title":"Library API","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"This page describes the public types and functions exported by Sunny. This documentation can be also be accessed using the Julia help system (enter ? at the Julia command prompt).","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"Sunny.plot_spins\nSunny.view_crystal\nSunny.export_vtk","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"Modules = [Sunny]\nPrivate = false","category":"page"},{"location":"library.html#Sunny.Site","page":"Library API","title":"Sunny.Site","text":"(cell1, cell2, cell3, i) :: Site\n\nFour indices identifying a single site in a System. The first three indices select the lattice cell and the last selects the sublattice (i.e., the atom within the unit cell).\n\nThis object can be used to index dipoles and coherents fields of a System. A Site is also required to specify inhomogeneous interactions via functions such as set_external_field_at! or set_exchange_at!.\n\nNote that the definition of a cell may change when a system is reshaped. In this case, it is convenient to construct the Site using position_to_site, which always takes a position in fractional coordinates of the original lattice vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Units","page":"Library API","title":"Sunny.Units","text":"Units.meV\nUnits.theory\n\nThe unit system is implicitly determined by the definition of two physical constants: the vacuum permeability μ₀ and the Bohr magneton μ_B. Temperatures are effectively measured in units of energy (k_B = 1) and time is effectively measured in units of inverse energy (ħ = 1). The default unit system, Units.meV, employs (meV, Å, tesla). Select alternatively Units.theory for a units system defined so that μ₀ = μ_B = 1.\n\nSee also meV_per_K\n\n\n\n\n\n","category":"constant"},{"location":"library.html#Sunny.meV_per_K","page":"Library API","title":"Sunny.meV_per_K","text":"meV_per_K = 0.086173332621451774\n\nA physical constant. Useful for converting kelvin into the default energy units, meV.\n\n\n\n\n\n","category":"constant"},{"location":"library.html#Sunny.BinningParameters","page":"Library API","title":"Sunny.BinningParameters","text":"BinningParameters(binstart,binend,binwidth;covectors = I(4))\nBinningParameters(binstart,binend;numbins,covectors = I(4))\n\nDescribes a 4D parallelepided histogram in a format compatible with experimental Inelasitic Neutron Scattering data. See generate_mantid_script_from_binning_parameters to convert BinningParameters to a format understandable by the Mantid software, or load_nxs to load BinningParameters from a Mantid .nxs file.\n\nThe coordinates of the histogram axes are specified by multiplication of (q,ω) with each row of the covectors matrix, with q given in [R.L.U.]. Since the default covectors matrix is the identity matrix, the default axes are (qx,qy,qz,ω) in absolute units.\n\nThe convention for the binning scheme is that:\n\nThe left edge of the first bin starts at binstart\nThe bin width is binwidth\nThe last bin contains binend\nThere are no \"partial bins;\" the last bin may contain values greater than binend. C.f. count_bins.\n\nA value can be binned by computing its bin index:\n\ncoords = covectors * value\nbin_ix = 1 .+ floor.(Int64,(coords .- binstart) ./ binwidth)\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Bond","page":"Library API","title":"Sunny.Bond","text":"Bond(i, j, n)\n\nRepresents a bond between atom indices i and j. n is a vector of three integers specifying unit cell displacement in terms of lattice vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Crystal","page":"Library API","title":"Sunny.Crystal","text":"An object describing a crystallographic unit cell and its space group symmetry. Constructors are as follows:\n\nCrystal(filename; symprec=1e-5)\n\nReads the crystal from a .cif file located at the path filename. The optional parameter symprec controls the precision tolerance for spacegroup symmetries.\n\nCrystal(latvecs, positions; types=nothing, symprec=1e-5)\n\nConstructs a crystal from the complete list of atom positions positions, with coordinates (between 0 and 1) in units of lattice vectors latvecs. Spacegroup symmetry information is automatically inferred. The optional parameter types is a list of strings, one for each atom, and can be used to break symmetry-equivalence between atoms.\n\nCrystal(latvecs, positions, spacegroup_number; types=nothing, setting=nothing, symprec=1e-5)\n\nBuilds a crystal by applying symmetry operators for a given international spacegroup number. For certain spacegroups, there are multiple possible unit cell settings; in this case, a warning message will be printed, and a list of crystals will be returned, one for every possible setting. Alternatively, the optional setting string will disambiguate between unit cell conventions.\n\nCurrently, crystals built using only the spacegroup number will be missing some symmetry information. It is generally preferred to build a crystal from a .cif file or from the full specification of the unit cell.\n\nExamples\n\n# Read a Crystal from a .cif file\nCrystal(\"filename.cif\")\n\n# Build an FCC crystal using the primitive unit cell. The spacegroup number\n# 225 is inferred.\nlatvecs = [1 1 0;\n 1 0 1;\n 0 1 1] / 2\npositions = [[0, 0, 0]]\nCrystal(latvecs, positions)\n\n# Build a CsCl crystal (two cubic sublattices). By providing distinct type\n# strings, the spacegroup number 221 is inferred.\nlatvecs = lattice_vectors(1, 1, 1, 90, 90, 90)\npositions = [[0,0,0], [0.5,0.5,0.5]]\ntypes = [\"Na\", \"Cl\"]\ncryst = Crystal(latvecs, positions; types)\n\n# Build a diamond cubic crystal from its spacegroup number 227. This\n# spacegroup has two possible settings (\"1\" or \"2\"), which determine an\n# overall unit cell translation.\nlatvecs = lattice_vectors(1, 1, 1, 90, 90, 90)\npositions = [[1, 1, 1] / 4]\ncryst = Crystal(latvecs, positions, 227; setting=\"1\")\n\nSee also lattice_vectors.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.FormFactor-Tuple{String}","page":"Library API","title":"Sunny.FormFactor","text":"FormFactor(ion::String; g_lande=2)\n\nThe magnetic form factor for a given magnetic ion and charge state. When passed to an intensity_formula, determines a 𝐪-dependent scaling of the structure factor.\n\nThe parameter ion must be one of the following strings:\n\nAm2, Am3, Am4, Am5, Am6, Am7, Au1, Au2, Au3, Au4, Au5, Ce2, Co0, Co1, Co2, Co3,\nCo4, Cr0, Cr1, Cr2, Cr3, Cr4, Cu0, Cu1, Cu2, Cu3, Cu4, Dy2, Dy3, Er2, Er3, Eu2,\nEu3, Fe0, Fe1, Fe2, Fe3, Fe4, Gd2, Gd3, Hf2, Hf3, Ho2, Ho3, Ir0a, Ir0b, Ir0c,\nIr1a, Ir1b, Ir2, Ir3, Ir4, Ir5, Ir6, Mn0, Mn1, Mn2, Mn3, Mn4, Mo0, Mo1, Nb0,\nNb1, Nd2, Nd3, Ni0, Ni1, Ni2, Ni3, Ni4, Np3, Np4, Np5, Np6, Os0a, Os0b, Os0c,\nOs1a, Os1b, Os2, Os3, Os4, Os5, Os6, Os7, Pd0, Pd1, Pr3, Pt1, Pt2, Pt3, Pt4,\nPt5, Pt6, Pu3, Pu4, Pu5, Pu6, Re0a, Re0b, Re0c, Re1a, Re1b, Re2, Re3, Re4, Re5,\nRe6, Rh0, Rh1, Ru0, Ru1, Sc0, Sc1, Sc2, Sm2, Sm3, Ta2, Ta3, Ta4, Tb2, Tb3, Tc0,\nTc1, Ti0, Ti1, Ti2, Ti3, Tm2, Tm3, U3, U4, U5, V0, V1, V2, V3, V4, W0a, W0b,\nW0c, W1a, W1b, W2c, W3, W4, W5, Y0, Yb2, Yb3, Zr0, Zr1\n\nThe trailing number denotes ionization state. For example, \"Fe0\" denotes a neutral iron atom, while \"Fe2\" denotes Fe²⁺. If multiple electronic configurations are possible, they will be distinguished by a trailing letter (a, b, ...). Omitting this letter will print an informative error,\n\nFormFactor(\"Ir0\")\n\nERROR: Disambiguate form factor according to electronic configuration:\n \"Ir0a\" -- 6s⁰5d⁹\n \"Ir0b\" -- 6s¹5d⁸\n \"Ir0c\" -- 6s²5d⁷\n\nThe form factor is approximated as\n\nF(s) = j_0(s) + frac2-gg j_2(s) s^2,\n\ninvolving the Landé g-factor. The j_l(s) are radial integrals associated with the lth Bessel function of the magnetic dipole, where s = k4π, and k is the magnitude of momentum transfer. \n\nThe radial integrals have been calculated using Hartree-Fock for transition metals, or Dirac-Fock for the rare earths and actinide series [1–3]. Sunny uses approximate fits as a sum of Gaussians,\n\nj_0(s) = A e^-as^2 + B e^-bs^2 + C e^-cs^2 + D e^-ds^2 + E \nj_l(s) = (A e^-as^2 + B e^-bs^2 + C e^-cs^2 + D e^-ds^2 + E) s^2\n\nReferences:\n\nhttps://www.ill.eu/sites/ccsl/ffacts/ffachtml.html\nJ. Brown, The Neutron Data Booklet, 2nd ed., Sec. 2.5 Magnetic Form Factors (2003)\nK. Kobayashi, T. Nagao, M. Ito, Acta Cryst. A, 67 pp 473–480 (2011)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.ImplicitMidpoint","page":"Library API","title":"Sunny.ImplicitMidpoint","text":"ImplicitMidpoint(Δt::Float64; atol=1e-12) where N\n\nEnergy-conserving spin dynamics. One call to the step! function will advance a System by Δt units of time.\n\nUses the spherical midpoint integration scheme for dipole systems and the Schrödinger midpoint integration scheme for SU(N) spin systems. Both integration schemes are symplectic, and therefore avoid energy drift over long periods of simulation time.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.Langevin","page":"Library API","title":"Sunny.Langevin","text":"Langevin(Δt::Float64; λ::Float64, kT::Float64)\n\nSpin dynamics with coupling to a Langevin thermostat, which includes damping and noise terms. One call to the step! function will advance a System by Δt units of time.\n\nAssuming ergodicity, the Langevin dynamics will sample from thermal equilibrium for the target temperature kT. The empirical parameter λ determines the strength of the coupling to the thermal bath. In other words, 1/λ is the decorrelation time-scale. If λ = 0, then the spin dynamics coincides with ImplicitMidpoint.\n\nAn alternative approach to sampling is LocalSampler, which may be preferred when the allowed spin values become effective discrete (e.g. Ising spins).\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.LocalSampler","page":"Library API","title":"Sunny.LocalSampler","text":"LocalSampler(; kT, nsweeps=1.0, propose=propose_uniform)\n\nMonte Carlo simulation involving Metropolis updates to individual spins. One call to the step! function will perform nsweeps of MCMC sampling for a provided System. The default value of 1.0 means that step! performs, on average, one trial update per spin.\n\nAssuming ergodicity, the LocalSampler will sample from thermal equilibrium for the target temperature kT. \n\nThe trial spin updates are sampled using the propose function. Built-in options include propose_uniform, propose_flip, and propose_delta. Multiple proposals can be mixed with the macro @mix_proposals.\n\nThe returned object stores fields ΔE and Δs, which represent the cumulative change to the net energy and dipole, respectively.\n\nAn alternative approach to sampling is Langevin, which may be preferred for simulating continuous spins, especially in the presence of long-range dipole-dipole interactions (cf. enable_dipole_dipole!).\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SampledCorrelations","page":"Library API","title":"Sunny.SampledCorrelations","text":"SampledCorrelations\n\nBasic data type for storing sampled correlation data. A SampleCorrelations is initialized by calling either dynamical_correlations or instant_correlations.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SpinInfo","page":"Library API","title":"Sunny.SpinInfo","text":"SpinInfo(atom::Int; S, g=2)\n\nCharacterizes the spin at a given atom index within the crystal unit cell. S is an integer multiple of 1/2 and gives the spin angular momentum in units of ħ. g is the g-factor or tensor, such that an angular momentum dipole s produces a magnetic moment g s in units of the Bohr magneton.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.SpinWaveTheory","page":"Library API","title":"Sunny.SpinWaveTheory","text":"SpinWaveTheory(sys, energy_ϵ::Float64=1e-8)\n\nConstructs an object to perform linear spin wave theory. Use it with dispersion and dssf functions.\n\nThe optional parameter energy_ϵ adds a small positive shift to the diagonal of the dynamical matrix D to avoid numerical issues with zero-energy quasi-particle modes.\n\n\n\n\n\n","category":"type"},{"location":"library.html#Sunny.System-Tuple{Crystal, Tuple{Int64, Int64, Int64}, Vector{SpinInfo}, Symbol}","page":"Library API","title":"Sunny.System","text":"System(crystal::Crystal, latsize, infos, mode; units=Units.meV, seed::Int)\n\nConstruct a System of spins for a given Crystal symmetry. The latsize parameter determines the number of unit cells in each lattice vector direction. The infos parameter is a list of SpinInfo objects, which determine the magnitude S and g-tensor of each spin.\n\nThe two primary options for mode are :SUN and :dipole. In the former, each spin-S degree of freedom is described as an SU(N) coherent state, i.e. a quantum superposition of N = 2S + 1 levels. This formalism can be useful to capture multipolar spin fluctuations or local entanglement effects. \n\nMode :dipole projects the SU(N) dynamics onto the restricted space of pure dipoles. In practice this means that Sunny will simulate Landau-Lifshitz dynamics, but single-ion anisotropy and biquadratic exchange interactions will be renormalized to improve accuracy. To disable this renormalization, use the mode :dipole_large_S which applies the S classical limit. For details, see the documentation page: Interaction Strength Renormalization.\n\nThe default units system of (meV, Å, tesla) can be overridden by with the units parameter; see Units. \n\nAn optional seed may be provided to achieve reproducible random number generation.\n\nAll spins are initially polarized in the z-direction.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.add_sample!-Tuple{SampledCorrelations, System}","page":"Library API","title":"Sunny.add_sample!","text":"add_sample!(sc::SampledCorrelations, sys::System)\n\nadd_trajectory uses the spin configuration contained in the System to generate a correlation data and accumulate it into sc. For static structure factors, this involves analyzing the spin-spin correlations of the spin configuration provided. For a dynamic structure factor, a trajectory is calculated using the given spin configuration as an initial condition. The spin-spin correlations are then calculating in time and accumulated into sc. \n\nThis function will change the state of sys when calculating dynamical structure factor data. To preserve the initial state of sys, it must be saved separately prior to calling add_sample!. Alternatively, the initial spin configuration may be copied into a new System and this new System can be passed to add_sample!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.available_energies-Tuple{SampledCorrelations}","page":"Library API","title":"Sunny.available_energies","text":"available_energies(sc::SampledCorrelations; negative_energies=false)\n\nReturn the ω values for the energy index of a SampledCorrelations. By default, only returns values for non-negative energies, which corresponds to the default output of intensities. Set negative_energies to true to retrieve all ω values.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.available_wave_vectors-Tuple{SampledCorrelations}","page":"Library API","title":"Sunny.available_wave_vectors","text":"available_wave_vectors(sc::SampledCorrelations; bzsize=(1,1,1))\n\nReturns all wave vectors for which sc contains exact values. bsize specifies the number of Brillouin zones to be included.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.axes_bincenters-Tuple{Any, Any, Any}","page":"Library API","title":"Sunny.axes_bincenters","text":"axes_bincenters(params::BinningParameters)\n\nReturns tick marks which label the bins of the histogram described by BinningParameters by their bin centers.\n\nThe following alternative syntax can be used to compute bin centers for a single axis:\n\naxes_bincenters(binstart,binend,binwidth)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.broaden_energy-Tuple{SampledCorrelations, Any, Function}","page":"Library API","title":"Sunny.broaden_energy","text":"broaden_energy(sc::SampledCorrelations, vals, kernel::Function; negative_energies=false)\n\nPerforms a real-space convolution along the energy axis of an array of intensities. Assumes the format of the intensities array corresponds to what would be returned by intensities_interpolated. kernel must be a function that takes two numbers: kernel(ω, ω₀), where ω is a frequency, and ω₀ is the center frequency of the kernel. Sunny provides lorentzian for the most common use case:\n\nnewvals = broaden_energy(sc, vals, (ω, ω₀) -> lorentzian(ω-ω₀, 0.2))\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.count_bins-Tuple{Any, Any, Any}","page":"Library API","title":"Sunny.count_bins","text":"count_bins(binstart,binend,binwidth)\n\nReturns the number of bins in the binning scheme implied by binstart, binend, and binwidth. To count the bins in a BinningParameters, use params.numbins.\n\nThis function defines how partial bins are handled, so it should be used preferentially over computing the number of bins manually.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dispersion-Tuple{SpinWaveTheory, Any}","page":"Library API","title":"Sunny.dispersion","text":"dispersion(swt::SpinWaveTheory, qs)\n\nComputes the spin excitation energy dispersion relations given a SpinWaveTheory and an array of wave vectors qs. Each element q of qs must be a 3-vector in units of reciprocal lattice units. I.e., qᵢ is given in 2πaᵢ with aᵢ the lattice constant of the original chemical lattice.\n\nThe first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of the array is an energy.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dmvec-Tuple{Any}","page":"Library API","title":"Sunny.dmvec","text":"dmvec(D)\n\nAntisymmetric matrix representation of the Dzyaloshinskii-Moriya pseudo-vector,\n\n [ 0 D[3] -D[2]\n -D[3] 0 D[1]\n D[2] -D[1] 0 ]\n\nUseful in the context of set_exchange!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dssf-Tuple{SpinWaveTheory, Any}","page":"Library API","title":"Sunny.dssf","text":"dssf(swt::SpinWaveTheory, qs)\n\nGiven a SpinWaveTheory object, computes the dynamical spin structure factor,\n\n 𝒮^αβ(𝐤 ω) = 1(2πN)dt _𝐫 expi(ωt - 𝐤𝐫) S^α(𝐫 t)S^β(0 0)\n\nusing the result from linear spin-wave theory,\n\n 𝒮^αβ(𝐤 ω) = _n A_n^αβ(𝐤)^2 δω-ω_n(𝐤)\n\nqs is an array of wave vectors of arbitrary dimension. Each element q of qs must be a 3-vector in reciprocal lattice units (RLU), i.e., in the basis of reciprocal lattice vectors.\n\nThe first indices of the returned array correspond to those of qs. A final index, corresponding to mode, is added to these. Each entry of this array is a tensor (3×3 matrix) corresponding to the indices α and β.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.dynamical_correlations-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.dynamical_correlations","text":"dynamical_correlations(sys::System; Δt, nω, ωmax, \n process_trajectory=:none, observables=nothing, correlations=nothing)\n\nCreates an empty SampledCorrelations object for calculating and storing dynamical structure factor intensities 𝒮(𝐪ω). Call add_sample! to accumulate data for the given configuration of a spin system. Internally, this will run a dynamical trajectory and measure time correlations. The 𝒮(𝐪ω) data can be retrieved by calling intensities_interpolated. Alternatively, instant_intensities_interpolated will integrate out ω to obtain 𝒮(𝐪), optionally applying classical-to-quantum correction factors.\n\nThree keywords are required to specify the dynamics used for the trajectory calculation.\n\nΔt: The time step used for calculating the trajectory from which dynamic spin-spin correlations are calculated. The trajectories are calculated with an ImplicitMidpoint integrator.\nωmax: The maximum energy, ω, that will be resolved.\nnω: The number of energy bins to calculated between 0 and ωmax.\n\nAdditional keyword options are the following:\n\nobservables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.\ncorrelations: Specify which correlation functions are calculated, i.e. which matrix elements αβ of 𝒮^αβ(qω) are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.eachsite-Tuple{System}","page":"Library API","title":"Sunny.eachsite","text":"eachsite(sys::System)\n\nAn iterator over all Sites in the system. \n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.enable_dipole_dipole!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.enable_dipole_dipole!","text":"enable_dipole_dipole!(sys::System)\n\nEnables long-range dipole-dipole interactions,\n\n -(μ_04π) _ij (3 (𝐌_j𝐫_ij)(𝐌_i𝐫_ij) - 𝐌_i𝐌_j) 𝐫_ij^3\n\nwhere the sum is over all pairs of spins (singly counted), including periodic images, regularized using the Ewald summation convention. The magnetic moments are 𝐌_i = μ_B g 𝐒_i where g is the g-factor or g-tensor, and 𝐒_i is the spin angular momentum dipole in units of ħ. The Bohr magneton μ_B and vacuum permeability μ_0 are physical constants, with numerical values determined by the unit system.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.energy-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.energy","text":"energy(sys::System)\n\nThe total system energy. See also energy_per_site.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.energy_per_site-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.energy_per_site","text":"energy_per_site(sys::System)\n\nThe total system energy divided by the number of sites.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.generate_mantid_script_from_binning_parameters-Tuple{Any}","page":"Library API","title":"Sunny.generate_mantid_script_from_binning_parameters","text":"generate_mantid_script_from_binning_parameters(params::BinningParameters)\n\nGenerate a Mantid script which bins data according to the given BinningParameters.\n\nwarning: Units\nTake care to ensure the units are correct (R.L.U. or absolute). You may want to call Sunny.bin_rlu_as_absolute_units! or Sunny.bin_absolute_units_as_rlu! first.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.global_position-Tuple{System, Any}","page":"Library API","title":"Sunny.global_position","text":"global_position(sys::System, site::Site)\n\nPosition of a Site in global coordinates.\n\nTo precompute a full list of positions, one can use eachsite as below:\n\npos = [global_position(sys, site) for site in eachsite(sys)]\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.instant_correlations-Tuple{System}","page":"Library API","title":"Sunny.instant_correlations","text":"instant_correlations(sys::System; process_trajectory=:none, observables=nothing, correlations=nothing)\n\nCreates an empty SampledCorrelations object for calculating and storing instantaneous structure factor intensities 𝒮(𝐪). Call add_sample! to accumulate data for the given configuration of a spin system. Call instant_intensities_interpolated to retrieve averaged 𝒮(𝐪) data.\n\nImportant note: When dealing with continuous (non-Ising) spins, consider creating using dynamical_correlations instead of instant_correlations. The former will provide full 𝒮(𝐪ω) data, from which 𝒮(𝐪) can be obtained by integrating out ω. During this integration step, Sunny can incorporate temperature- and ω-dependent classical-to-quantum correction factors to produce more accurate 𝒮(𝐪) estimates. See instant_intensities_interpolated for more information.\n\nThe following optional keywords are available:\n\nobservables: Allows the user to specify custom observables. The observables must be given as a list of complex N×N matrices or LinearMaps. It's recommended to name each observable, for example: observables = [:A => a_observable_matrix, :B => b_map, ...]. By default, Sunny uses the 3 components of the dipole, :Sx, :Sy and :Sz.\ncorrelations: Specify which correlation functions are calculated, i.e. which matrix elements αβ of 𝒮^αβ(qω) are calculated and stored. Specified with a vector of tuples. By default Sunny records all auto- and cross-correlations generated by all observables. To retain only the xx and xy correlations, one would set correlations=[(:Sx,:Sx), (:Sx,:Sy)] or correlations=[(1,1),(1,2)].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.instant_intensities_interpolated-Tuple{SampledCorrelations, Any, Any}","page":"Library API","title":"Sunny.instant_intensities_interpolated","text":"instant_intensities_interpolated(sc::SampledCorrelations, qs; kwargs...)\n\nReturn 𝒮(𝐪) intensities at wave vectors qs. The functionality is very similar to intensities_interpolated, except the returned array has dimensions identical to qs. If called on a SampledCorrelations with dynamical information, i.e., 𝒮(𝐪ω), the ω information is integrated out.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.integrate_axes!-Tuple{BinningParameters}","page":"Library API","title":"Sunny.integrate_axes!","text":"integrate_axes!(params::BinningParameters; axes)\n\nIntegrate over one or more axes of the histogram by setting the number of bins in that axis to 1. Examples:\n\nintegrate_axes!(params; axes = [2,3])\nintegrate_axes!(params; axes = 2)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.integrated_lorentzian-Tuple{Float64}","page":"Library API","title":"Sunny.integrated_lorentzian","text":"integrated_lorentzian(η)\n\nReturns x mapsto atan(xη)π for use with intensities_binned.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_bands-Tuple{SpinWaveTheory, Any, Sunny.SpinWaveIntensityFormula}","page":"Library API","title":"Sunny.intensities_bands","text":"dispersion, intensities = intensities_bands(swt::SpinWaveTheory, ks, formula::SpinWaveIntensityFormula)\n\nComputes the scattering intensities at each energy band for each momentum transfer k in ks, according to Linear Spin Wave Theory and the given intensity formula. The formula must have a delta-function kernel, e.g.:\n\nformula = intensity_formula(swt, :perp, formula; kernel = delta_function_kernel)\n\nor else the bands will be broadened, and their intensity can not be computed.\n\nThe outputs will be arrays with indices identical to ks, with the last index giving the band index. dispersions reports the energy of each band, while intensities reports the scattering intensity.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_binned-Tuple{SampledCorrelations, BinningParameters, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.intensities_binned","text":"intensity, counts = intensities_binned(sc::SampledCorrelations, params::BinningParameters, formula; integrated_kernel)\n\nGiven correlation data contained in a SampledCorrelations and BinningParameters describing the shape of a histogram, compute the intensity and normalization for each histogram bin using a given intensity_formula.\n\nThe BinningParameters are expected to accept (q,ω) in R.L.U. for the (possibly reshaped) crystal associated with sc.\n\nThis is an alternative to intensities_interpolated which bins the scattering intensities into a histogram instead of interpolating between them at specified qs values. See unit_resolution_binning_parameters for a reasonable default choice of BinningParameters which roughly emulates intensities_interpolated with interpolation = :round.\n\nIf a function integrated_kernel(Δω) is passed, it will be used as the CDF of a kernel function for energy broadening. For example, integrated_kernel = Δω -> atan(Δω/η)/pi (c.f. integrated_lorentzian implements Lorentzian broadening with parameter η. Energy-dependent energy broadening can be achieved by providing an integrated_kernel(ω,Δω) whose first argument is the energy transfer ω.\n\nCurrently, energy broadening is only supported if the BinningParameters are such that the first three axes are purely spatial and the last (energy) axis is [0,0,0,1].\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_broadened-Tuple{SpinWaveTheory, Any, Any, Any}","page":"Library API","title":"Sunny.intensities_broadened","text":"intensities_broadened(swt::SpinWaveTheory, ks, ωvals, formula)\n\nComputes the scattering intensities at each (Q,ω) according to Linear Spin Wave Theory and the given intensity formula. The required formula must have a non-delta-function kernel, e.g.:\n\nformula = intensity_formula(swt, :perp; kernel = lorentzian(0.05))\n\nor else the intensity at ωvals which are not exactly on the dispersion curve can not be calculated.\n\nThe intensity is computed at each wave vector in ks and each energy in ωvals. The output will be an array with indices identical to ks, with the last index matching ωvals.\n\nNote that ks is an array of wave vectors of arbitrary dimension. Each element k of ks must be a 3-wavevector in absolute units.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensities_interpolated-Tuple{SampledCorrelations, Any, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.intensities_interpolated","text":"intensities_interpolated(sc::SampledCorrelations, qs, formula:ClassicalIntensityFormula; interpolation=nothing, negative_energies=false)\n\nThe basic function for retrieving 𝒮(𝐪ω) information from a SampledCorrelations. Maps an array of wave vectors qs to an array of structure factor intensities, including an additional energy index. The values of ω associated with the energy index can be retrieved by calling available_energies. The three coordinates of each wave vector are measured in reciprocal lattice units, i.e., multiples of the reciprocal lattice vectors.\n\ninterpolation: Since 𝒮(𝐪 ω) is calculated on a finite lattice, data is only available at discrete wave vectors. By default, Sunny will round a requested q to the nearest available wave vector. Linear interpolation can be applied by setting interpolation=:linear.\nnegative_energies: If set to true, Sunny will return the periodic extension of the energy axis. Most users will not want this.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SampledCorrelations, AbstractVector{Int64}}","page":"Library API","title":"Sunny.intensity_formula","text":"formula = intensity_formula(sc::SampledCorrelations)\n\nEstablish a formula for computing the intensity of the discrete scattering modes (q,ω) using the correlation data 𝒮^αβ(qω) stored in the SampledCorrelations. The formula returned from intensity_formula can be passed to intensities_interpolated or intensities_binned.\n\nintensity_formula(sc,...; kT = Inf, formfactors = ...)\n\nThere are keyword arguments providing temperature and form factor corrections:\n\nkT: If a temperature is provided, the intensities will be rescaled by a temperature- and ω-dependent classical-to-quantum factor. kT should be specified when making comparisons with spin wave calculations or experimental data. If kT is not specified, infinite temperature (no correction) is assumed.\nformfactors: To apply form factor corrections, provide this keyword with a list of FormFactors, one for each symmetry-distinct site in the crystal. The order of FormFactors must correspond to the order of site symmetry classes, e.g., as they appear when printed in display(crystal).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SampledCorrelations, Any}","page":"Library API","title":"Sunny.intensity_formula","text":"A custom intensity formula can be specifed by providing a function intensity = f(q,ω,correlations) and specifying which correlations it requires:\n\nintensity_formula(f,sc::SampledCorrelations, required_correlations; kwargs...)\n\nThe function is intended to be specified using do notation. For example, this custom formula sums the off-diagonal correlations:\n\nrequired = [(:Sx,:Sy),(:Sy,:Sz),(:Sx,:Sz)]\nintensity_formula(sc,required,return_type = ComplexF64) do k, ω, off_diagonal_correlations\n sum(off_diagonal_correlations)\nend\n\nIf your custom formula returns a type other than Float64, use the return_type keyword argument to flag this.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{Function, SpinWaveTheory, AbstractVector{Int64}}","page":"Library API","title":"Sunny.intensity_formula","text":"formula = intensity_formula(swt::SpinWaveTheory; kernel = ...)\n\nEstablish a formula for computing the scattering intensity by diagonalizing the hamiltonian H(q) using Linear Spin Wave Theory.\n\nIf kernel = delta_function_kernel, then the resulting formula can be used with intensities_bands.\n\nIf kernel is an energy broadening kernel function, then the resulting formula can be used with intensities_broadened. Energy broadening kernel functions can either be a function of Δω only, e.g.:\n\nkernel = Δω -> ...\n\nor a function of both the energy transfer ω and of Δω, e.g.:\n\nkernel = (ω,Δω) -> ...\n\nThe integral of a properly normalized kernel function over all Δω is one.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.intensity_formula-Tuple{SpinWaveTheory, Symbol}","page":"Library API","title":"Sunny.intensity_formula","text":"intensity_formula([swt or sc], contraction_mode::Symbol)\n\nSunny has several built-in formulas that can be selected by setting contraction_mode to one of these values:\n\n:trace (default), which yields operatornametr 𝒮(qω) = _α 𝒮^αα(qω)\n:perp, which contracts 𝒮^αβ(qω) with the dipole factor δ_αβ - q_αq_β, returning the unpolarized intensity.\n:full, which will return all elements 𝒮^αβ(𝐪ω) without contraction.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lattice_params-Tuple{StaticArraysCore.SMatrix{3, 3, Float64, 9}}","page":"Library API","title":"Sunny.lattice_params","text":"lattice_params(latvecs::Mat3)\n\nCompute the lattice parameters (a b c α β γ) for the three lattice vectors provided as columns of latvecs. The inverse mapping is lattice_vectors.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lattice_vectors-NTuple{6, Any}","page":"Library API","title":"Sunny.lattice_vectors","text":"lattice_vectors(a, b, c, α, β, γ)\n\nReturn the lattice vectors, as columns of the 33 output matrix, that correspond to the conventional unit cell defined by the lattice constants (a b c) and the angles (α β γ) in degrees. The inverse mapping is lattice_params.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.load_nxs-Tuple{Any}","page":"Library API","title":"Sunny.load_nxs","text":"params, signal = load_nxs(filename)\n\nGiven the name of a Mantid-exported MDHistoWorkspace file, load the BinningParameters and the signal from that file.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.lorentzian-Tuple{Any, Any}","page":"Library API","title":"Sunny.lorentzian","text":"lorentzian(x, η)\n\nReturns η(π(x^2 + η^2)).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.magnetic_moment-Tuple{System, Any}","page":"Library API","title":"Sunny.magnetic_moment","text":"magnetic_moment(sys::System, site::Site)\n\nGet the magnetic moment for a Site. This is the spin dipole multiplied by the Bohr magneton and the local g-tensor.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.merge_correlations-Union{Tuple{Array{SampledCorrelations{N}, 1}}, Tuple{N}} where N","page":"Library API","title":"Sunny.merge_correlations","text":"merge_correlations(scs::Vector{SampledCorrelations)\n\nAccumulate a list of SampledCorrelations into a single, summary SampledCorrelations. Useful for reducing the results of parallel computations.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.minimize_energy!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.minimize_energy!","text":"minimize_energy!(sys::System{N}; maxiters=100, subiters=20,\n method=Optim.ConjugateGradient(), kwargs...) where N\n\nOptimizes the spin configuration in sys to minimize energy. A total of maxiters iterations will be attempted, with restarts after every subiters iterations. The remaining kwargs will be forwarded to the optimize method of the Optim.jl package.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.polarize_spins!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.polarize_spins!","text":"polarize_spins!(sys::System, dir)\n\nPolarize all spins in the system along the direction dir.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.position_to_site-Tuple{System, Any}","page":"Library API","title":"Sunny.position_to_site","text":"position_to_site(sys::System, r)\n\nConverts a position r to four indices of a Site. The coordinates of r are given in units of the lattice vectors for the original crystal. This function can be useful for working with systems that have been reshaped using reshape_supercell.\n\nExample\n\n# Find the `site` at the center of a unit cell which is displaced by four\n# multiples of the first lattice vector\nsite = position_to_site(sys, [4.5, 0.5, 0.5])\n\n# Print the dipole at this site\nprintln(sys.dipoles[site])\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.powder_average_binned-Tuple{SampledCorrelations, Any, Sunny.ClassicalIntensityFormula}","page":"Library API","title":"Sunny.powder_average_binned","text":"powder_average_binned(sc::SampledCorrelations, radial_binning_parameters; formula\n ω_binning_parameters, integrated_kernel = nothing, bzsize = nothing)\n\nThis function emulates the experimental situation of \"powder averaging,\" where only the magnitude (and not the direction) of the momentum transfer is resolvable. The intensities are binned similarly to intensities_binned, but the histogram x-axis is |k| in absolute units, which is a nonlinear function of kx,ky,kz. The y-axis is energy.\n\nRadial binning parameters are specified as tuples (start,end,bin_width), e.g. radial_binning_parameters = (0,6π,6π/55).\n\nEnergy broadening is supported in the same way as intensities_binned, and this function accepts the same kind of intensity_formula.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.primitive_cell_shape-Tuple{Crystal}","page":"Library API","title":"Sunny.primitive_cell_shape","text":"primitive_cell_shape(cryst::Crystal)\n\nReturns the shape of the primitive cell as a 3×3 matrix, in fractional coordinates of the conventional lattice vectors. May be useful for constructing inputs to reshape_supercell.\n\nExamples\n\n# Valid if `cryst` has not been reshaped\n@assert cryst.prim_latvecs ≈ cryst.latvecs * primitive_cell_shape(cryst)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_bond-Tuple{Crystal, Bond}","page":"Library API","title":"Sunny.print_bond","text":"print_bond(cryst::Crystal, bond::Bond; b_ref::Bond)\n\nPrints symmetry information for bond bond. A symmetry-equivalent reference bond b_ref can optionally be provided to fix the meaning of the coefficients A, B, ...\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_site-Tuple{Any, Any}","page":"Library API","title":"Sunny.print_site","text":"print_site(cryst, i; R=I)\n\nPrint symmetry information for the site i, including allowed g-tensor and allowed anisotropy operator. An optional rotation matrix R can be provided to define the reference frame for expression of the anisotropy.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_stevens_expansion-Tuple{AbstractMatrix}","page":"Library API","title":"Sunny.print_stevens_expansion","text":"function print_stevens_expansion(op)\n\nPrints a local Hermitian operator as a linear combination of Stevens operators. The operator op may be a finite-dimensional matrix or an abstract spin polynomial in the large-S limit.\n\nExamples\n\nS = spin_matrices(2)\nprint_stevens_expansion(S[1]^4 + S[2]^4 + S[3]^4)\n# Prints: (1/20)𝒪₄₀ + (1/4)𝒪₄₄ + 102/5\n\nS = spin_matrices(Inf)\nprint_stevens_expansion(S[1]^4 + S[2]^4 + S[3]^4)\n# Prints: (1/20)𝒪₄₀ + (1/4)𝒪₄₄ + (3/5)𝒮⁴\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_suggested_frame-Tuple{Crystal, Int64}","page":"Library API","title":"Sunny.print_suggested_frame","text":"print_suggested_frame(cryst, i; digits=4)\n\nPrint a suggested reference frame, as a rotation matrix R, that can be used as input to print_site(). The purpose is to simplify the description of allowed anisotropies.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_symmetry_table-Tuple{Crystal, Any}","page":"Library API","title":"Sunny.print_symmetry_table","text":"print_symmetry_table(cryst::Crystal, max_dist)\n\nPrint symmetry information for all equivalence classes of sites and bonds, up to a maximum bond distance of max_dist. Equivalent to calling print_bond(cryst, b) for every bond b in reference_bonds(cryst, max_dist), where Bond(i, i, [0,0,0]) refers to a single site i.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.print_wrapped_intensities-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.print_wrapped_intensities","text":"print_wrapped_intensities(sys::System; nmax=10)\n\nFor Bravais lattices: Prints up to nmax wavevectors according to their instantaneous (static) structure factor intensities, listed in descending order. For non-Bravais lattices: Performs the same analysis for each spin sublattice independently; the output weights are naïvely averaged over sublattices, without incorporating phase shift information. This procedure therefore wraps all wavevectors into the first Brillouin zone. Each wavevector coordinate is given between -12 and 12 in reciprocal lattice units (RLU). The output from this function will typically be used as input to suggest_magnetic_supercell.\n\nBecause this function does not incorporate phase information in its averaging over sublattices, the printed weights are not directly comparable with experiment. For that purpose, use instant_correlations instead.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_delta-Tuple{Any}","page":"Library API","title":"Sunny.propose_delta","text":"propose_delta(magnitude)\n\nGenerate a proposal function that adds a Gaussian perturbation to the existing spin state. In :dipole mode, the procedure is to first introduce a random three-vector perturbation 𝐬 = 𝐬 + 𝐬 ξ and then return the properly normalized spin 𝐬 (𝐬𝐬). Each component of the random vector ξ is Gaussian distributed with a standard deviation of magnitude; the latter is dimensionless and typically smaller than one. \n\nIn :SUN mode, the procedure is analogous, but now involving Gaussian perturbations to each of the N complex components of an SU(N) coherent state.\n\nIn the limit of very large magnitude, this function coincides with propose_uniform.\n\nFor use with LocalSampler.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_flip-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.propose_flip","text":"propose_flip\n\nFunction to propose pure spin flip updates in the context of a LocalSampler. Dipoles are flipped as 𝐬 -𝐬. SU(N) coherent states are flipped using the time-reversal operator.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.propose_uniform","page":"Library API","title":"Sunny.propose_uniform","text":"propose_uniform\n\nFunction to propose a uniformly random spin update in the context of a LocalSampler. In :dipole mode, the result is a random three-vector with appropriate normalization. In :SUN mode, the result is a random SU(N) coherent state with appropriate normalization.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.randomize_spins!-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.randomize_spins!","text":"randomize_spins!(sys::System)\n\nRandomizes all spins under appropriate the uniform distribution.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_path-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.reciprocal_space_path","text":"reciprocal_space_path(cryst::Crystal, qs, density)\n\nReturns a pair (path, xticks). The path return value is a list of wavevectors that samples linearly between the provided wavevectors qs. The xticks return value can be used to label the special 𝐪 values on the x-axis of a plot.\n\nSpecial note about units: the wavevectors qs must be provided in reciprocal lattice units (RLU) for the given crystal, but the sampling density must be specified in the global frame. Specifically, the density is given as number of sample points per unit of radian inverse length, where the unit of length is the same as that used to specify the lattice vectors of the Crystal. The path will therefore include more samples between q-points that are further apart in absolute Fourier distance.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_path_bins-Tuple{Any, Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.reciprocal_space_path_bins","text":"reciprocal_space_path_bins(sc,qs,density,args...;kwargs...)\n\nTakes a list of wave vectors, qs in R.L.U., and builds a series of histogram BinningParameters whose first axis traces a path through the provided points. The second and third axes are integrated over according to the args and kwargs, which are passed through to slice_2D_binning_parameters.\n\nAlso returned is a list of marker indices corresponding to the input points, and a list of ranges giving the indices of each histogram x-axis within a concatenated histogram. The density parameter is given in samples per reciprocal lattice unit (R.L.U.).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reciprocal_space_shell-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.reciprocal_space_shell","text":"reciprocal_space_shell(cryst::Crystal, radius, n)\n\nSample n points on the reciprocal space sphere with a given radius (units of inverse length).\n\nExamples\n\n# Sample wavevectors on the sphere at fixed density\nreciprocal_space_shell(cryst, r, 4π*r^2*density)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reference_bonds-Tuple{Crystal, Float64}","page":"Library API","title":"Sunny.reference_bonds","text":"reference_bonds(cryst::Crystal, max_dist)\n\nReturns a full list of bonds, one for each symmetry equivalence class, up to distance max_dist. The reference bond b for each equivalence class is selected according to a scoring system that prioritizes simplification of the elements in basis_for_symmetry_allowed_couplings(cryst, b).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.remove_periodicity!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.remove_periodicity!","text":"remove_periodicity!(sys::System, dims)\n\nRemove periodic interactions along the dimensions where dims is true. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nExample\n\n# Remove periodic boundaries along the 1st and 3rd dimensions\nremove_periodicity!(sys::System, (true, false, true))\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.repeat_periodically-Union{Tuple{N}, Tuple{System{N}, Tuple{Int64, Int64, Int64}}} where N","page":"Library API","title":"Sunny.repeat_periodically","text":"repeat_periodically(sys::System{N}, counts::NTuple{3,Int}) where N\n\nCreates a System identical to sys but repeated a given number of times in each dimension, specified by the tuple counts.\n\nSee also reshape_supercell.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.reshape_supercell-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.reshape_supercell","text":"reshape_supercell(sys::System, shape)\n\nMaps an existing System to a new one that has the shape and periodicity of a requested supercell. The columns of the 33 integer matrix shape represent the supercell lattice vectors measured in units of the original crystal lattice vectors.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.resize_supercell-Union{Tuple{N}, Tuple{System{N}, Tuple{Int64, Int64, Int64}}} where N","page":"Library API","title":"Sunny.resize_supercell","text":"resize_supercell(sys::System{N}, latsize::NTuple{3,Int}) where N\n\nCreates a System with a given number of conventional unit cells in each lattice vector direction. Interactions and other settings will be inherited from sys.\n\nConvenience function for:\n\nreshape_supercell(sys, [latsize[1] 0 0; 0 latsize[2] 0; 0 0 latsize[3]])\n\nSee also reshape_supercell.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.rotate_operator-Tuple{LinearAlgebra.Hermitian{ComplexF64, Matrix{ComplexF64}}, Any}","page":"Library API","title":"Sunny.rotate_operator","text":"rotate_operator(A, R)\n\nRotates the local quantum operator A according to the 33 rotation matrix R.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.rotation_in_rlu-Tuple{Crystal, Any, Any}","page":"Library API","title":"Sunny.rotation_in_rlu","text":"rotation_in_rlu(cryst::Crystal, axis, angle)\n\nReturns a 33 matrix that rotates wavevectors in reciprocal lattice units (RLU). The axis vector is a real-space direction in absolute units (but arbitrary magnitude), and the angle is in radians.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_coherent!-Union{Tuple{N}, Tuple{System{N}, Any, Any}} where N","page":"Library API","title":"Sunny.set_coherent!","text":"set_coherent!(sys::System, Z, site::Site)\n\nSet a coherent spin state at a Site using the N complex amplitudes in Z.\n\nFor a standard SpinInfo, these amplitudes will be interpreted in the eigenbasis of 𝒮ᶻ. That is, Z[1] represents the amplitude for the basis state fully polarized along the z-direction, and subsequent components represent states with decreasing angular momentum along this axis (m = S S-1 -S).\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_dipole!-Union{Tuple{N}, Tuple{System{N}, Any, Any}} where N","page":"Library API","title":"Sunny.set_dipole!","text":"set_dipole!(sys::System, dir, site::Site)\n\nPolarize the spin at a Site along the direction dir.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_exchange!-Union{Tuple{N}, Tuple{System{N}, Any, Bond}} where N","page":"Library API","title":"Sunny.set_exchange!","text":"set_exchange!(sys::System, J, bond::Bond)\n\nSets a 3×3 spin-exchange matrix J along bond, yielding a pairwise interaction energy 𝐒_iJ 𝐒_j. This interaction will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells.\n\nThe parameter J may be scalar or matrix-valued. As a convenience, dmvec(D) can be used to construct the antisymmetric part of the exchange, where D is the Dzyaloshinskii-Moriya pseudo-vector. The resulting interaction will be 𝐃(𝐒_i𝐒_j).\n\nFor more general interactions, such as biquadratic, use set_pair_coupling! instead.\n\nExamples\n\n# An explicit exchange matrix\nJ1 = [2 3 0;\n -3 2 0;\n 0 0 2]\nset_exchange!(sys, J1, bond)\n\n# An equivalent Heisenberg + DM exchange \nJ2 = 2*I + dmvec([0,0,3])\nset_exchange!(sys, J2, bond)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_exchange_at!-Union{Tuple{N}, Tuple{System{N}, Any, Union{NTuple{4, Int64}, CartesianIndex{4}}, Union{NTuple{4, Int64}, CartesianIndex{4}}}} where N","page":"Library API","title":"Sunny.set_exchange_at!","text":"set_exchange_at!(sys::System, J, site1::Site, site2::Site; offset=nothing)\n\nSets an exchange interaction `𝐒_i⋅J 𝐒_j along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nIf the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.\n\nFor more general interactions, such as biquadratic, use set_pair_coupling_at! instead.\n\nSee also set_exchange!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_external_field!-Tuple{System, Any}","page":"Library API","title":"Sunny.set_external_field!","text":"set_external_field!(sys::System, B::Vec3)\n\nSets the external field B that couples to all spins.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_external_field_at!-Tuple{System, Any, Any}","page":"Library API","title":"Sunny.set_external_field_at!","text":"set_external_field_at!(sys::System, B::Vec3, site::Site)\n\nSets a Zeeman coupling between a field B and a single spin. Site includes a unit cell and a sublattice index.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_onsite_coupling!-Tuple{System, Any, Int64}","page":"Library API","title":"Sunny.set_onsite_coupling!","text":"set_onsite_coupling!(sys::System, op, i::Int)\n\nSet the single-ion anisotropy for the ith atom of every unit cell, as well as all symmetry-equivalent atoms. The operator op may be provided as an abstract function of the local spin operators, as a polynomial of spin_matrices, or as a linear combination of stevens_matrices.\n\nExamples\n\n# An easy axis anisotropy in the z-direction\nset_onsite_coupling!(sys, S -> -D*S[3]^3, i)\n\n# The unique quartic single-ion anisotropy for a site with cubic point group\n# symmetry\nset_onsite_coupling!(sys, S -> 20*(S[1]^4 + S[2]^4 + S[3]^4), i)\n\n# An equivalent expression of this quartic anisotropy, up to a constant shift\nO = stevens_matrices(spin_label(sys, i))\nset_onsite_coupling!(sys, O[4,0] + 5*O[4,4], i)\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_onsite_coupling_at!-Tuple{System, Any, Union{NTuple{4, Int64}, CartesianIndex{4}}}","page":"Library API","title":"Sunny.set_onsite_coupling_at!","text":"set_onsite_coupling_at!(sys::System, op, site::Site)\n\nSets the single-ion anisotropy operator op for a single Site, ignoring crystal symmetry. The system must support inhomogeneous interactions via to_inhomogeneous.\n\nSee also set_onsite_coupling!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_pair_coupling!-Union{Tuple{N}, Tuple{System{N}, AbstractMatrix, Any}} where N","page":"Library API","title":"Sunny.set_pair_coupling!","text":"set_pair_coupling!(sys::System, op, bond)\n\nSets an arbitrary coupling op along bond. This coupling will be propagated to equivalent bonds in consistency with crystal symmetry. Any previous interactions on these bonds will be overwritten. The parameter bond has the form Bond(i, j, offset), where i and j are atom indices within the unit cell, and offset is a displacement in unit cells. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites.\n\nExamples\n\n# Bilinear+biquadratic exchange involving 3×3 matrices J1 and J2\nset_pair_coupling!(sys, (Si, Sj) -> Si'*J1*Sj + (Si'*J2*Sj)^2, bond)\n\n# Equivalent expression using an appropriate fixed matrix representation\nS = spin_matrices(1/2)\nSi, Sj = to_product_space(S, S)\nset_pair_coupling!(sys, Si'*J1*Sj + (Si'*J2*Sj)^2, bond)\n\nSee also spin_matrices, to_product_space.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_pair_coupling_at!-Union{Tuple{N}, Tuple{System{N}, AbstractMatrix, Union{NTuple{4, Int64}, CartesianIndex{4}}, Union{NTuple{4, Int64}, CartesianIndex{4}}}} where N","page":"Library API","title":"Sunny.set_pair_coupling_at!","text":"set_pair_coupling_at!(sys::System, op, bond)\n\nSets an arbitrary coupling along the single bond connecting two Sites, ignoring crystal symmetry. Any previous coupling on this bond will be overwritten. The operator op may be provided as an anonymous function that accepts two spin dipole operators, or as a matrix that acts in the tensor product space of the two sites. The documentation for set_pair_coupling! provides examples constructing op.\n\nThe system must support inhomogeneous interactions via to_inhomogeneous.\n\nIf the system is relatively small, the direction of the bond can be ambiguous due to possible periodic wrapping. Resolve this ambiguity by passing an explicit offset vector, in multiples of unit cells.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_spiral_order!-Tuple{Any}","page":"Library API","title":"Sunny.set_spiral_order!","text":"set_spiral_order!(sys; q, axis, S0)\n\nInitializes the system with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0 at the real-space origin. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.\n\nExample\n\n# Spiral order for a wavevector propagating in the direction of the first\n# reciprocal lattice vector (i.e., orthogonal to the lattice vectors ``𝐚_2``\n# and ``𝐚_3``), repeating with a period of 10 lattice constants, and spiraling\n# about the ``ẑ``-axis. The spin at the origin will point in the direction\n# ``𝐒_0 = ŷ + ẑ``. Here, ``(x̂, ŷ, ẑ)`` are the axes of Cartesian coordinate\n# system in the global frame.\nset_spiral_order!(sys; q=[1/10, 0, 0], axis=[0, 0, 1], S0=[0, 1, 1])\n\nSee also set_spiral_order_on_sublattice!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_spiral_order_on_sublattice!-Tuple{Any, Any}","page":"Library API","title":"Sunny.set_spiral_order_on_sublattice!","text":"set_spiral_order_on_sublattice!(sys, i; q, axis, S0)\n\nInitializes sublattice i with a spiral order described by the wavevector q, an axis of rotation axis, and an initial dipole direction S0. The phase is selected such that the spin at sys.dipole[1,1,1,i] will point in the direction of S0. The wavevector is expected in repicrocal lattice units (RLU), while the direction vectors axis and S0 are expected in global Cartesian coordinates.\n\nThis function is not available for systems with reshaped unit cells.\n\nSee also set_spiral_order!.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.set_vacancy_at!-Union{Tuple{N}, Tuple{System{N}, Any}} where N","page":"Library API","title":"Sunny.set_vacancy_at!","text":"set_vacancy_at!(sys::System, site::Site)\n\nMake a single site nonmagnetic. Site includes a unit cell and a sublattice index.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.slice_2D_binning_parameters-Tuple{Vector{Float64}, Any, Any, Int64, Any}","page":"Library API","title":"Sunny.slice_2D_binning_parameters","text":"slice_2D_binning_parameter(sc::SampledCorrelations, cut_from_q, cut_to_q, cut_bins::Int64, cut_width::Float64; plane_normal = [0,0,1],cut_height = cutwidth)\n\nCreates BinningParameters which make a cut along one dimension of Q-space.\n\nThe x-axis of the resulting histogram consists of cut_bins-many bins ranging from cut_from_q to cut_to_q. The width of the bins in the transverse direciton is controlled by cut_width and cut_height.\n\nThe binning in the transverse directions is defined in the following way, which sets their normalization and orthogonality properties:\n\ncut_covector = normalize(cut_to_q - cut_from_q)\ntransverse_covector = normalize(plane_normal × cut_covector)\ncotransverse_covector = normalize(transverse_covector × cut_covector)\n\nIn other words, the axes are orthonormal with respect to the Euclidean metric.\n\nIf the cut is too narrow, there will be very few scattering vectors per bin, or the number per bin will vary substantially along the cut. If the output appears under-resolved, try increasing cut_width.\n\nThe four axes of the resulting histogram are:\n\nAlong the cut\nFist transverse Q direction\nSecond transverse Q direction\nEnergy\n\nThis function can be used without reference to a SampledCorrelations using this alternate syntax to manually specify the bin centers for the energy axis:\n\nslice_2D_binning_parameter(ω_bincenters, cut_from, cut_to,...)\n\nwhere ω_bincenters specifies the energy axis, and both cut_from and cut_to are arbitrary covectors, in any units.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.spin_label-Tuple{System, Int64}","page":"Library API","title":"Sunny.spin_label","text":"spin_label(sys::System, i::Int)\n\nIf atom i carries a single spin-S moment, then returns the half-integer label S. Otherwise, throws an error.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.spin_matrices-Tuple{Any}","page":"Library API","title":"Sunny.spin_matrices","text":"spin_matrices(S)\n\nReturns a triple of NN spin matrices, where N = 2S+1. These are the generators of SU(2) in the spin-S representation.\n\nIf S == Inf, then the return values are abstract symbols denoting infinite-dimensional matrices that commute. These can be useful for repeating historical studies, or modeling micromagnetic systems. A technical discussion appears in the Sunny documentation page: Interaction Strength Renormalization.\n\nExample\n\nS = spin_matrices(3/2)\n@assert S'*S ≈ (3/2)*(3/2+1)*I\n@assert S[1]*S[2] - S[2]*S[1] ≈ im*S[3]\n\nS = spin_matrices(Inf)\n@assert S[1]*S[2] - S[2]*S[1] == 0\n\nSee also print_stevens_expansion.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.step!","page":"Library API","title":"Sunny.step!","text":"step!(sys::System, dynamics)\n\nAdvance the spin configuration one dynamical time-step. The dynamics object may be a continuous spin dynamics, such as Langevin or ImplicitMidpoint, or it may be a discrete Monte Carlo sampling scheme such as LocalSampler.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.stevens_matrices-Tuple{Any}","page":"Library API","title":"Sunny.stevens_matrices","text":"stevens_matrices(S)\n\nReturns a generator of Stevens operators in the spin-S representation. The return value O can be indexed as O[k,q], where 0 k 6 labels an irrep of SO(3) and -k q k. This will produce an NN matrix where ``N = 2S\n\n1``. Linear combinations of Stevens operators can be used as a \"physical\n\nbasis\" for decomposing local observables. To see this decomposition, use print_stevens_expansion.\n\nIf S == Inf, then symbolic operators will be returned. In this infinite dimensional limit, the Stevens operators become homogeneous polynomials of commuting spin operators.\n\nExample\n\nO = stevens_matrices(2)\nS = spin_matrices(2)\n\nA = (1/20)O[4,0] + (1/4)O[4,4] + (102/5)I\nB = S[1]^4 + S[2]^4 + S[3]^4\n@assert A ≈ B\n\nSee also spin_matrices and Interaction Strength Renormalization.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.subcrystal-Union{Tuple{N}, Tuple{Crystal, Vararg{String, N}}} where N","page":"Library API","title":"Sunny.subcrystal","text":"subcrystal(cryst, types) :: Crystal\n\nFilters sublattices of a Crystal by atom types, keeping the space group unchanged.\n\nsubcrystal(cryst, classes) :: Crystal\n\nFilters sublattices of Crystal by equivalence classes, keeping the space group unchanged.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.suggest_magnetic_supercell-Tuple{Any}","page":"Library API","title":"Sunny.suggest_magnetic_supercell","text":"suggest_magnetic_supercell(qs; tol=1e-12, maxsize=100)\n\nSuggests a magnetic supercell, in units of the crystal lattice vectors, that is consistent with periodicity of the wavevectors qs in RLU. If the wavevectors are incommensurate (with respect to the maximum supercell size maxsize), one can select a larger error tolerance tol to find a supercell that is almost commensurate.\n\nPrints a 33 matrix of integers that is suitable for use in reshape_supercell.\n\nExamples\n\n# A magnetic supercell for a single-Q structure. Will print\nq1 = [0, -1/4, 1/4]\nsuggest_magnetic_supercell([q1]) # [1 0 0; 0 2 1; 0 -2 1]\n\n# A larger magnetic supercell for a double-Q structure\nq2 = [1/4, 0, 1/4]\nsuggest_magnetic_supercell([q1, q2]) # [1 2 2; -1 2 -2; -1 2 2]\n\n# If given incommensurate wavevectors, find an approximate supercell that\n# is exactly commensurate for nearby wavevectors.\nsuggest_magnetic_supercell([[0, 0, 1/√5], [0, 0, 1/√7]]; tol=1e-2)\n\n# This prints [1 0 0; 0 1 0; 0 0 16], which becomes commensurate under the\n# approximations `1/√5 ≈ 7/16` and `1/√7 ≈ 3/8`.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.symmetry_equivalent_bonds-Tuple{System, Bond}","page":"Library API","title":"Sunny.symmetry_equivalent_bonds","text":"symmetry_equivalent_bonds(sys::System, bond::Bond)\n\nGiven a Bond for the original (unreshaped) crystal, return all symmetry equivalent bonds in the System. Each returned bond is represented as a pair of Sites, which may be used as input to set_exchange_at!. Reverse bonds are not included (no double counting).\n\nExample\n\nfor (site1, site2, offset) in symmetry_equivalent_bonds(sys, bond)\n @assert site1 < site2\n set_exchange_at!(sys, J, site1, site2; offset)\nend\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.to_inhomogeneous-Union{Tuple{System{N}}, Tuple{N}} where N","page":"Library API","title":"Sunny.to_inhomogeneous","text":"to_inhomogeneous(sys::System)\n\nReturns a copy of the system that allows for inhomogeneous interactions, which can be set using set_onsite_coupling_at!, set_exchange_at!, and set_vacancy_at!.\n\nInhomogeneous systems do not support symmetry-propagation of interactions or system reshaping.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.to_product_space-Tuple{Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.to_product_space","text":"to_product_space(A, B, Cs...)\n\nGiven lists of operators acting on local Hilbert spaces individually, return the corresponding operators that act on the tensor product space. In typical usage, the inputs will represent local physical observables and the outputs will be used to define quantum couplings.\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.unit_resolution_binning_parameters-Tuple{Any, Any, Vararg{Any}}","page":"Library API","title":"Sunny.unit_resolution_binning_parameters","text":"unit_resolution_binning_parameters(sc::SampledCorrelations)\n\nCreate BinningParameters which place one histogram bin centered at each possible (q,ω) scattering vector of the crystal. This is the finest possible binning without creating bins with zero scattering vectors in them.\n\nThis function can be used without reference to a SampledCorrelations using an alternate syntax to manually specify the bin centers for the energy axis and the lattice size:\n\nunit_resolution_binning_parameters(ω_bincenters,latsize,[reciprocal lattice vectors])\n\nThe last argument may be a 3x3 matrix specifying the reciprocal lattice vectors, or a Crystal.\n\nLastly, binning parameters for a single axis may be specifed by their bin centers:\n\n(binstart,binend,binwidth) = unit_resolution_binning_parameters(bincenters::Vector{Float64})\n\n\n\n\n\n","category":"method"},{"location":"library.html#Sunny.@mix_proposals-Tuple","page":"Library API","title":"Sunny.@mix_proposals","text":"@mix_proposals weight1 propose1 weight2 propose2 ...\n\nMacro to generate a proposal function that randomly selects among the provided functions according to the provided probability weights. For use with LocalSampler.\n\nExample\n\n# A proposal function that proposes a spin flip 40% of the time, and a\n# Gaussian perturbation 60% of the time.\n@mix_proposals 0.4 propose_flip 0.6 propose_delta(0.2)\n\n\n\n\n\n","category":"macro"},{"location":"library.html#Optional-Makie-extensions","page":"Library API","title":"Optional Makie extensions","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"The following will be enabled through a package extension if either GLMakie or WGLMakie is loaded.","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"plot_spins\nview_crystal","category":"page"},{"location":"library.html#Sunny.plot_spins","page":"Library API","title":"Sunny.plot_spins","text":"plot_spins(sys::System; arrowscale=1.0, color=:red, colorfn=nothing,\n colormap=:viridis, colorrange=nothing, show_cell=true, orthographic=false,\n ghost_radius=0, dims=3\n\nPlot the spin configuration defined by sys. Optional parameters are:\n\narrowscale: Scale all arrows by dimensionless factor.\ncolor: Arrow colors. May be symbolic or numeric. If scalar, will be shared among all sites.\ncolorfn: Function that dynamically maps from a site index to a numeric color value. Useful for animations.\ncolormap, colorrange: Used to populate colors from numbers following Makie conventions.\nshow_cell: Show original crystallographic unit cell.\northographic: Use camera with orthographic projection.\nghost_radius: Show translucent periodic images up to a given distance (length units).\ndims: Spatial dimensions of system (1, 2, or 3).\n\nCalling notify on the return value will animate the figure.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Sunny.view_crystal","page":"Library API","title":"Sunny.view_crystal","text":"view_crystal(crystal::Crystal, max_dist::Real; show_axis=true, orthographic=false)\n\nAn interactive crystal viewer, with bonds up to max_dist.\n\n\n\n\n\n","category":"function"},{"location":"library.html#Optional-WriteVTK-extensions","page":"Library API","title":"Optional WriteVTK extensions","text":"","category":"section"},{"location":"library.html","page":"Library API","title":"Library API","text":"The following will be enabled through a package extension if WriteVTK is loaded.","category":"page"},{"location":"library.html","page":"Library API","title":"Library API","text":"export_vtk","category":"page"},{"location":"library.html#Sunny.export_vtk","page":"Library API","title":"Sunny.export_vtk","text":"export_vtk(filename,params::BinningParameters,data)\n\nExport a VTK-compatible file to filename (do not include file extension when specifying the file name) which contains the data as VTK Cell Data on a grid parameterized by params.\n\nAt least one axis of the BinningParameters must be integrated over, since VTK does not support 4D data. See integrate_axes!.\n\n\n\n\n\n","category":"function"},{"location":"examples/contributed/MgCr2O4-tutorial.html#MgCr2O4-at-Finite-Temperature","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"","category":"section"},{"location":"examples/contributed/MgCr2O4-tutorial.html","page":"MgCr2O4 at Finite Temperature","title":"MgCr2O4 at Finite Temperature","text":"Author: Martin Mourigal 