Download this example as Jupyter notebook or Julia script.
FeI₂ is an effective spin-1 material with strong single-ion anisotropy. Quadrupolar fluctuations give rise to a single-ion bound state that cannot be described by a dipole-only model. This tutorial illustrates how to use the linear spin wave theory of SU(3) coherent states (i.e. 2-flavor bosons) to model the magnetic behavior in FeI₂. The original study was performed in Bai et al., Nature Physics 17, 467–472 (2021).
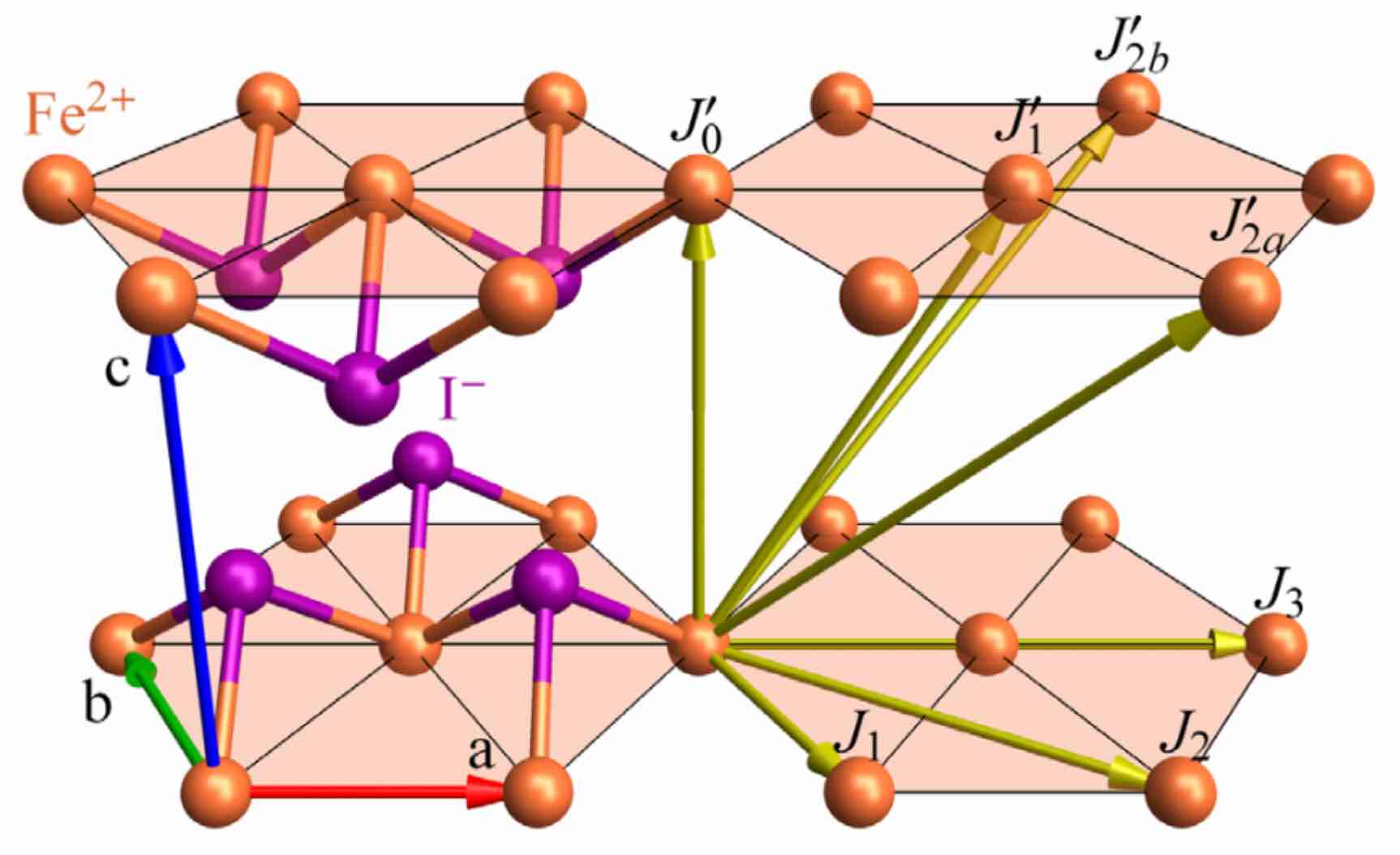
The Fe atoms are arranged in stacked triangular layers. The effective spin Hamiltonian takes the form,
\[\mathcal{H}=\sum_{(i,j)} 𝐒_i ⋅ J_{ij} 𝐒_j - D\sum_i \left(S_i^z\right)^2,\]
where the set of exchange matrices $J_{ij}$ between bonded sites $(i,j)$ includes competing ferromagnetic and antiferromagnetic interactions. This model also includes a strong easy axis anisotropy, $D > 0$.
We will formulate this Hamiltonian in Sunny and then calculate its dynamic structure factor.
Sunny is implemented in Julia. This is a relatively new programming language that allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users may wish to take a look at our Getting Started with Julia guide. Sunny requires Julia 1.9 or later.
From the Julia prompt, load Sunny. For plotting, one can choose either GLMakie (a pop-up window) or WGLMakie (inline plots for a Jupyter notebook or VSCode).
using Sunny, GLMakie
If these packages are not yet installed, Julia should offer to install them using its built-in package management system. If old versions are installed, you may need to update them to run this tutorial.
A Crystal describes the crystallographic unit cell and will usually be loaded from a .cif file. Here, we instead build a crystal by listing all atoms and their types.
a = b = 4.05012 # Lattice constants for triangular lattice
+c = 6.75214 # Spacing in the z-direction
+
+latvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that
+ # define the conventional unit cell
+positions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions
+ # of lattice vectors
+types = ["Fe", "I", "I"]
+FeI2 = Crystal(latvecs, positions; types)
Crystal
+HM symbol 'P -3 m 1' (164)
+Lattice params a=4.05, b=4.05, c=6.752, α=90°, β=90°, γ=120°
+Cell volume 95.92
+Type 'Fe', Wyckoff 1a (point group '-3m.'):
+ 1. [0, 0, 0]
+Type 'I', Wyckoff 2d (point group '3m.'):
+ 2. [1/3, 2/3, 1/4]
+ 3. [2/3, 1/3, 3/4]
+
Observe that Sunny inferred the space group, 'P -3 m 1' (164) and labeled the atoms according to their point group symmetries.
Only the Fe atoms are magnetic, so we discard the I ions using subcrystal.
cryst = subcrystal(FeI2, "Fe")
Crystal
+HM symbol 'P -3 m 1' (164)
+Lattice params a=4.05, b=4.05, c=6.752, α=90°, β=90°, γ=120°
+Cell volume 95.92
+Type 'Fe', Wyckoff 1a (point group '-3m.'):
+ 1. [0, 0, 0]
+
Observe that cryst retains the spacegroup symmetry of the full FeI₂ crystal. This information will be used, for example, to propagate exchange interactions between symmetry-equivalent bonds.
In a running Julia environment, the crystal can be viewed interactively using view_crystal.
view_crystal(cryst, 8.0)

The command print_symmetry_table provides a list of all the symmetry-allowed interactions up to a cutoff distance.
print_symmetry_table(cryst, 8.0)
Atom 1
+Type 'Fe', position [0, 0, 0], multiplicity 1
+Allowed g-tensor: [A 0 0
+ 0 A 0
+ 0 0 B]
+Allowed anisotropy in Stevens operators:
+ c₁*𝒪[2,0] +
+ c₂*𝒪[4,-3] + c₃*𝒪[4,0] +
+ c₄*𝒪[6,-3] + c₅*𝒪[6,0] + c₆*𝒪[6,6]
+
+Bond(1, 1, [1, 0, 0])
+Distance 4.05012, coordination 6
+Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 0, 0]
+Allowed exchange matrix:[A 0 0
+ 0 B D
+ 0 D C]
+
+Bond(1, 1, [0, 0, 1])
+Distance 6.75214, coordination 2
+Connects 'Fe' at [0, 0, 0] to 'Fe' at [0, 0, 1]
+Allowed exchange matrix:[A 0 0
+ 0 A 0
+ 0 0 B]
+
+Bond(1, 1, [1, 2, 0])
+Distance 7.01501361675086, coordination 6
+Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 2, 0]
+Allowed exchange matrix:[A 0 0
+ 0 B D
+ 0 D C]
+
+Bond(1, 1, [1, 0, 1])
+Distance 7.8736818956572, coordination 12
+Connects 'Fe' at [0, 0, 0] to 'Fe' at [1, 0, 1]
+Allowed exchange matrix:[A F E
+ F B D
+ E D C]
The allowed $g$-tensor is expressed as a 3×3 matrix in the free coefficients A, B, ... The allowed single-ion anisotropy is expressed as a linear combination of Stevens operators. The latter correspond to polynomials of the spin operators, as we will describe below.
The allowed exchange interactions are given as a 3×3 matrix for representative bonds. The notation Bond(i, j, n) indicates a bond between atom indices i and j, with cell offset n. In the general case, it will be necessary to associate atom indices with their positions in the unit cell; these can be viewed with display(cryst). Note that the order of the pair $(i, j)$ is significant if the exchange tensor contains antisymmetric Dzyaloshinskii–Moriya (DM) interactions.
In the case of FeI₂, Bond(1, 1, [1,0,0]) is one of the 6 nearest-neighbor Fe-Fe bonds on a triangular lattice layer, and Bond(1, 1, [0,0,1]) is an Fe-Fe bond between layers.
In constructing a spin System, we must provide several additional details about the spins.
sys = System(cryst, (4, 4, 4), [SpinInfo(1, S=1, g=2)], :SUN, seed=2)
System [SU(3)]
+Lattice: (4, 4, 4)×1
+
This system includes $4×4×4$ unit cells, i.e. 64 Fe atoms, each with spin $S=1$ and a $g$-factor of 2. Quantum mechanically, spin $S=1$ involves a superposition of $2S+1=3$ distinct angular momentum states. In :SUN mode, this superposition will be modeled explicitly using the formalism of SU(3) coherent states, which captures both dipolar and quadrupolar fluctuations. For the more traditional dipole dynamics, use :dipole mode instead.
Next we will use set_exchange! to assign interaction to bonds. Sunny will automatically propagate each interaction to all symmetry-equivalent bonds in the unit cell. The FeI₂ interactions below follow Bai et al.
J1pm = -0.236
+J1pmpm = -0.161
+J1zpm = -0.261
+J2pm = 0.026
+J3pm = 0.166
+J′0pm = 0.037
+J′1pm = 0.013
+J′2apm = 0.068
+
+J1zz = -0.236
+J2zz = 0.113
+J3zz = 0.211
+J′0zz = -0.036
+J′1zz = 0.051
+J′2azz = 0.073
+
+J1xx = J1pm + J1pmpm
+J1yy = J1pm - J1pmpm
+J1yz = J1zpm
+
+set_exchange!(sys, [J1xx 0.0 0.0;
+ 0.0 J1yy J1yz;
+ 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))
+set_exchange!(sys, [J2pm 0.0 0.0;
+ 0.0 J2pm 0.0;
+ 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))
+set_exchange!(sys, [J3pm 0.0 0.0;
+ 0.0 J3pm 0.0;
+ 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))
+set_exchange!(sys, [J′0pm 0.0 0.0;
+ 0.0 J′0pm 0.0;
+ 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))
+set_exchange!(sys, [J′1pm 0.0 0.0;
+ 0.0 J′1pm 0.0;
+ 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))
+set_exchange!(sys, [J′2apm 0.0 0.0;
+ 0.0 J′2apm 0.0;
+ 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))
The function set_onsite_coupling! assigns a single-ion anisotropy operator. It can be constructed, e.g., from the matrices given by spin_operators or stevens_operators. Here we construct an easy-axis anisotropy along the direction $\hat{z}$.
D = 2.165
+S = spin_operators(sys, 1)
+set_onsite_coupling!(sys, -D*S[3]^2, 1)
Any anisotropy operator can be converted to a linear combination of Stevens operators with print_stevens_expansion.
In the remainder of this tutorial, we will examine Sunny's tools for calculating the dynamical structure factor using a multi-boson generalization of linear spin wave theory (LSWT). This theory describes non-interacting quasi-particle excitations that hybridize dipolar and quadrupolar modes.
Begin with a random configuration and use minimize_energy! to find a configuration of the SU(3) coherent states (i.e. spin dipoles and quadrupoles) that locally minimizes energy.
randomize_spins!(sys)
+minimize_energy!(sys)
49
A positive number above indicates that the procedure has converged to a local energy minimum. The configuration, however, may still have defects. This can be checked by visualizing the spins, colored according to their $z$-components.
plot_spins(sys; color=[s[3] for s in sys.dipoles])

A different understanding of the magnetic ordering can be obtained by moving to Fourier space. The 'instant' structure factor $𝒮(𝐪)$ is an experimental observable. To investigate $𝒮(𝐪)$ as true 3D data, Sunny provides instant_correlations and related functions. Here, however, we will use print_wrapped_intensities, which gives average intensities for the individual Bravais sublattices (in effect, all wavevectors are wrapped to the first Brillouin zone).
print_wrapped_intensities(sys)
Dominant wavevectors for spin sublattices:
+
+ [-1/4, 1/4, 1/4] 37.33% weight
+ [1/4, -1/4, -1/4] 37.33%
+ [-1/4, 1/4, 0] 1.60%
+ [1/4, -1/4, 0] 1.60%
+ [-1/4, 1/4, 1/2] 1.60%
+ [1/4, -1/4, 1/2] 1.60%
+ [1/4, -1/4, 1/4] 1.59%
+ [-1/4, 1/4, -1/4] 1.59%
+ [0, -1/4, 1/4] 0.77%
+ [0, 1/4, -1/4] 0.77%
+ [1/4, 0, 1/4] 0.77%
+ ... ...
The result will likely be approximately consistent with the known zero-field energy-minimizing magnetic structure of FeI₂, which is single-$Q$ (two-up, two-down antiferromagnetic order). Mathematically, spontaneous symmetry breaking should select one of $±Q = [0, -1/4, 1/4]$, $[1/4, 0, 1/4]$, or $[-1/4,1/4,1/4]$, associated with the three-fold rotational symmetry of the crystal spacegroup. In nature, however, one will frequently encounter competing "domains" associated with the three possible orientations of the ground state.
If the desired ground state is already known, as with FeI₂, it could be entered by hand using set_dipole!. Alternatively, in the case of FeI₂, we could repeatedly employ the above randomization and minimization procedure until a defect-free configuration is found. Some systems will have more complicated ground states, which can be much more challenging to find. For this, Sunny provides experimental support for powerful simulated annealing via parallel tempering, but that is outside the scope of this tutorial.
Here, let's break the three-fold symmetry of FeI₂ by hand. Given one or more desired $Q$ modes, Sunny can suggest a magnetic supercell with appropriate periodicity. Let's arbitrarily select one of the three possible ordering wavevectors, $Q = [0, -1/4, 1/4]$. Sunny suggests a corresponding magnetic supercell in units of the crystal lattice vectors.
suggest_magnetic_supercell([[0, -1/4, 1/4]])
Suggested magnetic supercell in multiples of lattice vectors:
+
+ [1 0 0; 0 2 1; 0 -2 1]
+
+for wavevectors [[0, -1/4, 1/4]].
The system returned by reshape_supercell is smaller, and is sheared relative to the original system. This makes it much easier to find the global energy minimum.
sys_min = reshape_supercell(sys, [1 0 0; 0 2 1; 0 -2 1])
+randomize_spins!(sys_min)
+minimize_energy!(sys_min);
Plot the system again, now including "ghost" spins out to 12Å
plot_spins(sys_min; color=[s[3] for s in sys_min.dipoles], ghost_radius=12)

Now that we have found the ground state for a magnetic supercell, we can immediately proceed to perform zero-temperature calculations using linear spin wave theory. We begin by instantiating a SpinWaveTheory type using the supercell.
swt = SpinWaveTheory(sys_min)
SpinWaveTheory [Dipole correlations]
+Atoms in magnetic supercell: 4
+
Select a sequence of wavevectors that will define a piecewise linear interpolation in reciprocal lattice units (RLU).
q_points = [[0,0,0], [1,0,0], [0,1,0], [1/2,0,0], [0,1,0], [0,0,0]];
The function reciprocal_space_path will linearly sample a path between the provided $q$-points with a given density. The xticks return value provides labels for use in plotting.
density = 50
+path, xticks = reciprocal_space_path(cryst, q_points, density);
The dispersion function defines the quasiparticle excitation energies $ω_i(𝐪)$ for each point $𝐪$ along the reciprocal space path.
disp = dispersion(swt, path);
In addition to the band energies $ω_i(𝐪)$, Sunny can calculate the inelastic neutron scattering intensity $I_i(𝐪)$ for each band $i$ according to an intensity_formula. We choose to apply a polarization correction $(1 - 𝐪⊗𝐪)$ by setting the mode argument to :perp. Selecting delta_function_kernel specifies that we want the energy and intensity of each band individually.
formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)
Quantum Scattering Intensity Formula
+At any Q and for each band ωᵢ = εᵢ(Q), with S = S(Q,ωᵢ):
+
+ Intensity(Q,ω) = ∑ᵢ δ(ω-ωᵢ) ∑_ij (I - Q⊗Q){i,j} S{i,j}
+
+ (i,j = Sx,Sy,Sz)
+
+BandStructure information (ωᵢ and intensity) reported for each band
+
The function intensities_bands uses linear spin wave theory to calculate both the dispersion and intensity data for the provided path.
disp, intensity = intensities_bands(swt, path, formula);
These can be plotted in GLMakie.
fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+ylims!(ax, 0.0, 7.5)
+xlims!(ax, 1, size(disp, 1))
+colorrange = extrema(intensity)
+for i in axes(disp)[2]
+ lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange)
+end
+fig

To make comparisons with inelastic neutron scattering (INS) data, it is helpful to employ an empirical broadening kernel, e.g., a lorentzian.
γ = 0.15 # width in meV
+broadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))
Quantum Scattering Intensity Formula
+At any (Q,ω), with S = S(Q,ωᵢ):
+
+ Intensity(Q,ω) = ∑ᵢ Kernel(ω-ωᵢ) ∑_ij (I - Q⊗Q){i,j} S{i,j}
+
+ (i,j = Sx,Sy,Sz)
+
+Intensity(ω) reported
+
The intensities_broadened function requires an energy range in addition to the $𝐪$-space path.
energies = collect(0:0.01:10) # 0 < ω < 10 (meV).
+is1 = intensities_broadened(swt, path, energies, broadened_formula);
A real FeI₂ sample will exhibit competing magnetic domains associated with spontaneous symmetry breaking of the 6-fold rotational symmetry of the triangular lattice. Note that the wavevectors $𝐪$ and $-𝐪$ are equivalent in the structure factor, which leaves three distinct domain orientations, which are related by 120° rotations about the $ẑ$-axis. Rather than rotating the spin configuration directly, on can rotate the $𝐪$-space path. Below, we use rotation_in_rlu to average the intensities over all three possible orientations.
R = rotation_in_rlu(cryst, [0, 0, 1], 2π/3)
+is2 = intensities_broadened(swt, [R*q for q in path], energies, broadened_formula)
+is3 = intensities_broadened(swt, [R*R*q for q in path], energies, broadened_formula)
+is_averaged = (is1 + is2 + is3) / 3
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+heatmap!(ax, 1:size(is_averaged, 1), energies, is_averaged)
+fig

This result can be directly compared to experimental neutron scattering data from Bai et al.
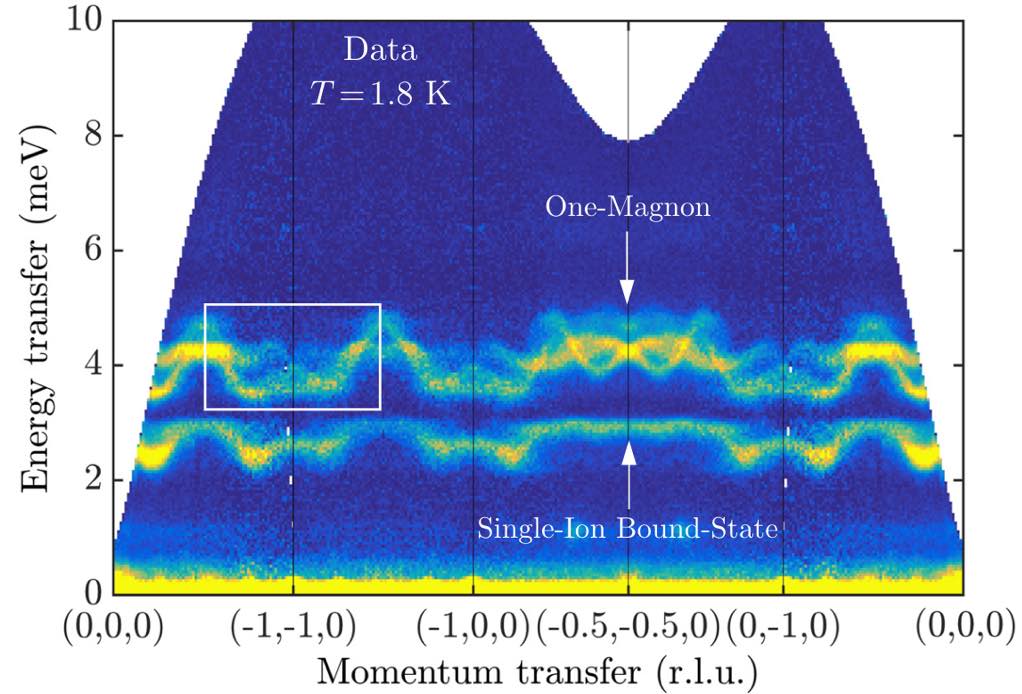
(The publication figure accidentally used a non-standard coordinate system to label the wave vectors.)
To get this agreement, the use of SU(3) coherent states is essential. In other words, we needed a theory of multi-flavored bosons. The lower band has large quadrupolar character, and arises from the strong easy-axis anisotropy of FeI₂. By setting mode = :SUN, the calculation captures this coupled dipole-quadrupole dynamics.
An interesting exercise is to repeat the same study, but using mode = :dipole instead of :SUN. That alternative choice would constrain the coherent state dynamics to the space of dipoles only.
The full dynamical spin structure factor (DSSF) can be retrieved as a $3×3$ matrix with the dssf function, for a given path of $𝐪$-vectors.
disp, is = dssf(swt, path);
The first output disp is identical to that obtained from dispersion. The second output is contains a list of $3×3$ matrix of intensities. For example, is[q,n][2,3] yields the $(ŷ,ẑ)$ component of the structure factor intensity for nth mode at the qth wavevector in the path.
The multi-boson linear spin wave theory, applied above, can be understood as the quantization of a certain generalization of the Landau-Lifshitz spin dynamics. Rather than dipoles, this dynamics takes places on the space of SU(N) coherent states.
The full SU(N) coherent state dynamics, with appropriate quantum correction factors, can be useful to model finite temperature scattering data. In particular, it captures certain anharmonic effects due to thermal fluctuations. This is the subject of our FeI₂ at Finite Temperature tutorial.
The classical dynamics is also a good starting point to study non-equilibrium phenomena. Empirical noise and damping terms can be used to model coupling to a thermal bath. This yields a Langevin dynamics of SU(N) coherent states. Our CP² Skyrmion Quench tutorial shows how this dynamics gives rise to the formation of novel topological defects in a temperature quench.
Relative to LSWT calculations, it can take much more time to estimate $\mathcal{S}(𝐪,ω)$ intensities using classical dynamics simulation. See the SunnyTutorials notebooks for examples of "production-scale" simulations.

 \n",
+ "\n",
+ "\n",
+ "(The publication figure accidentally used a non-standard coordinate system to\n",
+ "label the wave vectors.)\n",
+ "\n",
+ "To get this agreement, the use of SU(3) coherent states is essential. In other\n",
+ "words, we needed a theory of multi-flavored bosons. The lower band has large\n",
+ "quadrupolar character, and arises from the strong easy-axis anisotropy of\n",
+ "FeI₂. By setting `mode = :SUN`, the calculation captures this coupled\n",
+ "dipole-quadrupole dynamics.\n",
+ "\n",
+ "An interesting exercise is to repeat the same study, but using `mode =\n",
+ ":dipole` instead of `:SUN`. That alternative choice would constrain the\n",
+ "coherent state dynamics to the space of dipoles only."
+ ],
+ "metadata": {}
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The full dynamical spin structure factor (DSSF) can be retrieved as a $3×3$\n",
+ "matrix with the `dssf` function, for a given path of $𝐪$-vectors."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "disp, is = dssf(swt, path);"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The first output `disp` is identical to that obtained from `dispersion`. The\n",
+ "second output `is` contains a list of $3×3$ matrix of intensities. For\n",
+ "example, `is[q,n][2,3]` yields the $(ŷ,ẑ)$ component of the structure factor\n",
+ "intensity for `nth` mode at the `q`th wavevector in the `path`."
+ ],
+ "metadata": {}
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "## What's next?\n",
+ "\n",
+ "The multi-boson linear spin wave theory, applied above, can be understood as\n",
+ "the quantization of a certain generalization of the Landau-Lifshitz spin\n",
+ "dynamics. Rather than dipoles, this dynamics takes places on the space of\n",
+ "[SU(_N_) coherent states](https://arxiv.org/abs/2106.14125).\n",
+ "\n",
+ "The full SU(_N_) coherent state dynamics, with appropriate quantum correction\n",
+ "factors, can be useful to model finite temperature scattering data. In\n",
+ "particular, it captures certain anharmonic effects due to thermal\n",
+ "fluctuations. This is the subject of our FeI₂ at Finite Temperature\n",
+ "tutorial.\n",
+ "\n",
+ "The classical dynamics is also a good starting point to study non-equilibrium\n",
+ "phenomena. Empirical noise and damping terms can be used to model [coupling to\n",
+ "a thermal bath](https://arxiv.org/abs/2209.01265). This yields a Langevin\n",
+ "dynamics of SU(_N_) coherent states. Our CP² Skyrmion Quench\n",
+ "tutorial shows how this dynamics gives rise to the formation of novel\n",
+ "topological defects in a temperature quench.\n",
+ "\n",
+ "Relative to LSWT calculations, it can take much more time to estimate\n",
+ "$\\mathcal{S}(𝐪,ω)$ intensities using classical dynamics simulation. See the\n",
+ "[SunnyTutorials\n",
+ "notebooks](https://nbviewer.org/github/SunnySuite/SunnyTutorials/tree/main/Tutorials/)\n",
+ "for examples of \"production-scale\" simulations."
+ ],
+ "metadata": {}
+ }
+ ],
+ "nbformat_minor": 3,
+ "metadata": {
+ "language_info": {
+ "file_extension": ".jl",
+ "mimetype": "application/julia",
+ "name": "julia",
+ "version": "1.9.3"
+ },
+ "kernelspec": {
+ "name": "julia-1.9",
+ "display_name": "Julia 1.9.3",
+ "language": "julia"
+ }
+ },
+ "nbformat": 4
+}
diff --git a/previews/PR176/assets/notebooks/ising2d.ipynb b/previews/PR176/assets/notebooks/ising2d.ipynb
new file mode 100644
index 000000000..3d9a59ddd
--- /dev/null
+++ b/previews/PR176/assets/notebooks/ising2d.ipynb
@@ -0,0 +1,179 @@
+{
+ "cells": [
+ {
+ "cell_type": "markdown",
+ "source": [
+ "# Classical Ising model\n",
+ "\n",
+ "This tutorial illustrates simulation of the classical 2D Ising model."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "using Sunny, WGLMakie"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Sunny expects a 3D `Crystal` unit cell. To model a square lattice, we\n",
+ "create an orthogonal unit cell where the $z$-spacing is distinct from the $x$\n",
+ "and $y$ spacing."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "a = 1\n",
+ "latvecs = lattice_vectors(a,a,10a,90,90,90)\n",
+ "crystal = Crystal(latvecs, [[0,0,0]])"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Create a `System` of spins with linear size `L` in the $x$ and $y$\n",
+ "directions, and only one layer in the $z$ direction. The option `:dipole`\n",
+ "means that the system will store Heisenberg spins, as opposed to SU($N$)\n",
+ "coherent states. Polarize the initial spin configuration using\n",
+ "`polarize_spins!`. Following the Ising convention, we will restrict\n",
+ "these spins to the $z$-axis and give them magnitude $S=1$.\n",
+ "\n",
+ "By default, Sunny uses physical units, e.g. magnetic field in tesla. Here we\n",
+ "specify an alternative `Units` system, so that the Zeeman coupling\n",
+ "between the spin dipole $𝐬$ and an external field $𝐁$ has the dimensionless\n",
+ "form $-𝐁⋅𝐬$."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "L = 128\n",
+ "sys = System(crystal, (L,L,1), [SpinInfo(1, S=1, g=1)], :dipole, units=Units.theory, seed=0)\n",
+ "polarize_spins!(sys, (0,0,1))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Use `set_exchange!` to include a ferromagnetic Heisenberg interaction\n",
+ "along nearest-neighbor bonds. The `Bond` below connects two spins\n",
+ "displaced by one lattice constant in the $x$-direction. This interaction will\n",
+ "be propagated to all nearest-neighbors bonds in the system, consistent with\n",
+ "the symmetries of the square lattice."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "set_exchange!(sys, -1.0, Bond(1,1,(1,0,0)))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "If an external field is desired, it can be set using\n",
+ "`set_external_field!`."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "B = 0\n",
+ "set_external_field!(sys, (0,0,B))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The critical temperature for the Ising model is known analytically."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "Tc = 2/log(1+√2)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Use a `LocalSampler` to perform `nsweeps` Monte Carlo sweeps. A sweep\n",
+ "consists of, on average, one trial update per spin in the system. Each\n",
+ "proposed update is accepted or rejected according to the Metropolis acceptance\n",
+ "probability. As its name suggests, the `propose_flip` function will\n",
+ "only propose pure spin flips, $𝐬 \\rightarrow -𝐬$."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "nsweeps = 4000\n",
+ "sampler = LocalSampler(kT=Tc, propose=propose_flip)\n",
+ "for i in 1:nsweeps\n",
+ " step!(sys, sampler)\n",
+ "end"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Plot the Ising spins by extracting the $z$-component of the dipoles"
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "heatmap(reshape([s.z for s in sys.dipoles], (L,L)))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ }
+ ],
+ "nbformat_minor": 3,
+ "metadata": {
+ "language_info": {
+ "file_extension": ".jl",
+ "mimetype": "application/julia",
+ "name": "julia",
+ "version": "1.9.3"
+ },
+ "kernelspec": {
+ "name": "julia-1.9",
+ "display_name": "Julia 1.9.3",
+ "language": "julia"
+ }
+ },
+ "nbformat": 4
+}
diff --git a/previews/PR176/assets/notebooks/out_of_equilibrium.ipynb b/previews/PR176/assets/notebooks/out_of_equilibrium.ipynb
new file mode 100644
index 000000000..d0c8a2c63
--- /dev/null
+++ b/previews/PR176/assets/notebooks/out_of_equilibrium.ipynb
@@ -0,0 +1,279 @@
+{
+ "cells": [
+ {
+ "cell_type": "markdown",
+ "source": [
+ "# CP² Skyrmion Quench\n",
+ "\n",
+ "This example demonstrates Sunny's ability to simulate the out-of-equilibrium\n",
+ "dynamics of generalized spin systems. We will implement the model Hamiltonian\n",
+ "of [Zhang et al., Nature Communications **14**, 3626\n",
+ "(2023)](https://www.nature.com/articles/s41467-023-39232-8), which supports a\n",
+ "novel type of topological defect, a CP² skyrmion, that involves both the\n",
+ "dipolar and quadrupolar parts of a quantum spin.\n",
+ "\n",
+ "Beginning from an initial high-temperature state, a disordered gas of CP²\n",
+ "skyrmions can be formed by rapidly quenching to low temperature. To model the\n",
+ "coupled dynamics of dipoles and quadrupoles, Sunny uses a recently developed\n",
+ "generalization of the Landau-Lifshitz spin dynamics, [Dahlbom et al., Phys.\n",
+ "Rev. B **106**, 235154 (2022)](https://doi.org/10.1103/PhysRevB.106.235154)."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "using Sunny, WGLMakie"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The Hamiltonian we will implement,\n",
+ "$$\n",
+ "\\mathcal{H} = \\sum_{\\langle i,j \\rangle} J_{ij}( \\hat{S}_i^x \\hat{S}_j^x + \\hat{S}_i^y \\hat{S}_j^y + \\Delta\\hat{S}_i^z \\hat{S}_j^z) - h\\sum_{i}\\hat{S}_i^z + D\\sum_{i}(\\hat{S}_i^z)^2\n",
+ "$$\n",
+ "contains competing ferromagnetic nearest-neightbor and antiferromagnetic\n",
+ "next-nearest-neighbor exchange terms on a triangular lattice. Both exchanges\n",
+ "exhibit anisotropy on the z-term. Additionally, there is an external magnetic\n",
+ "field, $h$, and easy-plane single-ion anisotropy, $D > 0$. We begin by\n",
+ "implementing the `Crystal`."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "lat_vecs = lattice_vectors(1.0, 1.0, 2.0, 90, 90, 120)\n",
+ "basis_vecs = [[0,0,0]]\n",
+ "cryst = Crystal(lat_vecs, basis_vecs)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The crystal is then used to create a spin `System`. All parameters in\n",
+ "this model system are dimensionless, so we select \"theory\" units and set the\n",
+ "g-factor to one."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "L = 40\n",
+ "dims = (L, L, 1)\n",
+ "sys = System(cryst, dims, [SpinInfo(1, S=1, g=1)], :SUN; seed=101, units=Units.theory)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "We proceed to implement each term of the Hamiltonian, selecting our parameters\n",
+ "so that the system occupies a region of the phase diagram that supports\n",
+ "skyrmions. The exchange interactions are set as follows."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "J1 = -1 # Nearest-neighbor ferromagnetic\n",
+ "J2 = (2.0/(1+√5)) # Tune competing exchange to set skyrmion scale length\n",
+ "Δ = 2.6 # Exchange anisotropy\n",
+ "\n",
+ "ex1 = J1 * [1.0 0.0 0.0;\n",
+ " 0.0 1.0 0.0;\n",
+ " 0.0 0.0 Δ]\n",
+ "ex2 = J2 * [1.0 0.0 0.0;\n",
+ " 0.0 1.0 0.0;\n",
+ " 0.0 0.0 Δ]\n",
+ "set_exchange!(sys, ex1, Bond(1, 1, [1, 0, 0]))\n",
+ "set_exchange!(sys, ex2, Bond(1, 1, [1, 2, 0]))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Next we add the external field,"
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "h = 15.5\n",
+ "field = set_external_field!(sys, [0.0 0.0 h])"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "and finally the single-ion anisotropy,"
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "D = 19.0\n",
+ "Sz = spin_operators(sys, 1)[3]\n",
+ "set_onsite_coupling!(sys, D*Sz^2, 1)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Initialize system to an infinite temperature (fully randomized) initial\n",
+ "condition."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "randomize_spins!(sys)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "We are now ready to simulate the quenching process using a generalized\n",
+ "`Langevin` spin dynamics. If we were working with spin dipoles only,\n",
+ "then `Langevin` dynamics would be the usual Landau-Lifshitz spin dynamics,\n",
+ "augmented with damping and noise terms. In the present study, we are instead\n",
+ "working with quantum spin-1 (an ($N=3$)-level system that includes both\n",
+ "dipoles and quadrupoles). Here, `Langevin` captures the coupled\n",
+ "dipole-quadrupole dynamics using the formalism of SU($N$) coherent states.\n",
+ "\n",
+ "Selecting `kT = 0` in the Langevin dynamics will effective disable the noise\n",
+ "term. Then the parameter `λ` effectively determines the damping time-scale."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "Δt = 0.2/D # Integration time step (inverse meV). Typically this will be\n",
+ " # inversely proportional to the largest energy scale in the\n",
+ " # system. We can use a fairly large time-step here because\n",
+ " # accuracy isn't critical.\n",
+ "kT = 0 # Target equilibrium temperature (meV)\n",
+ "λ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)\n",
+ "integrator = Langevin(Δt; kT, λ)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Finally we run the dynamics. We will record the state of the system at three\n",
+ "different times during the quenching process by copying the `coherents` field\n",
+ "of the `System`."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "τs = [4., 16, 256] # Times to record snapshots\n",
+ "frames = [] # Empty array to store snapshots\n",
+ "for i in eachindex(τs)\n",
+ " dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate\n",
+ " numsteps = round(Int, dur/Δt)\n",
+ " for _ in 1:numsteps # Perform the integration\n",
+ " step!(sys, integrator)\n",
+ " end\n",
+ " push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration\n",
+ "end"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "To visualize the state of the system contained in each snapshot, we will\n",
+ "calculate and plot the skyrmion density on each plaquette of our lattice. The\n",
+ "function `plot_triangular_plaquettes` is not part of the core Sunny package,\n",
+ "but rather something you could define yourself. We are using the definition in\n",
+ "`plotting2d.jl` from the Sunny [`examples/extra`\n",
+ "directory](https://github.com/SunnySuite/Sunny.jl/tree/main/examples/extra)."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "include(pkgdir(Sunny, \"examples\", \"extra\", \"plotting2d.jl\"))\n",
+ "\n",
+ "function sun_berry_curvature(z₁, z₂, z₃)\n",
+ " z₁, z₂, z₃ = normalize.((z₁, z₂, z₃))\n",
+ " n₁ = z₁ ⋅ z₂\n",
+ " n₂ = z₂ ⋅ z₃\n",
+ " n₃ = z₃ ⋅ z₁\n",
+ " return angle(n₁ * n₂ * n₃)\n",
+ "end\n",
+ "\n",
+ "plot_triangular_plaquettes(sun_berry_curvature, frames; resolution=(1800,600),\n",
+ " offset_spacing=10, texts = [\"\\tt = \"*string(τ) for τ in τs], text_offset = (0.0, 6.0)\n",
+ ")"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The times are given in $\\hbar/|J_1|$. The white\n",
+ "background corresponds to a quantum paramagnetic state, where the local spin\n",
+ "exhibits a strong quadrupole moment and little or no dipole moment. Observe\n",
+ "that the process has generated a number of well-formed skyrmions of both\n",
+ "positive (red) and negative (blue) charge in addition to a number of other\n",
+ "metastable spin configurations. A full-sized version of this figure is\n",
+ "available in [Dahlbom et al.](https://doi.org/10.1103/PhysRevB.106.235154)."
+ ],
+ "metadata": {}
+ }
+ ],
+ "nbformat_minor": 3,
+ "metadata": {
+ "language_info": {
+ "file_extension": ".jl",
+ "mimetype": "application/julia",
+ "name": "julia",
+ "version": "1.9.3"
+ },
+ "kernelspec": {
+ "name": "julia-1.9",
+ "display_name": "Julia 1.9.3",
+ "language": "julia"
+ }
+ },
+ "nbformat": 4
+}
diff --git a/previews/PR176/assets/notebooks/powder_averaging.ipynb b/previews/PR176/assets/notebooks/powder_averaging.ipynb
new file mode 100644
index 000000000..0f84c42b8
--- /dev/null
+++ b/previews/PR176/assets/notebooks/powder_averaging.ipynb
@@ -0,0 +1,264 @@
+{
+ "cells": [
+ {
+ "cell_type": "markdown",
+ "source": [
+ "# Powder Averaged CoRh₂O₄\n",
+ "\n",
+ "This tutorial illustrates the calculation of the powder-averaged structure\n",
+ "factor by performing an orientational average. We consider a simple model of\n",
+ "the diamond-cubic crystal CoRh₂O₄, with parameters extracted from [Ge et al.,\n",
+ "Phys. Rev. B 96, 064413](https://doi.org/10.1103/PhysRevB.96.064413)."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "using Sunny, WGLMakie"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Construct a diamond `Crystal` in the conventional (non-primitive)\n",
+ "cubic unit cell. Sunny will populate all eight symmetry-equivalent sites when\n",
+ "given the international spacegroup number 227 (\"Fd-3m\") and the appropriate\n",
+ "setting. For this spacegroup, there are two conventional translations of the\n",
+ "unit cell, and it is necessary to disambiguate through the `setting` keyword\n",
+ "argument. (On your own: what happens if `setting` is omitted?)"
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "a = 8.5031 # (Å)\n",
+ "latvecs = lattice_vectors(a, a, a, 90, 90, 90)\n",
+ "cryst = Crystal(latvecs, [[0,0,0]], 227, setting=\"1\")"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "In a running Julia environment, the crystal can be viewed interactively using\n",
+ "`view_crystal`."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "view_crystal(cryst, 8.0)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Construct a `System` with an antiferromagnetic nearest neighbor\n",
+ "interaction `J`. Because the diamond crystal is bipartite, the ground state\n",
+ "will have unfrustrated Néel order. Selecting `latsize=(1,1,1)` is sufficient\n",
+ "because the ground state is periodic over each cubic unit cell. By passing an\n",
+ "explicit `seed`, the system's random number generator will give repeatable\n",
+ "results."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "latsize = (2, 2, 2)\n",
+ "seed = 0\n",
+ "S = 3/2\n",
+ "J = 7.5413*meV_per_K # (~ 0.65 meV)\n",
+ "sys = System(cryst, latsize, [SpinInfo(1; S, g=2)], :dipole; seed=0)\n",
+ "set_exchange!(sys, J, Bond(1, 3, [0,0,0]))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "In the ground state, each spin is exactly anti-aligned with its 4\n",
+ "nearest-neighbors. Because every bond contributes an energy of $-JS^2$, the\n",
+ "energy per site is $-2JS^2$. In this calculation, a factor of 1/2 avoids\n",
+ "double-counting the bonds. Due to lack of frustration, direct energy\n",
+ "minimization is successful in finding the ground state."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "randomize_spins!(sys)\n",
+ "minimize_energy!(sys)\n",
+ "\n",
+ "@assert energy_per_site(sys) ≈ -2J*S^2"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Plotting the spins confirms the expected Néel order. Note that the overall,\n",
+ "global rotation of dipoles is arbitrary."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "s0 = sys.dipoles[1,1,1,1]\n",
+ "plot_spins(sys; color=[s'*s0 for s in sys.dipoles])"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "For numerical efficiency, it will be helpful to work with the smallest\n",
+ "possible magnetic supercell. Here, it is the primitive unit cell, which\n",
+ "contains just two sites. The variable `shape` below defines the primitive\n",
+ "lattice vectors `cryst.prim_latvecs` in units of the conventional lattice\n",
+ "vectors. This result is used as input to `reshape_supercell`. The\n",
+ "energy per site remains the same, which verifies that the magnetic supercell\n",
+ "is valid."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "shape = cryst.latvecs \\ cryst.prim_latvecs\n",
+ "sys_prim = reshape_supercell(sys, shape)\n",
+ "@assert energy_per_site(sys_prim) ≈ -2J*S^2"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Now estimate $𝒮(𝐪,ω)$ with `SpinWaveTheory` and an\n",
+ "`intensity_formula`. The mode `:perp` contracts with a dipole factor\n",
+ "to return the unpolarized intensity. The formula also employs\n",
+ "`lorentzian` broadening. The isotropic `FormFactor` for\n",
+ "Cobalt(2+) dampens intensities at large $𝐪$."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "swt = SpinWaveTheory(sys_prim)\n",
+ "η = 0.4 # (meV)\n",
+ "kernel = lorentzian(η)\n",
+ "formfactors = [FormFactor(\"Co2\")]\n",
+ "formula = intensity_formula(swt, :perp; kernel, formfactors)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "For the \"single crystal\" result, we may use `reciprocal_space_path` to\n",
+ "construct a path that connects high-symmetry points in reciprocal space. The\n",
+ "`intensities_broadened` function collects intensities along this path\n",
+ "for the given set of energy values."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "qpoints = [[0.0, 0.0, 0.0], [0.5, 0.0, 0.0], [0.5, 0.5, 0.0], [0.0, 0.0, 0.0]]\n",
+ "path, xticks = reciprocal_space_path(cryst, qpoints, 50)\n",
+ "energies = collect(0:0.01:6)\n",
+ "is = intensities_broadened(swt, path, energies, formula)\n",
+ "\n",
+ "fig = Figure()\n",
+ "ax = Axis(fig[1,1]; aspect=1.4, ylabel=\"ω (meV)\", xlabel=\"𝐪 (r.l.u.)\",\n",
+ " xticks, xticklabelrotation=π/10)\n",
+ "heatmap!(ax, 1:size(is, 1), energies, is, colormap=:gnuplot2)\n",
+ "fig"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "A powder measurement effectively involves an average over all possible crystal\n",
+ "orientations. We use the function `reciprocal_space_shell` to sample\n",
+ "`n` wavevectors on a sphere of a given radius (inverse angstroms), and then\n",
+ "calculate the spherically-averaged intensity."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "radii = 0.01:0.02:3 # (1/Å)\n",
+ "output = zeros(Float64, length(radii), length(energies))\n",
+ "for (i, radius) in enumerate(radii)\n",
+ " n = 300\n",
+ " qs = reciprocal_space_shell(cryst, radius, n)\n",
+ " is = intensities_broadened(swt, qs, energies, formula)\n",
+ " output[i, :] = sum(is, dims=1) / size(is, 1)\n",
+ "end\n",
+ "\n",
+ "fig = Figure()\n",
+ "ax = Axis(fig[1,1]; xlabel=\"Q (Å⁻¹)\", ylabel=\"ω (meV)\")\n",
+ "heatmap!(ax, radii, energies, output, colormap=:gnuplot2)\n",
+ "fig"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "This result can be compared to experimental neutron scattering data\n",
+ "from Fig. 5 of [Ge et al.](https://doi.org/10.1103/PhysRevB.96.064413)\n",
+ "\n",
+ "
\n",
+ "\n",
+ "\n",
+ "(The publication figure accidentally used a non-standard coordinate system to\n",
+ "label the wave vectors.)\n",
+ "\n",
+ "To get this agreement, the use of SU(3) coherent states is essential. In other\n",
+ "words, we needed a theory of multi-flavored bosons. The lower band has large\n",
+ "quadrupolar character, and arises from the strong easy-axis anisotropy of\n",
+ "FeI₂. By setting `mode = :SUN`, the calculation captures this coupled\n",
+ "dipole-quadrupole dynamics.\n",
+ "\n",
+ "An interesting exercise is to repeat the same study, but using `mode =\n",
+ ":dipole` instead of `:SUN`. That alternative choice would constrain the\n",
+ "coherent state dynamics to the space of dipoles only."
+ ],
+ "metadata": {}
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The full dynamical spin structure factor (DSSF) can be retrieved as a $3×3$\n",
+ "matrix with the `dssf` function, for a given path of $𝐪$-vectors."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "disp, is = dssf(swt, path);"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The first output `disp` is identical to that obtained from `dispersion`. The\n",
+ "second output `is` contains a list of $3×3$ matrix of intensities. For\n",
+ "example, `is[q,n][2,3]` yields the $(ŷ,ẑ)$ component of the structure factor\n",
+ "intensity for `nth` mode at the `q`th wavevector in the `path`."
+ ],
+ "metadata": {}
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "## What's next?\n",
+ "\n",
+ "The multi-boson linear spin wave theory, applied above, can be understood as\n",
+ "the quantization of a certain generalization of the Landau-Lifshitz spin\n",
+ "dynamics. Rather than dipoles, this dynamics takes places on the space of\n",
+ "[SU(_N_) coherent states](https://arxiv.org/abs/2106.14125).\n",
+ "\n",
+ "The full SU(_N_) coherent state dynamics, with appropriate quantum correction\n",
+ "factors, can be useful to model finite temperature scattering data. In\n",
+ "particular, it captures certain anharmonic effects due to thermal\n",
+ "fluctuations. This is the subject of our FeI₂ at Finite Temperature\n",
+ "tutorial.\n",
+ "\n",
+ "The classical dynamics is also a good starting point to study non-equilibrium\n",
+ "phenomena. Empirical noise and damping terms can be used to model [coupling to\n",
+ "a thermal bath](https://arxiv.org/abs/2209.01265). This yields a Langevin\n",
+ "dynamics of SU(_N_) coherent states. Our CP² Skyrmion Quench\n",
+ "tutorial shows how this dynamics gives rise to the formation of novel\n",
+ "topological defects in a temperature quench.\n",
+ "\n",
+ "Relative to LSWT calculations, it can take much more time to estimate\n",
+ "$\\mathcal{S}(𝐪,ω)$ intensities using classical dynamics simulation. See the\n",
+ "[SunnyTutorials\n",
+ "notebooks](https://nbviewer.org/github/SunnySuite/SunnyTutorials/tree/main/Tutorials/)\n",
+ "for examples of \"production-scale\" simulations."
+ ],
+ "metadata": {}
+ }
+ ],
+ "nbformat_minor": 3,
+ "metadata": {
+ "language_info": {
+ "file_extension": ".jl",
+ "mimetype": "application/julia",
+ "name": "julia",
+ "version": "1.9.3"
+ },
+ "kernelspec": {
+ "name": "julia-1.9",
+ "display_name": "Julia 1.9.3",
+ "language": "julia"
+ }
+ },
+ "nbformat": 4
+}
diff --git a/previews/PR176/assets/notebooks/ising2d.ipynb b/previews/PR176/assets/notebooks/ising2d.ipynb
new file mode 100644
index 000000000..3d9a59ddd
--- /dev/null
+++ b/previews/PR176/assets/notebooks/ising2d.ipynb
@@ -0,0 +1,179 @@
+{
+ "cells": [
+ {
+ "cell_type": "markdown",
+ "source": [
+ "# Classical Ising model\n",
+ "\n",
+ "This tutorial illustrates simulation of the classical 2D Ising model."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "using Sunny, WGLMakie"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Sunny expects a 3D `Crystal` unit cell. To model a square lattice, we\n",
+ "create an orthogonal unit cell where the $z$-spacing is distinct from the $x$\n",
+ "and $y$ spacing."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "a = 1\n",
+ "latvecs = lattice_vectors(a,a,10a,90,90,90)\n",
+ "crystal = Crystal(latvecs, [[0,0,0]])"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Create a `System` of spins with linear size `L` in the $x$ and $y$\n",
+ "directions, and only one layer in the $z$ direction. The option `:dipole`\n",
+ "means that the system will store Heisenberg spins, as opposed to SU($N$)\n",
+ "coherent states. Polarize the initial spin configuration using\n",
+ "`polarize_spins!`. Following the Ising convention, we will restrict\n",
+ "these spins to the $z$-axis and give them magnitude $S=1$.\n",
+ "\n",
+ "By default, Sunny uses physical units, e.g. magnetic field in tesla. Here we\n",
+ "specify an alternative `Units` system, so that the Zeeman coupling\n",
+ "between the spin dipole $𝐬$ and an external field $𝐁$ has the dimensionless\n",
+ "form $-𝐁⋅𝐬$."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "L = 128\n",
+ "sys = System(crystal, (L,L,1), [SpinInfo(1, S=1, g=1)], :dipole, units=Units.theory, seed=0)\n",
+ "polarize_spins!(sys, (0,0,1))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Use `set_exchange!` to include a ferromagnetic Heisenberg interaction\n",
+ "along nearest-neighbor bonds. The `Bond` below connects two spins\n",
+ "displaced by one lattice constant in the $x$-direction. This interaction will\n",
+ "be propagated to all nearest-neighbors bonds in the system, consistent with\n",
+ "the symmetries of the square lattice."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "set_exchange!(sys, -1.0, Bond(1,1,(1,0,0)))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "If an external field is desired, it can be set using\n",
+ "`set_external_field!`."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "B = 0\n",
+ "set_external_field!(sys, (0,0,B))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The critical temperature for the Ising model is known analytically."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "Tc = 2/log(1+√2)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Use a `LocalSampler` to perform `nsweeps` Monte Carlo sweeps. A sweep\n",
+ "consists of, on average, one trial update per spin in the system. Each\n",
+ "proposed update is accepted or rejected according to the Metropolis acceptance\n",
+ "probability. As its name suggests, the `propose_flip` function will\n",
+ "only propose pure spin flips, $𝐬 \\rightarrow -𝐬$."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "nsweeps = 4000\n",
+ "sampler = LocalSampler(kT=Tc, propose=propose_flip)\n",
+ "for i in 1:nsweeps\n",
+ " step!(sys, sampler)\n",
+ "end"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Plot the Ising spins by extracting the $z$-component of the dipoles"
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "heatmap(reshape([s.z for s in sys.dipoles], (L,L)))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ }
+ ],
+ "nbformat_minor": 3,
+ "metadata": {
+ "language_info": {
+ "file_extension": ".jl",
+ "mimetype": "application/julia",
+ "name": "julia",
+ "version": "1.9.3"
+ },
+ "kernelspec": {
+ "name": "julia-1.9",
+ "display_name": "Julia 1.9.3",
+ "language": "julia"
+ }
+ },
+ "nbformat": 4
+}
diff --git a/previews/PR176/assets/notebooks/out_of_equilibrium.ipynb b/previews/PR176/assets/notebooks/out_of_equilibrium.ipynb
new file mode 100644
index 000000000..d0c8a2c63
--- /dev/null
+++ b/previews/PR176/assets/notebooks/out_of_equilibrium.ipynb
@@ -0,0 +1,279 @@
+{
+ "cells": [
+ {
+ "cell_type": "markdown",
+ "source": [
+ "# CP² Skyrmion Quench\n",
+ "\n",
+ "This example demonstrates Sunny's ability to simulate the out-of-equilibrium\n",
+ "dynamics of generalized spin systems. We will implement the model Hamiltonian\n",
+ "of [Zhang et al., Nature Communications **14**, 3626\n",
+ "(2023)](https://www.nature.com/articles/s41467-023-39232-8), which supports a\n",
+ "novel type of topological defect, a CP² skyrmion, that involves both the\n",
+ "dipolar and quadrupolar parts of a quantum spin.\n",
+ "\n",
+ "Beginning from an initial high-temperature state, a disordered gas of CP²\n",
+ "skyrmions can be formed by rapidly quenching to low temperature. To model the\n",
+ "coupled dynamics of dipoles and quadrupoles, Sunny uses a recently developed\n",
+ "generalization of the Landau-Lifshitz spin dynamics, [Dahlbom et al., Phys.\n",
+ "Rev. B **106**, 235154 (2022)](https://doi.org/10.1103/PhysRevB.106.235154)."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "using Sunny, WGLMakie"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The Hamiltonian we will implement,\n",
+ "$$\n",
+ "\\mathcal{H} = \\sum_{\\langle i,j \\rangle} J_{ij}( \\hat{S}_i^x \\hat{S}_j^x + \\hat{S}_i^y \\hat{S}_j^y + \\Delta\\hat{S}_i^z \\hat{S}_j^z) - h\\sum_{i}\\hat{S}_i^z + D\\sum_{i}(\\hat{S}_i^z)^2\n",
+ "$$\n",
+ "contains competing ferromagnetic nearest-neightbor and antiferromagnetic\n",
+ "next-nearest-neighbor exchange terms on a triangular lattice. Both exchanges\n",
+ "exhibit anisotropy on the z-term. Additionally, there is an external magnetic\n",
+ "field, $h$, and easy-plane single-ion anisotropy, $D > 0$. We begin by\n",
+ "implementing the `Crystal`."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "lat_vecs = lattice_vectors(1.0, 1.0, 2.0, 90, 90, 120)\n",
+ "basis_vecs = [[0,0,0]]\n",
+ "cryst = Crystal(lat_vecs, basis_vecs)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The crystal is then used to create a spin `System`. All parameters in\n",
+ "this model system are dimensionless, so we select \"theory\" units and set the\n",
+ "g-factor to one."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "L = 40\n",
+ "dims = (L, L, 1)\n",
+ "sys = System(cryst, dims, [SpinInfo(1, S=1, g=1)], :SUN; seed=101, units=Units.theory)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "We proceed to implement each term of the Hamiltonian, selecting our parameters\n",
+ "so that the system occupies a region of the phase diagram that supports\n",
+ "skyrmions. The exchange interactions are set as follows."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "J1 = -1 # Nearest-neighbor ferromagnetic\n",
+ "J2 = (2.0/(1+√5)) # Tune competing exchange to set skyrmion scale length\n",
+ "Δ = 2.6 # Exchange anisotropy\n",
+ "\n",
+ "ex1 = J1 * [1.0 0.0 0.0;\n",
+ " 0.0 1.0 0.0;\n",
+ " 0.0 0.0 Δ]\n",
+ "ex2 = J2 * [1.0 0.0 0.0;\n",
+ " 0.0 1.0 0.0;\n",
+ " 0.0 0.0 Δ]\n",
+ "set_exchange!(sys, ex1, Bond(1, 1, [1, 0, 0]))\n",
+ "set_exchange!(sys, ex2, Bond(1, 1, [1, 2, 0]))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Next we add the external field,"
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "h = 15.5\n",
+ "field = set_external_field!(sys, [0.0 0.0 h])"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "and finally the single-ion anisotropy,"
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "D = 19.0\n",
+ "Sz = spin_operators(sys, 1)[3]\n",
+ "set_onsite_coupling!(sys, D*Sz^2, 1)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Initialize system to an infinite temperature (fully randomized) initial\n",
+ "condition."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "randomize_spins!(sys)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "We are now ready to simulate the quenching process using a generalized\n",
+ "`Langevin` spin dynamics. If we were working with spin dipoles only,\n",
+ "then `Langevin` dynamics would be the usual Landau-Lifshitz spin dynamics,\n",
+ "augmented with damping and noise terms. In the present study, we are instead\n",
+ "working with quantum spin-1 (an ($N=3$)-level system that includes both\n",
+ "dipoles and quadrupoles). Here, `Langevin` captures the coupled\n",
+ "dipole-quadrupole dynamics using the formalism of SU($N$) coherent states.\n",
+ "\n",
+ "Selecting `kT = 0` in the Langevin dynamics will effective disable the noise\n",
+ "term. Then the parameter `λ` effectively determines the damping time-scale."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "Δt = 0.2/D # Integration time step (inverse meV). Typically this will be\n",
+ " # inversely proportional to the largest energy scale in the\n",
+ " # system. We can use a fairly large time-step here because\n",
+ " # accuracy isn't critical.\n",
+ "kT = 0 # Target equilibrium temperature (meV)\n",
+ "λ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)\n",
+ "integrator = Langevin(Δt; kT, λ)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Finally we run the dynamics. We will record the state of the system at three\n",
+ "different times during the quenching process by copying the `coherents` field\n",
+ "of the `System`."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "τs = [4., 16, 256] # Times to record snapshots\n",
+ "frames = [] # Empty array to store snapshots\n",
+ "for i in eachindex(τs)\n",
+ " dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate\n",
+ " numsteps = round(Int, dur/Δt)\n",
+ " for _ in 1:numsteps # Perform the integration\n",
+ " step!(sys, integrator)\n",
+ " end\n",
+ " push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration\n",
+ "end"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "To visualize the state of the system contained in each snapshot, we will\n",
+ "calculate and plot the skyrmion density on each plaquette of our lattice. The\n",
+ "function `plot_triangular_plaquettes` is not part of the core Sunny package,\n",
+ "but rather something you could define yourself. We are using the definition in\n",
+ "`plotting2d.jl` from the Sunny [`examples/extra`\n",
+ "directory](https://github.com/SunnySuite/Sunny.jl/tree/main/examples/extra)."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "include(pkgdir(Sunny, \"examples\", \"extra\", \"plotting2d.jl\"))\n",
+ "\n",
+ "function sun_berry_curvature(z₁, z₂, z₃)\n",
+ " z₁, z₂, z₃ = normalize.((z₁, z₂, z₃))\n",
+ " n₁ = z₁ ⋅ z₂\n",
+ " n₂ = z₂ ⋅ z₃\n",
+ " n₃ = z₃ ⋅ z₁\n",
+ " return angle(n₁ * n₂ * n₃)\n",
+ "end\n",
+ "\n",
+ "plot_triangular_plaquettes(sun_berry_curvature, frames; resolution=(1800,600),\n",
+ " offset_spacing=10, texts = [\"\\tt = \"*string(τ) for τ in τs], text_offset = (0.0, 6.0)\n",
+ ")"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "The times are given in $\\hbar/|J_1|$. The white\n",
+ "background corresponds to a quantum paramagnetic state, where the local spin\n",
+ "exhibits a strong quadrupole moment and little or no dipole moment. Observe\n",
+ "that the process has generated a number of well-formed skyrmions of both\n",
+ "positive (red) and negative (blue) charge in addition to a number of other\n",
+ "metastable spin configurations. A full-sized version of this figure is\n",
+ "available in [Dahlbom et al.](https://doi.org/10.1103/PhysRevB.106.235154)."
+ ],
+ "metadata": {}
+ }
+ ],
+ "nbformat_minor": 3,
+ "metadata": {
+ "language_info": {
+ "file_extension": ".jl",
+ "mimetype": "application/julia",
+ "name": "julia",
+ "version": "1.9.3"
+ },
+ "kernelspec": {
+ "name": "julia-1.9",
+ "display_name": "Julia 1.9.3",
+ "language": "julia"
+ }
+ },
+ "nbformat": 4
+}
diff --git a/previews/PR176/assets/notebooks/powder_averaging.ipynb b/previews/PR176/assets/notebooks/powder_averaging.ipynb
new file mode 100644
index 000000000..0f84c42b8
--- /dev/null
+++ b/previews/PR176/assets/notebooks/powder_averaging.ipynb
@@ -0,0 +1,264 @@
+{
+ "cells": [
+ {
+ "cell_type": "markdown",
+ "source": [
+ "# Powder Averaged CoRh₂O₄\n",
+ "\n",
+ "This tutorial illustrates the calculation of the powder-averaged structure\n",
+ "factor by performing an orientational average. We consider a simple model of\n",
+ "the diamond-cubic crystal CoRh₂O₄, with parameters extracted from [Ge et al.,\n",
+ "Phys. Rev. B 96, 064413](https://doi.org/10.1103/PhysRevB.96.064413)."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "using Sunny, WGLMakie"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Construct a diamond `Crystal` in the conventional (non-primitive)\n",
+ "cubic unit cell. Sunny will populate all eight symmetry-equivalent sites when\n",
+ "given the international spacegroup number 227 (\"Fd-3m\") and the appropriate\n",
+ "setting. For this spacegroup, there are two conventional translations of the\n",
+ "unit cell, and it is necessary to disambiguate through the `setting` keyword\n",
+ "argument. (On your own: what happens if `setting` is omitted?)"
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "a = 8.5031 # (Å)\n",
+ "latvecs = lattice_vectors(a, a, a, 90, 90, 90)\n",
+ "cryst = Crystal(latvecs, [[0,0,0]], 227, setting=\"1\")"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "In a running Julia environment, the crystal can be viewed interactively using\n",
+ "`view_crystal`."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "view_crystal(cryst, 8.0)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Construct a `System` with an antiferromagnetic nearest neighbor\n",
+ "interaction `J`. Because the diamond crystal is bipartite, the ground state\n",
+ "will have unfrustrated Néel order. Selecting `latsize=(1,1,1)` is sufficient\n",
+ "because the ground state is periodic over each cubic unit cell. By passing an\n",
+ "explicit `seed`, the system's random number generator will give repeatable\n",
+ "results."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "latsize = (2, 2, 2)\n",
+ "seed = 0\n",
+ "S = 3/2\n",
+ "J = 7.5413*meV_per_K # (~ 0.65 meV)\n",
+ "sys = System(cryst, latsize, [SpinInfo(1; S, g=2)], :dipole; seed=0)\n",
+ "set_exchange!(sys, J, Bond(1, 3, [0,0,0]))"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "In the ground state, each spin is exactly anti-aligned with its 4\n",
+ "nearest-neighbors. Because every bond contributes an energy of $-JS^2$, the\n",
+ "energy per site is $-2JS^2$. In this calculation, a factor of 1/2 avoids\n",
+ "double-counting the bonds. Due to lack of frustration, direct energy\n",
+ "minimization is successful in finding the ground state."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "randomize_spins!(sys)\n",
+ "minimize_energy!(sys)\n",
+ "\n",
+ "@assert energy_per_site(sys) ≈ -2J*S^2"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Plotting the spins confirms the expected Néel order. Note that the overall,\n",
+ "global rotation of dipoles is arbitrary."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "s0 = sys.dipoles[1,1,1,1]\n",
+ "plot_spins(sys; color=[s'*s0 for s in sys.dipoles])"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "For numerical efficiency, it will be helpful to work with the smallest\n",
+ "possible magnetic supercell. Here, it is the primitive unit cell, which\n",
+ "contains just two sites. The variable `shape` below defines the primitive\n",
+ "lattice vectors `cryst.prim_latvecs` in units of the conventional lattice\n",
+ "vectors. This result is used as input to `reshape_supercell`. The\n",
+ "energy per site remains the same, which verifies that the magnetic supercell\n",
+ "is valid."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "shape = cryst.latvecs \\ cryst.prim_latvecs\n",
+ "sys_prim = reshape_supercell(sys, shape)\n",
+ "@assert energy_per_site(sys_prim) ≈ -2J*S^2"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "Now estimate $𝒮(𝐪,ω)$ with `SpinWaveTheory` and an\n",
+ "`intensity_formula`. The mode `:perp` contracts with a dipole factor\n",
+ "to return the unpolarized intensity. The formula also employs\n",
+ "`lorentzian` broadening. The isotropic `FormFactor` for\n",
+ "Cobalt(2+) dampens intensities at large $𝐪$."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "swt = SpinWaveTheory(sys_prim)\n",
+ "η = 0.4 # (meV)\n",
+ "kernel = lorentzian(η)\n",
+ "formfactors = [FormFactor(\"Co2\")]\n",
+ "formula = intensity_formula(swt, :perp; kernel, formfactors)"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "For the \"single crystal\" result, we may use `reciprocal_space_path` to\n",
+ "construct a path that connects high-symmetry points in reciprocal space. The\n",
+ "`intensities_broadened` function collects intensities along this path\n",
+ "for the given set of energy values."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "qpoints = [[0.0, 0.0, 0.0], [0.5, 0.0, 0.0], [0.5, 0.5, 0.0], [0.0, 0.0, 0.0]]\n",
+ "path, xticks = reciprocal_space_path(cryst, qpoints, 50)\n",
+ "energies = collect(0:0.01:6)\n",
+ "is = intensities_broadened(swt, path, energies, formula)\n",
+ "\n",
+ "fig = Figure()\n",
+ "ax = Axis(fig[1,1]; aspect=1.4, ylabel=\"ω (meV)\", xlabel=\"𝐪 (r.l.u.)\",\n",
+ " xticks, xticklabelrotation=π/10)\n",
+ "heatmap!(ax, 1:size(is, 1), energies, is, colormap=:gnuplot2)\n",
+ "fig"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "A powder measurement effectively involves an average over all possible crystal\n",
+ "orientations. We use the function `reciprocal_space_shell` to sample\n",
+ "`n` wavevectors on a sphere of a given radius (inverse angstroms), and then\n",
+ "calculate the spherically-averaged intensity."
+ ],
+ "metadata": {}
+ },
+ {
+ "outputs": [],
+ "cell_type": "code",
+ "source": [
+ "radii = 0.01:0.02:3 # (1/Å)\n",
+ "output = zeros(Float64, length(radii), length(energies))\n",
+ "for (i, radius) in enumerate(radii)\n",
+ " n = 300\n",
+ " qs = reciprocal_space_shell(cryst, radius, n)\n",
+ " is = intensities_broadened(swt, qs, energies, formula)\n",
+ " output[i, :] = sum(is, dims=1) / size(is, 1)\n",
+ "end\n",
+ "\n",
+ "fig = Figure()\n",
+ "ax = Axis(fig[1,1]; xlabel=\"Q (Å⁻¹)\", ylabel=\"ω (meV)\")\n",
+ "heatmap!(ax, radii, energies, output, colormap=:gnuplot2)\n",
+ "fig"
+ ],
+ "metadata": {},
+ "execution_count": null
+ },
+ {
+ "cell_type": "markdown",
+ "source": [
+ "This result can be compared to experimental neutron scattering data\n",
+ "from Fig. 5 of [Ge et al.](https://doi.org/10.1103/PhysRevB.96.064413)\n",
+ "\n",
+ " "
+ ],
+ "metadata": {}
+ }
+ ],
+ "nbformat_minor": 3,
+ "metadata": {
+ "language_info": {
+ "file_extension": ".jl",
+ "mimetype": "application/julia",
+ "name": "julia",
+ "version": "1.9.3"
+ },
+ "kernelspec": {
+ "name": "julia-1.9",
+ "display_name": "Julia 1.9.3",
+ "language": "julia"
+ }
+ },
+ "nbformat": 4
+}
diff --git a/previews/PR176/assets/paraviewcolormap.png b/previews/PR176/assets/paraviewcolormap.png
new file mode 100644
index 000000000..1d1073abe
Binary files /dev/null and b/previews/PR176/assets/paraviewcolormap.png differ
diff --git a/previews/PR176/assets/paraviewimport.png b/previews/PR176/assets/paraviewimport.png
new file mode 100644
index 000000000..dd6d0b1f5
Binary files /dev/null and b/previews/PR176/assets/paraviewimport.png differ
diff --git a/previews/PR176/assets/paraviewrender.jpg b/previews/PR176/assets/paraviewrender.jpg
new file mode 100644
index 000000000..975c20149
Binary files /dev/null and b/previews/PR176/assets/paraviewrender.jpg differ
diff --git a/previews/PR176/assets/paraviewvolume.png b/previews/PR176/assets/paraviewvolume.png
new file mode 100644
index 000000000..eed55ec5e
Binary files /dev/null and b/previews/PR176/assets/paraviewvolume.png differ
diff --git a/previews/PR176/assets/scripts/08_Kagome_AFM.jl b/previews/PR176/assets/scripts/08_Kagome_AFM.jl
new file mode 100644
index 000000000..5fdaecb1d
--- /dev/null
+++ b/previews/PR176/assets/scripts/08_Kagome_AFM.jl
@@ -0,0 +1,36 @@
+using Sunny, GLMakie
+
+a = b = 6.0 # (Å)
+c = 40.0
+latvecs = lattice_vectors(a, b, c, 90, 90, 120)
+crystal = Crystal(latvecs, [[1/2,0,0]], 147; types=["Cr"])
+
+S = 1
+sys = System(crystal, (3,3,1), [SpinInfo(1; S, g=2)], :dipole)
+J = 1.0
+set_exchange!(sys, J, Bond(2,3,[0,0,0]))
+
+q = -[1/3, 1/3, 0]
+axis = [0,0,1]
+set_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[cos(0),sin(0),0])
+set_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[cos(0),sin(0),0])
+set_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[cos(2π/3),sin(2π/3),0])
+plot_spins(sys; ghost_radius=30, orthographic=true)
+
+@assert energy_per_site(sys) ≈ (4/2)*J*S^2*cos(2π/3)
+
+points_rlu = [[-1/2, 0, 0], [0, 0, 0], [1/2, 1/2, 0]]
+density = 100
+path, xticks = reciprocal_space_path(crystal, points_rlu, density);
+
+swt = SpinWaveTheory(sys)
+formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)
+disp, intensity = intensities_bands(swt, path, formula);
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+ylims!(ax, -1e-1, 2.3)
+for i in axes(disp)[2]
+ lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange=(0,1e-2))
+end
+fig
diff --git a/previews/PR176/assets/scripts/15_Ba3NbFe3Si2O14.jl b/previews/PR176/assets/scripts/15_Ba3NbFe3Si2O14.jl
new file mode 100644
index 000000000..a6d7a837d
--- /dev/null
+++ b/previews/PR176/assets/scripts/15_Ba3NbFe3Si2O14.jl
@@ -0,0 +1,61 @@
+using Sunny, GLMakie
+
+a = b = 8.539 # (Å)
+c = 5.2414
+latvecs = lattice_vectors(a, b, c, 90, 90, 120)
+types = ["Fe","Nb","Ba","Si","O","O","O"]
+positions = [[0.24964,0,0.5],[0,0,0],[0.56598,0,0],[2/3,1/3,0.5220],[2/3,1/3,0.2162],[0.5259,0.7024,0.3536],[0.7840,0.9002,0.7760]]
+langasite = Crystal(latvecs, positions, 150; types)
+crystal = subcrystal(langasite, "Fe")
+view_crystal(crystal, 7)
+
+latsize = (1,1,7)
+S = 5/2
+seed = 5
+sys = System(crystal, latsize, [SpinInfo(1; S, g=2)], :dipole; seed)
+
+J₁ = 0.85
+J₂ = 0.24
+J₃ = 0.053
+J₄ = 0.017
+J₅ = 0.24
+set_exchange!(sys, J₁, Bond(3, 2, [1,1,0]))
+set_exchange!(sys, J₄, Bond(1, 1, [0,0,1]))
+set_exchange!(sys, J₂, Bond(1, 3, [0,0,0]))
+
+ϵD = -1
+ϵH = +1
+ϵT = ϵD * ϵH
+
+if ϵT == -1
+ set_exchange!(sys, J₃, Bond(2, 3, [-1,-1,1]))
+ set_exchange!(sys, J₅, Bond(3, 2, [1,1,1]))
+elseif ϵT == 1
+ set_exchange!(sys, J₅, Bond(2, 3, [-1,-1,1]))
+ set_exchange!(sys, J₃, Bond(3, 2, [1,1,1]))
+else
+ throw("Provide a valid chirality")
+end
+
+q = [0, 0, 1/7]
+axis = [0,0,1]
+set_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[1, 0, 0])
+set_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[-1/2, -sqrt(3)/2, 0])
+set_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[-1/2, +sqrt(3)/2, 0])
+
+plot_spins(sys; color=[s[1] for s in sys.dipoles])
+
+points_rlu = [[0,1,-1],[0,1,-1+1],[0,1,-1+2],[0,1,-1+3]];
+density = 100
+path, xticks = reciprocal_space_path(crystal, points_rlu, density);
+
+swt = SpinWaveTheory(sys)
+γ = 0.15 # width in meV
+broadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))
+energies = collect(0:0.01:6) # 0 < ω < 6 (meV).
+is = intensities_broadened(swt, path, energies, broadened_formula);
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+heatmap!(ax, 1:size(is,1), energies, is, colorrange=(0,5))
+fig
diff --git a/previews/PR176/assets/scripts/fei2_classical.jl b/previews/PR176/assets/scripts/fei2_classical.jl
new file mode 100644
index 000000000..a6940adbd
--- /dev/null
+++ b/previews/PR176/assets/scripts/fei2_classical.jl
@@ -0,0 +1,250 @@
+using Sunny, LinearAlgebra, GLMakie
+
+a = b = 4.05012#hide
+c = 6.75214#hide
+latvecs = lattice_vectors(a, b, c, 90, 90, 120)#hide
+positions = [[0,0,0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]]#hide
+types = ["Fe", "I", "I"]#hide
+FeI2 = Crystal(latvecs, positions; types)#hide
+cryst = subcrystal(FeI2, "Fe")#hide
+sys = System(cryst, (4,4,4), [SpinInfo(1,S=1,g=2)], :SUN, seed=2)#hide
+J1pm = -0.236#hide
+J1pmpm = -0.161#hide
+J1zpm = -0.261#hide
+J2pm = 0.026#hide
+J3pm = 0.166#hide
+J′0pm = 0.037#hide
+J′1pm = 0.013#hide
+J′2apm = 0.068#hide
+J1zz = -0.236#hide
+J2zz = 0.113#hide
+J3zz = 0.211#hide
+J′0zz = -0.036#hide
+J′1zz = 0.051#hide
+J′2azz = 0.073#hide
+J1xx = J1pm + J1pmpm#hide
+J1yy = J1pm - J1pmpm#hide
+J1yz = J1zpm#hide
+set_exchange!(sys, [J1xx 0.0 0.0; 0.0 J1yy J1yz; 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))#hide
+set_exchange!(sys, [J2pm 0.0 0.0; 0.0 J2pm 0.0; 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))#hide
+set_exchange!(sys, [J3pm 0.0 0.0; 0.0 J3pm 0.0; 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))#hide
+set_exchange!(sys, [J′0pm 0.0 0.0; 0.0 J′0pm 0.0; 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))#hide
+set_exchange!(sys, [J′1pm 0.0 0.0; 0.0 J′1pm 0.0; 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))#hide
+set_exchange!(sys, [J′2apm 0.0 0.0; 0.0 J′2apm 0.0; 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))#hide
+D = 2.165#hide
+S = spin_operators(sys, 1)#hide
+set_onsite_coupling!(sys, -D*S[3]^2, 1)#hide
+sys
+
+Δt = 0.05/D # Should be inversely proportional to the largest energy scale
+ # in the system. For FeI2, this is the easy-axis anisotropy,
+ # `D = 2.165` (meV). The prefactor 0.05 is relatively small,
+ # and achieves high accuracy.
+kT = 0.2 # Temperature of the thermal bath (meV).
+λ = 0.1 # This value is typically good for Monte Carlo sampling,
+ # independent of system details.
+
+langevin = Langevin(Δt; kT, λ);
+
+randomize_spins!(sys)
+for _ in 1:20_000
+ step!(sys, langevin)
+end
+
+plot_spins(sys; color=[s[3] for s in sys.dipoles])
+
+sys_large = resize_supercell(sys, (16,16,4)) # 16x16x4 copies of the original unit cell
+plot_spins(sys_large; color=[s[3] for s in sys_large.dipoles])
+
+kT = 3.5 * meV_per_K # 3.5K ≈ 0.30 meV
+langevin.kT = kT
+for _ in 1:10_000
+ step!(sys_large, langevin)
+end
+
+sc = dynamical_correlations(sys_large; Δt=2Δt, nω=120, ωmax=7.5)
+
+add_sample!(sc, sys_large) # Accumulate the sample into `sc`
+
+for _ in 1:2
+ for _ in 1:1000 # Enough steps to decorrelate spins
+ step!(sys_large, langevin)
+ end
+ add_sample!(sc, sys_large) # Accumulate the sample into `sc`
+end
+
+sc
+
+formula = intensity_formula(sc, :trace; kT)
+
+qs = [[0, 0, 0], [0.5, 0.5, 0.5]]
+is = intensities_interpolated(sc, qs, formula; interpolation = :round)
+
+ωs = available_energies(sc)
+fig = lines(ωs, is[1,:]; axis=(xlabel="meV", ylabel="Intensity"), label="(0,0,0)")
+lines!(ωs, is[2,:]; label="(π,π,π)")
+axislegend()
+fig
+
+formfactors = [FormFactor("Fe2"; g_lande=3/2)]
+new_formula = intensity_formula(sc, :perp; kT, formfactors = formfactors)
+
+points = [[0, 0, 0], # List of wave vectors that define a path
+ [1, 0, 0],
+ [0, 1, 0],
+ [1/2, 0, 0],
+ [0, 1, 0],
+ [0, 0, 0]]
+density = 40
+path, xticks = reciprocal_space_path(cryst, points, density);
+
+is_interpolated = intensities_interpolated(sc, path, new_formula;
+ interpolation = :linear, # Interpolate between available wave vectors
+);
+# Add artificial broadening
+is_interpolated_broadened = broaden_energy(sc, is, (ω, ω₀)->lorentzian(ω-ω₀, 0.05));
+
+cut_width = 0.3
+density = 15
+paramsList, markers, ranges = reciprocal_space_path_bins(sc,points,density,cut_width);
+
+total_bins = ranges[end][end]
+energy_bins = paramsList[1].numbins[4]
+is_binned = zeros(Float64,total_bins,energy_bins)
+integrated_kernel = integrated_lorentzian(0.05) # Lorentzian broadening
+for k in eachindex(paramsList)
+ bin_data, counts = intensities_binned(sc,paramsList[k], new_formula;
+ integrated_kernel = integrated_kernel
+ )
+ is_binned[ranges[k],:] = bin_data[:,1,1,:] ./ counts[:,1,1,:]
+end
+
+fig = Figure()
+ax_top = Axis(fig[1,1],ylabel = "meV",xticklabelrotation=π/8,xticklabelsize=12;xticks)
+ax_bottom = Axis(fig[2,1],ylabel = "meV",xticks = (markers, string.(points)),xticklabelrotation=π/8,xticklabelsize=12)
+
+heatmap!(ax_top,1:size(is_interpolated,1), ωs, is_interpolated;
+ colorrange=(0.0,0.07),
+)
+
+heatmap!(ax_bottom,1:size(is_binned,1), ωs, is_binned;
+ colorrange=(0.0,0.05),
+)
+
+fig
+
+ωidx = 60
+target_ω = ωs[ωidx]
+
+params = unit_resolution_binning_parameters(sc)
+params.binstart[1:2] .= -1 # Expand plot range slightly
+
+# Set energy integration range
+omega_width = 0.3
+params.binstart[4] = target_ω - (omega_width/2)
+params.binend[4] = target_ω # `binend` should be inside (e.g. at the center) of the range
+params.binwidth[4] = omega_width
+
+integrate_axes!(params, axes = 3) # Integrate out z direction entirely
+
+params
+
+is, counts = intensities_binned(sc,params,new_formula)
+
+fig = Figure()
+ax = Axis(fig[1,1];
+ title="Δω=0.3 meV (Binned)", aspect=true,
+ xlabel = "[H, 0, 0]",
+ ylabel = "[0, K, 0]"
+)
+bcs = axes_bincenters(params)
+hm = heatmap!(ax,bcs[1],bcs[2],is[:,:,1,1] ./ counts[:,:,1,1])
+function add_lines!(ax,params)#hide
+ bes = Sunny.axes_binedges(params)#hide
+ hrange = range(-2,2,length=17)#hide
+ linesegments!(ax,[(Point2f(params.covectors[1,1:3] ⋅ [h,-10,0],params.covectors[2,1:3] ⋅ [h,-10,0]),Point2f(params.covectors[1,1:3] ⋅ [h,10,0],params.covectors[2,1:3] ⋅ [h,10,0])) for h = hrange],linestyle=:dash,color=:black)#hide
+ krange = range(-2,2,length=17)#hide
+ linesegments!(ax,[(Point2f(params.covectors[1,1:3] ⋅ [-10,k,0],params.covectors[2,1:3] ⋅ [-10,k,0]),Point2f(params.covectors[1,1:3] ⋅ [10,k,0],params.covectors[2,1:3] ⋅ [10,k,0])) for k = krange],linestyle=:dash,color=:black)#hide
+ xlims!(ax,bes[1][1],bes[1][end])#hide
+ ylims!(ax,bes[2][1],bes[2][end])#hide
+end#hide
+add_lines!(ax,params)
+Colorbar(fig[1,2], hm);
+fig
+
+latvecs = sys.crystal.latvecs
+metric = latvecs' * I(3) * latvecs
+
+(latvecs * [1/2,1,0]) ⋅ (latvecs * [1,0,0]) == 0
+
+f = Figure()#hide
+ax = Axis(f[1,1])#hide
+arrows!(ax,[Point2f(0,0),Point2f(latvecs[1:2,1] ./ 2)],[Vec2f(latvecs[1:2,1] ./ 2), Vec2f(latvecs[1:2,2])],arrowcolor = :blue,arrowsize = 30.,linewidth = 5.,linecolor = :blue)#hide
+arrows!(ax,[Point2f(0,0)],[Vec2f(latvecs[1:2,:] * [1/2,1,0])],arrowcolor = :red,arrowsize = 30.,linewidth = 5.,linecolor = :red, linestyle = :dash)#hide
+scatter!(ax,[Point2f(latvecs[1:2,:] * [a,b,0]) for a in -1:1, b in -1:1][:],color = :black)#hide
+annotations!(ax,["0","0+b","0+a", "a/2", "b"],[Point2f(0,-0.3),Point2f(latvecs[1:2,2]) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1]) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1] ./ 4) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1] ./ 2) .+ Vec2f(latvecs[1:2,2] ./ 2) .+ Vec2f(0.3,0.3)],color=[:black,:black,:black,:blue,:blue])#hide
+f#hide
+
+params.covectors[2,1:3] = [1/2,1,0] # [1/2,1,0] times [a;b;c] is (a/2 + b)
+params#hide
+
+# Zoom out horizontal axis
+params.binstart[1], params.binend[1] = -2, 2
+
+# Adjust vertical axis bounds to account for
+# length of a/2 + b
+params.binstart[2], params.binend[2] = -2 * sqrt(3/4), 2 * sqrt(3/4)
+
+# Re-compute in the new coordinate system
+is, counts = intensities_binned(sc,params,new_formula)
+
+fig = Figure(; resolution=(1200,500))#hide
+ax_right = Axis(fig[1,3];#hide
+ title="ω≈$(round(target_ω, digits=2)) meV with Δω=0.3 meV (Binned)", aspect=true,#hide
+ xlabel = "[H, -1/2H, 0]"#hide
+)#hide
+bcs = axes_bincenters(params)#hide
+hm_right = heatmap!(ax_right,bcs[1],bcs[2],is[:,:,1,1] ./ counts[:,:,1,1])#hide
+add_lines!(ax_right,params)
+Colorbar(fig[1,4], hm_right);#hide
+
+# New basis matrix
+A = [1 0 0
+ -1/2 1 0
+ 0 0 1]
+
+# Define our grid of wave vectors
+npoints = 60
+as = range(-2, 2, npoints)
+bs = range(-3/√3, 3/√3, npoints)
+qs_ortho = [[a, b, 0] for a in as, b in bs]
+
+# Convert to original RLU system for input to Sunny
+qs = [A * q for q in qs_ortho]
+
+# Use interpolation to get intensities
+is = intensities_interpolated(sc, qs, new_formula; interpolation=:linear)
+
+ax_left = Axis(fig[1,2];#hide
+ title="ω≈$(round(ωs[ωidx], digits=2)) meV (Interpolated)", aspect=true,#hide
+ xlabel = "[H, -1/2H, 0]", ylabel = "[0, K, 0]"#hide
+)#hide
+hm_left = heatmap!(ax_left, as, bs, is[:,:,ωidx])#hide
+add_lines!(ax_left,params)
+Colorbar(fig[1,1], hm_left);#hide
+fig
+
+metric = (latvecs * inv(A'))' * I(3) * (latvecs * inv(A'))
+
+is_static = instant_intensities_interpolated(sc, qs, new_formula; interpolation = :linear)
+
+hm = heatmap(as, bs, is_static;
+ axis=(
+ title="Instantaneous Structure Factor",
+ xlabel = "[H, -1/2H, 0]",
+ ylabel = "[0, K, 0]",
+ aspect=true
+ )
+)
+Colorbar(hm.figure[1,2], hm.plot)
+hm
diff --git a/previews/PR176/assets/scripts/fei2_tutorial.jl b/previews/PR176/assets/scripts/fei2_tutorial.jl
new file mode 100644
index 000000000..5d1b97d8a
--- /dev/null
+++ b/previews/PR176/assets/scripts/fei2_tutorial.jl
@@ -0,0 +1,118 @@
+using Sunny, GLMakie
+
+a = b = 4.05012 # Lattice constants for triangular lattice
+c = 6.75214 # Spacing in the z-direction
+
+latvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that
+ # define the conventional unit cell
+positions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions
+ # of lattice vectors
+types = ["Fe", "I", "I"]
+FeI2 = Crystal(latvecs, positions; types)
+
+cryst = subcrystal(FeI2, "Fe")
+
+view_crystal(cryst, 8.0)
+
+print_symmetry_table(cryst, 8.0)
+
+sys = System(cryst, (4, 4, 4), [SpinInfo(1, S=1, g=2)], :SUN, seed=2)
+
+J1pm = -0.236
+J1pmpm = -0.161
+J1zpm = -0.261
+J2pm = 0.026
+J3pm = 0.166
+J′0pm = 0.037
+J′1pm = 0.013
+J′2apm = 0.068
+
+J1zz = -0.236
+J2zz = 0.113
+J3zz = 0.211
+J′0zz = -0.036
+J′1zz = 0.051
+J′2azz = 0.073
+
+J1xx = J1pm + J1pmpm
+J1yy = J1pm - J1pmpm
+J1yz = J1zpm
+
+set_exchange!(sys, [J1xx 0.0 0.0;
+ 0.0 J1yy J1yz;
+ 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))
+set_exchange!(sys, [J2pm 0.0 0.0;
+ 0.0 J2pm 0.0;
+ 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))
+set_exchange!(sys, [J3pm 0.0 0.0;
+ 0.0 J3pm 0.0;
+ 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))
+set_exchange!(sys, [J′0pm 0.0 0.0;
+ 0.0 J′0pm 0.0;
+ 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))
+set_exchange!(sys, [J′1pm 0.0 0.0;
+ 0.0 J′1pm 0.0;
+ 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))
+set_exchange!(sys, [J′2apm 0.0 0.0;
+ 0.0 J′2apm 0.0;
+ 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))
+
+D = 2.165
+S = spin_operators(sys, 1)
+set_onsite_coupling!(sys, -D*S[3]^2, 1)
+
+randomize_spins!(sys)
+minimize_energy!(sys)
+
+plot_spins(sys; color=[s[3] for s in sys.dipoles])
+
+print_wrapped_intensities(sys)
+
+suggest_magnetic_supercell([[0, -1/4, 1/4]])
+
+sys_min = reshape_supercell(sys, [1 0 0; 0 2 1; 0 -2 1])
+randomize_spins!(sys_min)
+minimize_energy!(sys_min);
+
+plot_spins(sys_min; color=[s[3] for s in sys_min.dipoles], ghost_radius=12)
+
+swt = SpinWaveTheory(sys_min)
+
+q_points = [[0,0,0], [1,0,0], [0,1,0], [1/2,0,0], [0,1,0], [0,0,0]];
+
+density = 50
+path, xticks = reciprocal_space_path(cryst, q_points, density);
+
+disp = dispersion(swt, path);
+
+formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)
+
+disp, intensity = intensities_bands(swt, path, formula);
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+ylims!(ax, 0.0, 7.5)
+xlims!(ax, 1, size(disp, 1))
+colorrange = extrema(intensity)
+for i in axes(disp)[2]
+ lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange)
+end
+fig
+
+γ = 0.15 # width in meV
+broadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))
+
+energies = collect(0:0.01:10) # 0 < ω < 10 (meV).
+is1 = intensities_broadened(swt, path, energies, broadened_formula);
+
+R = rotation_in_rlu(cryst, [0, 0, 1], 2π/3)
+is2 = intensities_broadened(swt, [R*q for q in path], energies, broadened_formula)
+is3 = intensities_broadened(swt, [R*R*q for q in path], energies, broadened_formula)
+is_averaged = (is1 + is2 + is3) / 3
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+heatmap!(ax, 1:size(is_averaged, 1), energies, is_averaged)
+fig
+
+disp, is = dssf(swt, path);
diff --git a/previews/PR176/assets/scripts/ising2d.jl b/previews/PR176/assets/scripts/ising2d.jl
new file mode 100644
index 000000000..6ab158eb6
--- /dev/null
+++ b/previews/PR176/assets/scripts/ising2d.jl
@@ -0,0 +1,24 @@
+using Sunny, GLMakie
+
+a = 1
+latvecs = lattice_vectors(a,a,10a,90,90,90)
+crystal = Crystal(latvecs, [[0,0,0]])
+
+L = 128
+sys = System(crystal, (L,L,1), [SpinInfo(1, S=1, g=1)], :dipole, units=Units.theory, seed=0)
+polarize_spins!(sys, (0,0,1))
+
+set_exchange!(sys, -1.0, Bond(1,1,(1,0,0)))
+
+B = 0
+set_external_field!(sys, (0,0,B))
+
+Tc = 2/log(1+√2)
+
+nsweeps = 4000
+sampler = LocalSampler(kT=Tc, propose=propose_flip)
+for i in 1:nsweeps
+ step!(sys, sampler)
+end
+
+heatmap(reshape([s.z for s in sys.dipoles], (L,L)))
diff --git a/previews/PR176/assets/scripts/out_of_equilibrium.jl b/previews/PR176/assets/scripts/out_of_equilibrium.jl
new file mode 100644
index 000000000..60ce32a36
--- /dev/null
+++ b/previews/PR176/assets/scripts/out_of_equilibrium.jl
@@ -0,0 +1,64 @@
+using Sunny, GLMakie
+
+lat_vecs = lattice_vectors(1.0, 1.0, 2.0, 90, 90, 120)
+basis_vecs = [[0,0,0]]
+cryst = Crystal(lat_vecs, basis_vecs)
+
+L = 40
+dims = (L, L, 1)
+sys = System(cryst, dims, [SpinInfo(1, S=1, g=1)], :SUN; seed=101, units=Units.theory)
+
+J1 = -1 # Nearest-neighbor ferromagnetic
+J2 = (2.0/(1+√5)) # Tune competing exchange to set skyrmion scale length
+Δ = 2.6 # Exchange anisotropy
+
+ex1 = J1 * [1.0 0.0 0.0;
+ 0.0 1.0 0.0;
+ 0.0 0.0 Δ]
+ex2 = J2 * [1.0 0.0 0.0;
+ 0.0 1.0 0.0;
+ 0.0 0.0 Δ]
+set_exchange!(sys, ex1, Bond(1, 1, [1, 0, 0]))
+set_exchange!(sys, ex2, Bond(1, 1, [1, 2, 0]))
+
+h = 15.5
+field = set_external_field!(sys, [0.0 0.0 h])
+
+D = 19.0
+Sz = spin_operators(sys, 1)[3]
+set_onsite_coupling!(sys, D*Sz^2, 1)
+
+randomize_spins!(sys)
+
+Δt = 0.2/D # Integration time step (inverse meV). Typically this will be
+ # inversely proportional to the largest energy scale in the
+ # system. We can use a fairly large time-step here because
+ # accuracy isn't critical.
+kT = 0 # Target equilibrium temperature (meV)
+λ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)
+integrator = Langevin(Δt; kT, λ)
+
+τs = [4., 16, 256] # Times to record snapshots
+frames = [] # Empty array to store snapshots
+for i in eachindex(τs)
+ dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate
+ numsteps = round(Int, dur/Δt)
+ for _ in 1:numsteps # Perform the integration
+ step!(sys, integrator)
+ end
+ push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration
+end
+
+include(pkgdir(Sunny, "examples", "extra", "plotting2d.jl"))
+
+function sun_berry_curvature(z₁, z₂, z₃)
+ z₁, z₂, z₃ = normalize.((z₁, z₂, z₃))
+ n₁ = z₁ ⋅ z₂
+ n₂ = z₂ ⋅ z₃
+ n₃ = z₃ ⋅ z₁
+ return angle(n₁ * n₂ * n₃)
+end
+
+plot_triangular_plaquettes(sun_berry_curvature, frames; resolution=(1800,600),
+ offset_spacing=10, texts = ["\tt = "*string(τ) for τ in τs], text_offset = (0.0, 6.0)
+)
diff --git a/previews/PR176/assets/scripts/powder_averaging.jl b/previews/PR176/assets/scripts/powder_averaging.jl
new file mode 100644
index 000000000..5e18d9af5
--- /dev/null
+++ b/previews/PR176/assets/scripts/powder_averaging.jl
@@ -0,0 +1,57 @@
+using Sunny, GLMakie
+
+a = 8.5031 # (Å)
+latvecs = lattice_vectors(a, a, a, 90, 90, 90)
+cryst = Crystal(latvecs, [[0,0,0]], 227, setting="1")
+
+view_crystal(cryst, 8.0)
+
+latsize = (2, 2, 2)
+seed = 0
+S = 3/2
+J = 7.5413*meV_per_K # (~ 0.65 meV)
+sys = System(cryst, latsize, [SpinInfo(1; S, g=2)], :dipole; seed=0)
+set_exchange!(sys, J, Bond(1, 3, [0,0,0]))
+
+randomize_spins!(sys)
+minimize_energy!(sys)
+
+@assert energy_per_site(sys) ≈ -2J*S^2
+
+s0 = sys.dipoles[1,1,1,1]
+plot_spins(sys; color=[s'*s0 for s in sys.dipoles])
+
+shape = cryst.latvecs \ cryst.prim_latvecs
+sys_prim = reshape_supercell(sys, shape)
+@assert energy_per_site(sys_prim) ≈ -2J*S^2
+
+swt = SpinWaveTheory(sys_prim)
+η = 0.4 # (meV)
+kernel = lorentzian(η)
+formfactors = [FormFactor("Co2")]
+formula = intensity_formula(swt, :perp; kernel, formfactors)
+
+qpoints = [[0.0, 0.0, 0.0], [0.5, 0.0, 0.0], [0.5, 0.5, 0.0], [0.0, 0.0, 0.0]]
+path, xticks = reciprocal_space_path(cryst, qpoints, 50)
+energies = collect(0:0.01:6)
+is = intensities_broadened(swt, path, energies, formula)
+
+fig = Figure()
+ax = Axis(fig[1,1]; aspect=1.4, ylabel="ω (meV)", xlabel="𝐪 (r.l.u.)",
+ xticks, xticklabelrotation=π/10)
+heatmap!(ax, 1:size(is, 1), energies, is, colormap=:gnuplot2)
+fig
+
+radii = 0.01:0.02:3 # (1/Å)
+output = zeros(Float64, length(radii), length(energies))
+for (i, radius) in enumerate(radii)
+ n = 300
+ qs = reciprocal_space_shell(cryst, radius, n)
+ is = intensities_broadened(swt, qs, energies, formula)
+ output[i, :] = sum(is, dims=1) / size(is, 1)
+end
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Q (Å⁻¹)", ylabel="ω (meV)")
+heatmap!(ax, radii, energies, output, colormap=:gnuplot2)
+fig
diff --git a/previews/PR176/assets/themes/documenter-dark.css b/previews/PR176/assets/themes/documenter-dark.css
new file mode 100644
index 000000000..691b83abd
--- /dev/null
+++ b/previews/PR176/assets/themes/documenter-dark.css
@@ -0,0 +1,7 @@
+html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-ellipsis,html.theme--documenter-dark .file-cta,html.theme--documenter-dark .file-name,html.theme--documenter-dark .select select,html.theme--documenter-dark .textarea,html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark .button{-moz-appearance:none;-webkit-appearance:none;align-items:center;border:1px solid transparent;border-radius:.4em;box-shadow:none;display:inline-flex;font-size:1rem;height:2.5em;justify-content:flex-start;line-height:1.5;padding-bottom:calc(0.5em - 1px);padding-left:calc(0.75em - 1px);padding-right:calc(0.75em - 1px);padding-top:calc(0.5em - 1px);position:relative;vertical-align:top}html.theme--documenter-dark .pagination-previous:focus,html.theme--documenter-dark .pagination-next:focus,html.theme--documenter-dark .pagination-link:focus,html.theme--documenter-dark .pagination-ellipsis:focus,html.theme--documenter-dark .file-cta:focus,html.theme--documenter-dark .file-name:focus,html.theme--documenter-dark .select select:focus,html.theme--documenter-dark .textarea:focus,html.theme--documenter-dark .input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:focus,html.theme--documenter-dark .button:focus,html.theme--documenter-dark .is-focused.pagination-previous,html.theme--documenter-dark .is-focused.pagination-next,html.theme--documenter-dark .is-focused.pagination-link,html.theme--documenter-dark .is-focused.pagination-ellipsis,html.theme--documenter-dark .is-focused.file-cta,html.theme--documenter-dark .is-focused.file-name,html.theme--documenter-dark .select select.is-focused,html.theme--documenter-dark .is-focused.textarea,html.theme--documenter-dark .is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-focused.button,html.theme--documenter-dark .pagination-previous:active,html.theme--documenter-dark .pagination-next:active,html.theme--documenter-dark .pagination-link:active,html.theme--documenter-dark .pagination-ellipsis:active,html.theme--documenter-dark .file-cta:active,html.theme--documenter-dark .file-name:active,html.theme--documenter-dark .select select:active,html.theme--documenter-dark .textarea:active,html.theme--documenter-dark .input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:active,html.theme--documenter-dark .button:active,html.theme--documenter-dark .is-active.pagination-previous,html.theme--documenter-dark .is-active.pagination-next,html.theme--documenter-dark .is-active.pagination-link,html.theme--documenter-dark .is-active.pagination-ellipsis,html.theme--documenter-dark .is-active.file-cta,html.theme--documenter-dark .is-active.file-name,html.theme--documenter-dark .select select.is-active,html.theme--documenter-dark .is-active.textarea,html.theme--documenter-dark .is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active,html.theme--documenter-dark .is-active.button{outline:none}html.theme--documenter-dark .pagination-previous[disabled],html.theme--documenter-dark .pagination-next[disabled],html.theme--documenter-dark .pagination-link[disabled],html.theme--documenter-dark .pagination-ellipsis[disabled],html.theme--documenter-dark .file-cta[disabled],html.theme--documenter-dark .file-name[disabled],html.theme--documenter-dark .select select[disabled],html.theme--documenter-dark .textarea[disabled],html.theme--documenter-dark .input[disabled],html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled],html.theme--documenter-dark .button[disabled],fieldset[disabled] html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark fieldset[disabled] .pagination-previous,fieldset[disabled] html.theme--documenter-dark .pagination-next,html.theme--documenter-dark fieldset[disabled] .pagination-next,fieldset[disabled] html.theme--documenter-dark .pagination-link,html.theme--documenter-dark fieldset[disabled] .pagination-link,fieldset[disabled] html.theme--documenter-dark .pagination-ellipsis,html.theme--documenter-dark fieldset[disabled] .pagination-ellipsis,fieldset[disabled] html.theme--documenter-dark .file-cta,html.theme--documenter-dark fieldset[disabled] .file-cta,fieldset[disabled] html.theme--documenter-dark .file-name,html.theme--documenter-dark fieldset[disabled] .file-name,fieldset[disabled] html.theme--documenter-dark .select select,fieldset[disabled] html.theme--documenter-dark .textarea,fieldset[disabled] html.theme--documenter-dark .input,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark fieldset[disabled] .select select,html.theme--documenter-dark .select fieldset[disabled] select,html.theme--documenter-dark fieldset[disabled] .textarea,html.theme--documenter-dark fieldset[disabled] .input,html.theme--documenter-dark fieldset[disabled] #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar fieldset[disabled] form.docs-search>input,fieldset[disabled] html.theme--documenter-dark .button,html.theme--documenter-dark fieldset[disabled] .button{cursor:not-allowed}html.theme--documenter-dark .tabs,html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-ellipsis,html.theme--documenter-dark .breadcrumb,html.theme--documenter-dark .file,html.theme--documenter-dark .button,.is-unselectable{-webkit-touch-callout:none;-webkit-user-select:none;-moz-user-select:none;-ms-user-select:none;user-select:none}html.theme--documenter-dark .navbar-link:not(.is-arrowless)::after,html.theme--documenter-dark .select:not(.is-multiple):not(.is-loading)::after{border:3px solid rgba(0,0,0,0);border-radius:2px;border-right:0;border-top:0;content:" ";display:block;height:0.625em;margin-top:-0.4375em;pointer-events:none;position:absolute;top:50%;transform:rotate(-45deg);transform-origin:center;width:0.625em}html.theme--documenter-dark .admonition:not(:last-child),html.theme--documenter-dark .tabs:not(:last-child),html.theme--documenter-dark .pagination:not(:last-child),html.theme--documenter-dark .message:not(:last-child),html.theme--documenter-dark .level:not(:last-child),html.theme--documenter-dark .breadcrumb:not(:last-child),html.theme--documenter-dark .block:not(:last-child),html.theme--documenter-dark .title:not(:last-child),html.theme--documenter-dark .subtitle:not(:last-child),html.theme--documenter-dark .table-container:not(:last-child),html.theme--documenter-dark .table:not(:last-child),html.theme--documenter-dark .progress:not(:last-child),html.theme--documenter-dark .notification:not(:last-child),html.theme--documenter-dark .content:not(:last-child),html.theme--documenter-dark .box:not(:last-child){margin-bottom:1.5rem}html.theme--documenter-dark .modal-close,html.theme--documenter-dark .delete{-webkit-touch-callout:none;-webkit-user-select:none;-moz-user-select:none;-ms-user-select:none;user-select:none;-moz-appearance:none;-webkit-appearance:none;background-color:rgba(10,10,10,0.2);border:none;border-radius:9999px;cursor:pointer;pointer-events:auto;display:inline-block;flex-grow:0;flex-shrink:0;font-size:0;height:20px;max-height:20px;max-width:20px;min-height:20px;min-width:20px;outline:none;position:relative;vertical-align:top;width:20px}html.theme--documenter-dark .modal-close::before,html.theme--documenter-dark .delete::before,html.theme--documenter-dark .modal-close::after,html.theme--documenter-dark .delete::after{background-color:#fff;content:"";display:block;left:50%;position:absolute;top:50%;transform:translateX(-50%) translateY(-50%) rotate(45deg);transform-origin:center center}html.theme--documenter-dark .modal-close::before,html.theme--documenter-dark .delete::before{height:2px;width:50%}html.theme--documenter-dark .modal-close::after,html.theme--documenter-dark .delete::after{height:50%;width:2px}html.theme--documenter-dark .modal-close:hover,html.theme--documenter-dark .delete:hover,html.theme--documenter-dark .modal-close:focus,html.theme--documenter-dark .delete:focus{background-color:rgba(10,10,10,0.3)}html.theme--documenter-dark .modal-close:active,html.theme--documenter-dark .delete:active{background-color:rgba(10,10,10,0.4)}html.theme--documenter-dark .is-small.modal-close,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.modal-close,html.theme--documenter-dark .is-small.delete,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.delete{height:16px;max-height:16px;max-width:16px;min-height:16px;min-width:16px;width:16px}html.theme--documenter-dark .is-medium.modal-close,html.theme--documenter-dark .is-medium.delete{height:24px;max-height:24px;max-width:24px;min-height:24px;min-width:24px;width:24px}html.theme--documenter-dark .is-large.modal-close,html.theme--documenter-dark .is-large.delete{height:32px;max-height:32px;max-width:32px;min-height:32px;min-width:32px;width:32px}html.theme--documenter-dark .control.is-loading::after,html.theme--documenter-dark .select.is-loading::after,html.theme--documenter-dark .loader,html.theme--documenter-dark .button.is-loading::after{animation:spinAround 500ms infinite linear;border:2px solid #dbdee0;border-radius:9999px;border-right-color:transparent;border-top-color:transparent;content:"";display:block;height:1em;position:relative;width:1em}html.theme--documenter-dark .hero-video,html.theme--documenter-dark .modal-background,html.theme--documenter-dark .modal,html.theme--documenter-dark .image.is-square img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square img,html.theme--documenter-dark .image.is-square .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square .has-ratio,html.theme--documenter-dark .image.is-1by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1 img,html.theme--documenter-dark .image.is-1by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1 .has-ratio,html.theme--documenter-dark .image.is-5by4 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4 img,html.theme--documenter-dark .image.is-5by4 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4 .has-ratio,html.theme--documenter-dark .image.is-4by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3 img,html.theme--documenter-dark .image.is-4by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3 .has-ratio,html.theme--documenter-dark .image.is-3by2 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2 img,html.theme--documenter-dark .image.is-3by2 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2 .has-ratio,html.theme--documenter-dark .image.is-5by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3 img,html.theme--documenter-dark .image.is-5by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3 .has-ratio,html.theme--documenter-dark .image.is-16by9 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9 img,html.theme--documenter-dark .image.is-16by9 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9 .has-ratio,html.theme--documenter-dark .image.is-2by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1 img,html.theme--documenter-dark .image.is-2by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1 .has-ratio,html.theme--documenter-dark .image.is-3by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1 img,html.theme--documenter-dark .image.is-3by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1 .has-ratio,html.theme--documenter-dark .image.is-4by5 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5 img,html.theme--documenter-dark .image.is-4by5 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5 .has-ratio,html.theme--documenter-dark .image.is-3by4 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4 img,html.theme--documenter-dark .image.is-3by4 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4 .has-ratio,html.theme--documenter-dark .image.is-2by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3 img,html.theme--documenter-dark .image.is-2by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3 .has-ratio,html.theme--documenter-dark .image.is-3by5 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5 img,html.theme--documenter-dark .image.is-3by5 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5 .has-ratio,html.theme--documenter-dark .image.is-9by16 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16 img,html.theme--documenter-dark .image.is-9by16 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16 .has-ratio,html.theme--documenter-dark .image.is-1by2 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2 img,html.theme--documenter-dark .image.is-1by2 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2 .has-ratio,html.theme--documenter-dark .image.is-1by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3 img,html.theme--documenter-dark .image.is-1by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3 .has-ratio,.is-overlay{bottom:0;left:0;position:absolute;right:0;top:0}html.theme--documenter-dark .navbar-burger{-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;color:currentColor;font-family:inherit;font-size:1em;margin:0;padding:0}/*! minireset.css v0.0.6 | MIT License | github.com/jgthms/minireset.css */html,body,p,ol,ul,li,dl,dt,dd,blockquote,figure,fieldset,legend,textarea,pre,iframe,hr,h1,h2,h3,h4,h5,h6{margin:0;padding:0}h1,h2,h3,h4,h5,h6{font-size:100%;font-weight:normal}ul{list-style:none}button,input,select,textarea{margin:0}html{box-sizing:border-box}*,*::before,*::after{box-sizing:inherit}img,video{height:auto;max-width:100%}iframe{border:0}table{border-collapse:collapse;border-spacing:0}td,th{padding:0}td:not([align]),th:not([align]){text-align:inherit}.has-text-white{color:#fff !important}a.has-text-white:hover,a.has-text-white:focus{color:#e6e6e6 !important}.has-background-white{background-color:#fff !important}.has-text-black{color:#0a0a0a !important}a.has-text-black:hover,a.has-text-black:focus{color:#000 !important}.has-background-black{background-color:#0a0a0a !important}.has-text-light{color:#ecf0f1 !important}a.has-text-light:hover,a.has-text-light:focus{color:#cfd9db !important}.has-background-light{background-color:#ecf0f1 !important}.has-text-dark{color:#282f2f !important}a.has-text-dark:hover,a.has-text-dark:focus{color:#111414 !important}.has-background-dark{background-color:#282f2f !important}.has-text-primary{color:#375a7f !important}a.has-text-primary:hover,a.has-text-primary:focus{color:#28415b !important}.has-background-primary{background-color:#375a7f !important}.has-text-primary-light{color:#f1f5f9 !important}a.has-text-primary-light:hover,a.has-text-primary-light:focus{color:#cddbe9 !important}.has-background-primary-light{background-color:#f1f5f9 !important}.has-text-primary-dark{color:#4d7eb2 !important}a.has-text-primary-dark:hover,a.has-text-primary-dark:focus{color:#7198c1 !important}.has-background-primary-dark{background-color:#4d7eb2 !important}.has-text-link{color:#1abc9c !important}a.has-text-link:hover,a.has-text-link:focus{color:#148f77 !important}.has-background-link{background-color:#1abc9c !important}.has-text-link-light{color:#edfdf9 !important}a.has-text-link-light:hover,a.has-text-link-light:focus{color:#c0f6ec !important}.has-background-link-light{background-color:#edfdf9 !important}.has-text-link-dark{color:#15987e !important}a.has-text-link-dark:hover,a.has-text-link-dark:focus{color:#1bc5a4 !important}.has-background-link-dark{background-color:#15987e !important}.has-text-info{color:#024c7d !important}a.has-text-info:hover,a.has-text-info:focus{color:#012d4b !important}.has-background-info{background-color:#024c7d !important}.has-text-info-light{color:#ebf7ff !important}a.has-text-info-light:hover,a.has-text-info-light:focus{color:#b9e2fe !important}.has-background-info-light{background-color:#ebf7ff !important}.has-text-info-dark{color:#0e9dfb !important}a.has-text-info-dark:hover,a.has-text-info-dark:focus{color:#40b1fc !important}.has-background-info-dark{background-color:#0e9dfb !important}.has-text-success{color:#008438 !important}a.has-text-success:hover,a.has-text-success:focus{color:#005122 !important}.has-background-success{background-color:#008438 !important}.has-text-success-light{color:#ebfff3 !important}a.has-text-success-light:hover,a.has-text-success-light:focus{color:#b8ffd6 !important}.has-background-success-light{background-color:#ebfff3 !important}.has-text-success-dark{color:#00eb64 !important}a.has-text-success-dark:hover,a.has-text-success-dark:focus{color:#1fff7e !important}.has-background-success-dark{background-color:#00eb64 !important}.has-text-warning{color:#ad8100 !important}a.has-text-warning:hover,a.has-text-warning:focus{color:#7a5b00 !important}.has-background-warning{background-color:#ad8100 !important}.has-text-warning-light{color:#fffaeb !important}a.has-text-warning-light:hover,a.has-text-warning-light:focus{color:#ffedb8 !important}.has-background-warning-light{background-color:#fffaeb !important}.has-text-warning-dark{color:#d19c00 !important}a.has-text-warning-dark:hover,a.has-text-warning-dark:focus{color:#ffbf05 !important}.has-background-warning-dark{background-color:#d19c00 !important}.has-text-danger{color:#9e1b0d !important}a.has-text-danger:hover,a.has-text-danger:focus{color:#6f1309 !important}.has-background-danger{background-color:#9e1b0d !important}.has-text-danger-light{color:#fdeeec !important}a.has-text-danger-light:hover,a.has-text-danger-light:focus{color:#fac3bd !important}.has-background-danger-light{background-color:#fdeeec !important}.has-text-danger-dark{color:#ec311d !important}a.has-text-danger-dark:hover,a.has-text-danger-dark:focus{color:#f05c4c !important}.has-background-danger-dark{background-color:#ec311d !important}.has-text-black-bis{color:#121212 !important}.has-background-black-bis{background-color:#121212 !important}.has-text-black-ter{color:#242424 !important}.has-background-black-ter{background-color:#242424 !important}.has-text-grey-darker{color:#282f2f !important}.has-background-grey-darker{background-color:#282f2f !important}.has-text-grey-dark{color:#343c3d !important}.has-background-grey-dark{background-color:#343c3d !important}.has-text-grey{color:#5e6d6f !important}.has-background-grey{background-color:#5e6d6f !important}.has-text-grey-light{color:#8c9b9d !important}.has-background-grey-light{background-color:#8c9b9d !important}.has-text-grey-lighter{color:#dbdee0 !important}.has-background-grey-lighter{background-color:#dbdee0 !important}.has-text-white-ter{color:#ecf0f1 !important}.has-background-white-ter{background-color:#ecf0f1 !important}.has-text-white-bis{color:#fafafa !important}.has-background-white-bis{background-color:#fafafa !important}.is-flex-direction-row{flex-direction:row !important}.is-flex-direction-row-reverse{flex-direction:row-reverse !important}.is-flex-direction-column{flex-direction:column !important}.is-flex-direction-column-reverse{flex-direction:column-reverse !important}.is-flex-wrap-nowrap{flex-wrap:nowrap !important}.is-flex-wrap-wrap{flex-wrap:wrap !important}.is-flex-wrap-wrap-reverse{flex-wrap:wrap-reverse !important}.is-justify-content-flex-start{justify-content:flex-start !important}.is-justify-content-flex-end{justify-content:flex-end !important}.is-justify-content-center{justify-content:center !important}.is-justify-content-space-between{justify-content:space-between !important}.is-justify-content-space-around{justify-content:space-around !important}.is-justify-content-space-evenly{justify-content:space-evenly !important}.is-justify-content-start{justify-content:start !important}.is-justify-content-end{justify-content:end !important}.is-justify-content-left{justify-content:left !important}.is-justify-content-right{justify-content:right !important}.is-align-content-flex-start{align-content:flex-start !important}.is-align-content-flex-end{align-content:flex-end !important}.is-align-content-center{align-content:center !important}.is-align-content-space-between{align-content:space-between !important}.is-align-content-space-around{align-content:space-around !important}.is-align-content-space-evenly{align-content:space-evenly !important}.is-align-content-stretch{align-content:stretch !important}.is-align-content-start{align-content:start !important}.is-align-content-end{align-content:end !important}.is-align-content-baseline{align-content:baseline !important}.is-align-items-stretch{align-items:stretch !important}.is-align-items-flex-start{align-items:flex-start !important}.is-align-items-flex-end{align-items:flex-end !important}.is-align-items-center{align-items:center !important}.is-align-items-baseline{align-items:baseline !important}.is-align-items-start{align-items:start !important}.is-align-items-end{align-items:end !important}.is-align-items-self-start{align-items:self-start !important}.is-align-items-self-end{align-items:self-end !important}.is-align-self-auto{align-self:auto !important}.is-align-self-flex-start{align-self:flex-start !important}.is-align-self-flex-end{align-self:flex-end !important}.is-align-self-center{align-self:center !important}.is-align-self-baseline{align-self:baseline !important}.is-align-self-stretch{align-self:stretch !important}.is-flex-grow-0{flex-grow:0 !important}.is-flex-grow-1{flex-grow:1 !important}.is-flex-grow-2{flex-grow:2 !important}.is-flex-grow-3{flex-grow:3 !important}.is-flex-grow-4{flex-grow:4 !important}.is-flex-grow-5{flex-grow:5 !important}.is-flex-shrink-0{flex-shrink:0 !important}.is-flex-shrink-1{flex-shrink:1 !important}.is-flex-shrink-2{flex-shrink:2 !important}.is-flex-shrink-3{flex-shrink:3 !important}.is-flex-shrink-4{flex-shrink:4 !important}.is-flex-shrink-5{flex-shrink:5 !important}.is-clearfix::after{clear:both;content:" ";display:table}.is-pulled-left{float:left !important}.is-pulled-right{float:right !important}.is-radiusless{border-radius:0 !important}.is-shadowless{box-shadow:none !important}.is-clickable{cursor:pointer !important;pointer-events:all !important}.is-clipped{overflow:hidden !important}.is-relative{position:relative !important}.is-marginless{margin:0 !important}.is-paddingless{padding:0 !important}.m-0{margin:0 !important}.mt-0{margin-top:0 !important}.mr-0{margin-right:0 !important}.mb-0{margin-bottom:0 !important}.ml-0{margin-left:0 !important}.mx-0{margin-left:0 !important;margin-right:0 !important}.my-0{margin-top:0 !important;margin-bottom:0 !important}.m-1{margin:.25rem !important}.mt-1{margin-top:.25rem !important}.mr-1{margin-right:.25rem !important}.mb-1{margin-bottom:.25rem !important}.ml-1{margin-left:.25rem !important}.mx-1{margin-left:.25rem !important;margin-right:.25rem !important}.my-1{margin-top:.25rem !important;margin-bottom:.25rem !important}.m-2{margin:.5rem !important}.mt-2{margin-top:.5rem !important}.mr-2{margin-right:.5rem !important}.mb-2{margin-bottom:.5rem !important}.ml-2{margin-left:.5rem !important}.mx-2{margin-left:.5rem !important;margin-right:.5rem !important}.my-2{margin-top:.5rem !important;margin-bottom:.5rem !important}.m-3{margin:.75rem !important}.mt-3{margin-top:.75rem !important}.mr-3{margin-right:.75rem !important}.mb-3{margin-bottom:.75rem !important}.ml-3{margin-left:.75rem !important}.mx-3{margin-left:.75rem !important;margin-right:.75rem !important}.my-3{margin-top:.75rem !important;margin-bottom:.75rem !important}.m-4{margin:1rem !important}.mt-4{margin-top:1rem !important}.mr-4{margin-right:1rem !important}.mb-4{margin-bottom:1rem !important}.ml-4{margin-left:1rem !important}.mx-4{margin-left:1rem !important;margin-right:1rem !important}.my-4{margin-top:1rem !important;margin-bottom:1rem !important}.m-5{margin:1.5rem !important}.mt-5{margin-top:1.5rem !important}.mr-5{margin-right:1.5rem !important}.mb-5{margin-bottom:1.5rem !important}.ml-5{margin-left:1.5rem !important}.mx-5{margin-left:1.5rem !important;margin-right:1.5rem !important}.my-5{margin-top:1.5rem !important;margin-bottom:1.5rem !important}.m-6{margin:3rem !important}.mt-6{margin-top:3rem !important}.mr-6{margin-right:3rem !important}.mb-6{margin-bottom:3rem !important}.ml-6{margin-left:3rem !important}.mx-6{margin-left:3rem !important;margin-right:3rem !important}.my-6{margin-top:3rem !important;margin-bottom:3rem !important}.m-auto{margin:auto !important}.mt-auto{margin-top:auto !important}.mr-auto{margin-right:auto !important}.mb-auto{margin-bottom:auto !important}.ml-auto{margin-left:auto !important}.mx-auto{margin-left:auto !important;margin-right:auto !important}.my-auto{margin-top:auto !important;margin-bottom:auto !important}.p-0{padding:0 !important}.pt-0{padding-top:0 !important}.pr-0{padding-right:0 !important}.pb-0{padding-bottom:0 !important}.pl-0{padding-left:0 !important}.px-0{padding-left:0 !important;padding-right:0 !important}.py-0{padding-top:0 !important;padding-bottom:0 !important}.p-1{padding:.25rem !important}.pt-1{padding-top:.25rem !important}.pr-1{padding-right:.25rem !important}.pb-1{padding-bottom:.25rem !important}.pl-1{padding-left:.25rem !important}.px-1{padding-left:.25rem !important;padding-right:.25rem !important}.py-1{padding-top:.25rem !important;padding-bottom:.25rem !important}.p-2{padding:.5rem !important}.pt-2{padding-top:.5rem !important}.pr-2{padding-right:.5rem !important}.pb-2{padding-bottom:.5rem !important}.pl-2{padding-left:.5rem !important}.px-2{padding-left:.5rem !important;padding-right:.5rem !important}.py-2{padding-top:.5rem !important;padding-bottom:.5rem !important}.p-3{padding:.75rem !important}.pt-3{padding-top:.75rem !important}.pr-3{padding-right:.75rem !important}.pb-3{padding-bottom:.75rem !important}.pl-3{padding-left:.75rem !important}.px-3{padding-left:.75rem !important;padding-right:.75rem !important}.py-3{padding-top:.75rem !important;padding-bottom:.75rem !important}.p-4{padding:1rem !important}.pt-4{padding-top:1rem !important}.pr-4{padding-right:1rem !important}.pb-4{padding-bottom:1rem !important}.pl-4{padding-left:1rem !important}.px-4{padding-left:1rem !important;padding-right:1rem !important}.py-4{padding-top:1rem !important;padding-bottom:1rem !important}.p-5{padding:1.5rem !important}.pt-5{padding-top:1.5rem !important}.pr-5{padding-right:1.5rem !important}.pb-5{padding-bottom:1.5rem !important}.pl-5{padding-left:1.5rem !important}.px-5{padding-left:1.5rem !important;padding-right:1.5rem !important}.py-5{padding-top:1.5rem !important;padding-bottom:1.5rem !important}.p-6{padding:3rem !important}.pt-6{padding-top:3rem !important}.pr-6{padding-right:3rem !important}.pb-6{padding-bottom:3rem !important}.pl-6{padding-left:3rem !important}.px-6{padding-left:3rem !important;padding-right:3rem !important}.py-6{padding-top:3rem !important;padding-bottom:3rem !important}.p-auto{padding:auto !important}.pt-auto{padding-top:auto !important}.pr-auto{padding-right:auto !important}.pb-auto{padding-bottom:auto !important}.pl-auto{padding-left:auto !important}.px-auto{padding-left:auto !important;padding-right:auto !important}.py-auto{padding-top:auto !important;padding-bottom:auto !important}.is-size-1{font-size:3rem !important}.is-size-2{font-size:2.5rem !important}.is-size-3{font-size:2rem !important}.is-size-4{font-size:1.5rem !important}.is-size-5{font-size:1.25rem !important}.is-size-6{font-size:1rem !important}.is-size-7,html.theme--documenter-dark .docstring>section>a.docs-sourcelink{font-size:.75rem !important}@media screen and (max-width: 768px){.is-size-1-mobile{font-size:3rem !important}.is-size-2-mobile{font-size:2.5rem !important}.is-size-3-mobile{font-size:2rem !important}.is-size-4-mobile{font-size:1.5rem !important}.is-size-5-mobile{font-size:1.25rem !important}.is-size-6-mobile{font-size:1rem !important}.is-size-7-mobile{font-size:.75rem !important}}@media screen and (min-width: 769px),print{.is-size-1-tablet{font-size:3rem !important}.is-size-2-tablet{font-size:2.5rem !important}.is-size-3-tablet{font-size:2rem !important}.is-size-4-tablet{font-size:1.5rem !important}.is-size-5-tablet{font-size:1.25rem !important}.is-size-6-tablet{font-size:1rem !important}.is-size-7-tablet{font-size:.75rem !important}}@media screen and (max-width: 1055px){.is-size-1-touch{font-size:3rem !important}.is-size-2-touch{font-size:2.5rem !important}.is-size-3-touch{font-size:2rem !important}.is-size-4-touch{font-size:1.5rem !important}.is-size-5-touch{font-size:1.25rem !important}.is-size-6-touch{font-size:1rem !important}.is-size-7-touch{font-size:.75rem !important}}@media screen and (min-width: 1056px){.is-size-1-desktop{font-size:3rem !important}.is-size-2-desktop{font-size:2.5rem !important}.is-size-3-desktop{font-size:2rem !important}.is-size-4-desktop{font-size:1.5rem !important}.is-size-5-desktop{font-size:1.25rem !important}.is-size-6-desktop{font-size:1rem !important}.is-size-7-desktop{font-size:.75rem !important}}@media screen and (min-width: 1216px){.is-size-1-widescreen{font-size:3rem !important}.is-size-2-widescreen{font-size:2.5rem !important}.is-size-3-widescreen{font-size:2rem !important}.is-size-4-widescreen{font-size:1.5rem !important}.is-size-5-widescreen{font-size:1.25rem !important}.is-size-6-widescreen{font-size:1rem !important}.is-size-7-widescreen{font-size:.75rem !important}}@media screen and (min-width: 1408px){.is-size-1-fullhd{font-size:3rem !important}.is-size-2-fullhd{font-size:2.5rem !important}.is-size-3-fullhd{font-size:2rem !important}.is-size-4-fullhd{font-size:1.5rem !important}.is-size-5-fullhd{font-size:1.25rem !important}.is-size-6-fullhd{font-size:1rem !important}.is-size-7-fullhd{font-size:.75rem !important}}.has-text-centered{text-align:center !important}.has-text-justified{text-align:justify !important}.has-text-left{text-align:left !important}.has-text-right{text-align:right !important}@media screen and (max-width: 768px){.has-text-centered-mobile{text-align:center !important}}@media screen and (min-width: 769px),print{.has-text-centered-tablet{text-align:center !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-centered-tablet-only{text-align:center !important}}@media screen and (max-width: 1055px){.has-text-centered-touch{text-align:center !important}}@media screen and (min-width: 1056px){.has-text-centered-desktop{text-align:center !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-centered-desktop-only{text-align:center !important}}@media screen and (min-width: 1216px){.has-text-centered-widescreen{text-align:center !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-centered-widescreen-only{text-align:center !important}}@media screen and (min-width: 1408px){.has-text-centered-fullhd{text-align:center !important}}@media screen and (max-width: 768px){.has-text-justified-mobile{text-align:justify !important}}@media screen and (min-width: 769px),print{.has-text-justified-tablet{text-align:justify !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-justified-tablet-only{text-align:justify !important}}@media screen and (max-width: 1055px){.has-text-justified-touch{text-align:justify !important}}@media screen and (min-width: 1056px){.has-text-justified-desktop{text-align:justify !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-justified-desktop-only{text-align:justify !important}}@media screen and (min-width: 1216px){.has-text-justified-widescreen{text-align:justify !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-justified-widescreen-only{text-align:justify !important}}@media screen and (min-width: 1408px){.has-text-justified-fullhd{text-align:justify !important}}@media screen and (max-width: 768px){.has-text-left-mobile{text-align:left !important}}@media screen and (min-width: 769px),print{.has-text-left-tablet{text-align:left !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-left-tablet-only{text-align:left !important}}@media screen and (max-width: 1055px){.has-text-left-touch{text-align:left !important}}@media screen and (min-width: 1056px){.has-text-left-desktop{text-align:left !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-left-desktop-only{text-align:left !important}}@media screen and (min-width: 1216px){.has-text-left-widescreen{text-align:left !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-left-widescreen-only{text-align:left !important}}@media screen and (min-width: 1408px){.has-text-left-fullhd{text-align:left !important}}@media screen and (max-width: 768px){.has-text-right-mobile{text-align:right !important}}@media screen and (min-width: 769px),print{.has-text-right-tablet{text-align:right !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-right-tablet-only{text-align:right !important}}@media screen and (max-width: 1055px){.has-text-right-touch{text-align:right !important}}@media screen and (min-width: 1056px){.has-text-right-desktop{text-align:right !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-right-desktop-only{text-align:right !important}}@media screen and (min-width: 1216px){.has-text-right-widescreen{text-align:right !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-right-widescreen-only{text-align:right !important}}@media screen and (min-width: 1408px){.has-text-right-fullhd{text-align:right !important}}.is-capitalized{text-transform:capitalize !important}.is-lowercase{text-transform:lowercase !important}.is-uppercase{text-transform:uppercase !important}.is-italic{font-style:italic !important}.is-underlined{text-decoration:underline !important}.has-text-weight-light{font-weight:300 !important}.has-text-weight-normal{font-weight:400 !important}.has-text-weight-medium{font-weight:500 !important}.has-text-weight-semibold{font-weight:600 !important}.has-text-weight-bold{font-weight:700 !important}.is-family-primary{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-secondary{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-sans-serif{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-monospace{font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace !important}.is-family-code{font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace !important}.is-block{display:block !important}@media screen and (max-width: 768px){.is-block-mobile{display:block !important}}@media screen and (min-width: 769px),print{.is-block-tablet{display:block !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-block-tablet-only{display:block !important}}@media screen and (max-width: 1055px){.is-block-touch{display:block !important}}@media screen and (min-width: 1056px){.is-block-desktop{display:block !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-block-desktop-only{display:block !important}}@media screen and (min-width: 1216px){.is-block-widescreen{display:block !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-block-widescreen-only{display:block !important}}@media screen and (min-width: 1408px){.is-block-fullhd{display:block !important}}.is-flex{display:flex !important}@media screen and (max-width: 768px){.is-flex-mobile{display:flex !important}}@media screen and (min-width: 769px),print{.is-flex-tablet{display:flex !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-flex-tablet-only{display:flex !important}}@media screen and (max-width: 1055px){.is-flex-touch{display:flex !important}}@media screen and (min-width: 1056px){.is-flex-desktop{display:flex !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-flex-desktop-only{display:flex !important}}@media screen and (min-width: 1216px){.is-flex-widescreen{display:flex !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-flex-widescreen-only{display:flex !important}}@media screen and (min-width: 1408px){.is-flex-fullhd{display:flex !important}}.is-inline{display:inline !important}@media screen and (max-width: 768px){.is-inline-mobile{display:inline !important}}@media screen and (min-width: 769px),print{.is-inline-tablet{display:inline !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-tablet-only{display:inline !important}}@media screen and (max-width: 1055px){.is-inline-touch{display:inline !important}}@media screen and (min-width: 1056px){.is-inline-desktop{display:inline !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-desktop-only{display:inline !important}}@media screen and (min-width: 1216px){.is-inline-widescreen{display:inline !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-widescreen-only{display:inline !important}}@media screen and (min-width: 1408px){.is-inline-fullhd{display:inline !important}}.is-inline-block{display:inline-block !important}@media screen and (max-width: 768px){.is-inline-block-mobile{display:inline-block !important}}@media screen and (min-width: 769px),print{.is-inline-block-tablet{display:inline-block !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-block-tablet-only{display:inline-block !important}}@media screen and (max-width: 1055px){.is-inline-block-touch{display:inline-block !important}}@media screen and (min-width: 1056px){.is-inline-block-desktop{display:inline-block !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-block-desktop-only{display:inline-block !important}}@media screen and (min-width: 1216px){.is-inline-block-widescreen{display:inline-block !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-block-widescreen-only{display:inline-block !important}}@media screen and (min-width: 1408px){.is-inline-block-fullhd{display:inline-block !important}}.is-inline-flex{display:inline-flex !important}@media screen and (max-width: 768px){.is-inline-flex-mobile{display:inline-flex !important}}@media screen and (min-width: 769px),print{.is-inline-flex-tablet{display:inline-flex !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-flex-tablet-only{display:inline-flex !important}}@media screen and (max-width: 1055px){.is-inline-flex-touch{display:inline-flex !important}}@media screen and (min-width: 1056px){.is-inline-flex-desktop{display:inline-flex !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-flex-desktop-only{display:inline-flex !important}}@media screen and (min-width: 1216px){.is-inline-flex-widescreen{display:inline-flex !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-flex-widescreen-only{display:inline-flex !important}}@media screen and (min-width: 1408px){.is-inline-flex-fullhd{display:inline-flex !important}}.is-hidden{display:none !important}.is-sr-only{border:none !important;clip:rect(0, 0, 0, 0) !important;height:0.01em !important;overflow:hidden !important;padding:0 !important;position:absolute !important;white-space:nowrap !important;width:0.01em !important}@media screen and (max-width: 768px){.is-hidden-mobile{display:none !important}}@media screen and (min-width: 769px),print{.is-hidden-tablet{display:none !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-hidden-tablet-only{display:none !important}}@media screen and (max-width: 1055px){.is-hidden-touch{display:none !important}}@media screen and (min-width: 1056px){.is-hidden-desktop{display:none !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-hidden-desktop-only{display:none !important}}@media screen and (min-width: 1216px){.is-hidden-widescreen{display:none !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-hidden-widescreen-only{display:none !important}}@media screen and (min-width: 1408px){.is-hidden-fullhd{display:none !important}}.is-invisible{visibility:hidden !important}@media screen and (max-width: 768px){.is-invisible-mobile{visibility:hidden !important}}@media screen and (min-width: 769px),print{.is-invisible-tablet{visibility:hidden !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-invisible-tablet-only{visibility:hidden !important}}@media screen and (max-width: 1055px){.is-invisible-touch{visibility:hidden !important}}@media screen and (min-width: 1056px){.is-invisible-desktop{visibility:hidden !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-invisible-desktop-only{visibility:hidden !important}}@media screen and (min-width: 1216px){.is-invisible-widescreen{visibility:hidden !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-invisible-widescreen-only{visibility:hidden !important}}@media screen and (min-width: 1408px){.is-invisible-fullhd{visibility:hidden !important}}html.theme--documenter-dark{/*!
+ Theme: a11y-dark
+ Author: @ericwbailey
+ Maintainer: @ericwbailey
+
+ Based on the Tomorrow Night Eighties theme: https://github.com/isagalaev/highlight.js/blob/master/src/styles/tomorrow-night-eighties.css
+*/}html.theme--documenter-dark html{background-color:#1f2424;font-size:16px;-moz-osx-font-smoothing:grayscale;-webkit-font-smoothing:antialiased;min-width:300px;overflow-x:auto;overflow-y:scroll;text-rendering:optimizeLegibility;text-size-adjust:100%}html.theme--documenter-dark article,html.theme--documenter-dark aside,html.theme--documenter-dark figure,html.theme--documenter-dark footer,html.theme--documenter-dark header,html.theme--documenter-dark hgroup,html.theme--documenter-dark section{display:block}html.theme--documenter-dark body,html.theme--documenter-dark button,html.theme--documenter-dark input,html.theme--documenter-dark optgroup,html.theme--documenter-dark select,html.theme--documenter-dark textarea{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif}html.theme--documenter-dark code,html.theme--documenter-dark pre{-moz-osx-font-smoothing:auto;-webkit-font-smoothing:auto;font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace}html.theme--documenter-dark body{color:#fff;font-size:1em;font-weight:400;line-height:1.5}html.theme--documenter-dark a{color:#1abc9c;cursor:pointer;text-decoration:none}html.theme--documenter-dark a strong{color:currentColor}html.theme--documenter-dark a:hover{color:#1dd2af}html.theme--documenter-dark code{background-color:rgba(255,255,255,0.05);color:#ececec;font-size:.875em;font-weight:normal;padding:.1em}html.theme--documenter-dark hr{background-color:#282f2f;border:none;display:block;height:2px;margin:1.5rem 0}html.theme--documenter-dark img{height:auto;max-width:100%}html.theme--documenter-dark input[type="checkbox"],html.theme--documenter-dark input[type="radio"]{vertical-align:baseline}html.theme--documenter-dark small{font-size:.875em}html.theme--documenter-dark span{font-style:inherit;font-weight:inherit}html.theme--documenter-dark strong{color:#f2f2f2;font-weight:700}html.theme--documenter-dark fieldset{border:none}html.theme--documenter-dark pre{-webkit-overflow-scrolling:touch;background-color:#282f2f;color:#fff;font-size:.875em;overflow-x:auto;padding:1.25rem 1.5rem;white-space:pre;word-wrap:normal}html.theme--documenter-dark pre code{background-color:transparent;color:currentColor;font-size:1em;padding:0}html.theme--documenter-dark table td,html.theme--documenter-dark table th{vertical-align:top}html.theme--documenter-dark table td:not([align]),html.theme--documenter-dark table th:not([align]){text-align:inherit}html.theme--documenter-dark table th{color:#f2f2f2}html.theme--documenter-dark .box{background-color:#343c3d;border-radius:8px;box-shadow:none;color:#fff;display:block;padding:1.25rem}html.theme--documenter-dark a.box:hover,html.theme--documenter-dark a.box:focus{box-shadow:0 0.5em 1em -0.125em rgba(10,10,10,0.1),0 0 0 1px #1abc9c}html.theme--documenter-dark a.box:active{box-shadow:inset 0 1px 2px rgba(10,10,10,0.2),0 0 0 1px #1abc9c}html.theme--documenter-dark .button{background-color:#282f2f;border-color:#4c5759;border-width:1px;color:#375a7f;cursor:pointer;justify-content:center;padding-bottom:calc(0.5em - 1px);padding-left:1em;padding-right:1em;padding-top:calc(0.5em - 1px);text-align:center;white-space:nowrap}html.theme--documenter-dark .button strong{color:inherit}html.theme--documenter-dark .button .icon,html.theme--documenter-dark .button .icon.is-small,html.theme--documenter-dark .button #documenter .docs-sidebar form.docs-search>input.icon,html.theme--documenter-dark #documenter .docs-sidebar .button form.docs-search>input.icon,html.theme--documenter-dark .button .icon.is-medium,html.theme--documenter-dark .button .icon.is-large{height:1.5em;width:1.5em}html.theme--documenter-dark .button .icon:first-child:not(:last-child){margin-left:calc(-0.5em - 1px);margin-right:.25em}html.theme--documenter-dark .button .icon:last-child:not(:first-child){margin-left:.25em;margin-right:calc(-0.5em - 1px)}html.theme--documenter-dark .button .icon:first-child:last-child{margin-left:calc(-0.5em - 1px);margin-right:calc(-0.5em - 1px)}html.theme--documenter-dark .button:hover,html.theme--documenter-dark .button.is-hovered{border-color:#8c9b9d;color:#f2f2f2}html.theme--documenter-dark .button:focus,html.theme--documenter-dark .button.is-focused{border-color:#8c9b9d;color:#17a689}html.theme--documenter-dark .button:focus:not(:active),html.theme--documenter-dark .button.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .button:active,html.theme--documenter-dark .button.is-active{border-color:#343c3d;color:#f2f2f2}html.theme--documenter-dark .button.is-text{background-color:transparent;border-color:transparent;color:#fff;text-decoration:underline}html.theme--documenter-dark .button.is-text:hover,html.theme--documenter-dark .button.is-text.is-hovered,html.theme--documenter-dark .button.is-text:focus,html.theme--documenter-dark .button.is-text.is-focused{background-color:#282f2f;color:#f2f2f2}html.theme--documenter-dark .button.is-text:active,html.theme--documenter-dark .button.is-text.is-active{background-color:#1d2122;color:#f2f2f2}html.theme--documenter-dark .button.is-text[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-text{background-color:transparent;border-color:transparent;box-shadow:none}html.theme--documenter-dark .button.is-ghost{background:none;border-color:rgba(0,0,0,0);color:#1abc9c;text-decoration:none}html.theme--documenter-dark .button.is-ghost:hover,html.theme--documenter-dark .button.is-ghost.is-hovered{color:#1abc9c;text-decoration:underline}html.theme--documenter-dark .button.is-white{background-color:#fff;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .button.is-white:hover,html.theme--documenter-dark .button.is-white.is-hovered{background-color:#f9f9f9;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .button.is-white:focus,html.theme--documenter-dark .button.is-white.is-focused{border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .button.is-white:focus:not(:active),html.theme--documenter-dark .button.is-white.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}html.theme--documenter-dark .button.is-white:active,html.theme--documenter-dark .button.is-white.is-active{background-color:#f2f2f2;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .button.is-white[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-white{background-color:#fff;border-color:#fff;box-shadow:none}html.theme--documenter-dark .button.is-white.is-inverted{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .button.is-white.is-inverted:hover,html.theme--documenter-dark .button.is-white.is-inverted.is-hovered{background-color:#000}html.theme--documenter-dark .button.is-white.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-white.is-inverted{background-color:#0a0a0a;border-color:transparent;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-white.is-loading::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}html.theme--documenter-dark .button.is-white.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-white.is-outlined:hover,html.theme--documenter-dark .button.is-white.is-outlined.is-hovered,html.theme--documenter-dark .button.is-white.is-outlined:focus,html.theme--documenter-dark .button.is-white.is-outlined.is-focused{background-color:#fff;border-color:#fff;color:#0a0a0a}html.theme--documenter-dark .button.is-white.is-outlined.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-white.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-white.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-white.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-white.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}html.theme--documenter-dark .button.is-white.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-white.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-white.is-inverted.is-outlined{background-color:transparent;border-color:#0a0a0a;color:#0a0a0a}html.theme--documenter-dark .button.is-white.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-focused{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-white.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-white.is-inverted.is-outlined{background-color:transparent;border-color:#0a0a0a;box-shadow:none;color:#0a0a0a}html.theme--documenter-dark .button.is-black{background-color:#0a0a0a;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-black:hover,html.theme--documenter-dark .button.is-black.is-hovered{background-color:#040404;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-black:focus,html.theme--documenter-dark .button.is-black.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-black:focus:not(:active),html.theme--documenter-dark .button.is-black.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}html.theme--documenter-dark .button.is-black:active,html.theme--documenter-dark .button.is-black.is-active{background-color:#000;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-black[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-black{background-color:#0a0a0a;border-color:#0a0a0a;box-shadow:none}html.theme--documenter-dark .button.is-black.is-inverted{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-inverted:hover,html.theme--documenter-dark .button.is-black.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-black.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-black.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-black.is-outlined{background-color:transparent;border-color:#0a0a0a;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-outlined:hover,html.theme--documenter-dark .button.is-black.is-outlined.is-hovered,html.theme--documenter-dark .button.is-black.is-outlined:focus,html.theme--documenter-dark .button.is-black.is-outlined.is-focused{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}html.theme--documenter-dark .button.is-black.is-outlined.is-loading::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}html.theme--documenter-dark .button.is-black.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-black.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-black.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-black.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-black.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-black.is-outlined{background-color:transparent;border-color:#0a0a0a;box-shadow:none;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-black.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-focused{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}html.theme--documenter-dark .button.is-black.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-black.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-light{background-color:#ecf0f1;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light:hover,html.theme--documenter-dark .button.is-light.is-hovered{background-color:#e5eaec;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light:focus,html.theme--documenter-dark .button.is-light.is-focused{border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light:focus:not(:active),html.theme--documenter-dark .button.is-light.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(236,240,241,0.25)}html.theme--documenter-dark .button.is-light:active,html.theme--documenter-dark .button.is-light.is-active{background-color:#dde4e6;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-light{background-color:#ecf0f1;border-color:#ecf0f1;box-shadow:none}html.theme--documenter-dark .button.is-light.is-inverted{background-color:rgba(0,0,0,0.7);color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-inverted:hover,html.theme--documenter-dark .button.is-light.is-inverted.is-hovered{background-color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-light.is-inverted{background-color:rgba(0,0,0,0.7);border-color:transparent;box-shadow:none;color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-loading::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}html.theme--documenter-dark .button.is-light.is-outlined{background-color:transparent;border-color:#ecf0f1;color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-outlined:hover,html.theme--documenter-dark .button.is-light.is-outlined.is-hovered,html.theme--documenter-dark .button.is-light.is-outlined:focus,html.theme--documenter-dark .button.is-light.is-outlined.is-focused{background-color:#ecf0f1;border-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light.is-outlined.is-loading::after{border-color:transparent transparent #ecf0f1 #ecf0f1 !important}html.theme--documenter-dark .button.is-light.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-light.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-light.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-light.is-outlined.is-loading.is-focused::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}html.theme--documenter-dark .button.is-light.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-light.is-outlined{background-color:transparent;border-color:#ecf0f1;box-shadow:none;color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-focused{background-color:rgba(0,0,0,0.7);color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #ecf0f1 #ecf0f1 !important}html.theme--documenter-dark .button.is-light.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-light.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);box-shadow:none;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-dark,html.theme--documenter-dark .content kbd.button{background-color:#282f2f;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-dark:hover,html.theme--documenter-dark .content kbd.button:hover,html.theme--documenter-dark .button.is-dark.is-hovered,html.theme--documenter-dark .content kbd.button.is-hovered{background-color:#232829;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-dark:focus,html.theme--documenter-dark .content kbd.button:focus,html.theme--documenter-dark .button.is-dark.is-focused,html.theme--documenter-dark .content kbd.button.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-dark:focus:not(:active),html.theme--documenter-dark .content kbd.button:focus:not(:active),html.theme--documenter-dark .button.is-dark.is-focused:not(:active),html.theme--documenter-dark .content kbd.button.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(40,47,47,0.25)}html.theme--documenter-dark .button.is-dark:active,html.theme--documenter-dark .content kbd.button:active,html.theme--documenter-dark .button.is-dark.is-active,html.theme--documenter-dark .content kbd.button.is-active{background-color:#1d2122;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-dark[disabled],html.theme--documenter-dark .content kbd.button[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-dark,fieldset[disabled] html.theme--documenter-dark .content kbd.button{background-color:#282f2f;border-color:#282f2f;box-shadow:none}html.theme--documenter-dark .button.is-dark.is-inverted,html.theme--documenter-dark .content kbd.button.is-inverted{background-color:#fff;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-inverted:hover,html.theme--documenter-dark .content kbd.button.is-inverted:hover,html.theme--documenter-dark .button.is-dark.is-inverted.is-hovered,html.theme--documenter-dark .content kbd.button.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-dark.is-inverted[disabled],html.theme--documenter-dark .content kbd.button.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-dark.is-inverted,fieldset[disabled] html.theme--documenter-dark .content kbd.button.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-loading::after,html.theme--documenter-dark .content kbd.button.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-dark.is-outlined,html.theme--documenter-dark .content kbd.button.is-outlined{background-color:transparent;border-color:#282f2f;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-outlined:hover,html.theme--documenter-dark .content kbd.button.is-outlined:hover,html.theme--documenter-dark .button.is-dark.is-outlined.is-hovered,html.theme--documenter-dark .content kbd.button.is-outlined.is-hovered,html.theme--documenter-dark .button.is-dark.is-outlined:focus,html.theme--documenter-dark .content kbd.button.is-outlined:focus,html.theme--documenter-dark .button.is-dark.is-outlined.is-focused,html.theme--documenter-dark .content kbd.button.is-outlined.is-focused{background-color:#282f2f;border-color:#282f2f;color:#fff}html.theme--documenter-dark .button.is-dark.is-outlined.is-loading::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading::after{border-color:transparent transparent #282f2f #282f2f !important}html.theme--documenter-dark .button.is-dark.is-outlined.is-loading:hover::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-dark.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-dark.is-outlined.is-loading:focus::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-dark.is-outlined.is-loading.is-focused::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-dark.is-outlined[disabled],html.theme--documenter-dark .content kbd.button.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-dark.is-outlined,fieldset[disabled] html.theme--documenter-dark .content kbd.button.is-outlined{background-color:transparent;border-color:#282f2f;box-shadow:none;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined:hover,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined:focus,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-focused,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-focused{background-color:#fff;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-loading.is-focused::after,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #282f2f #282f2f !important}html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined[disabled],html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined,fieldset[disabled] html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-primary,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink{background-color:#375a7f;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-primary:hover,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-hovered.docs-sourcelink{background-color:#335476;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-primary:focus,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:focus,html.theme--documenter-dark .button.is-primary.is-focused,html.theme--documenter-dark .docstring>section>a.button.is-focused.docs-sourcelink{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-primary:focus:not(:active),html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:focus:not(:active),html.theme--documenter-dark .button.is-primary.is-focused:not(:active),html.theme--documenter-dark .docstring>section>a.button.is-focused.docs-sourcelink:not(:active){box-shadow:0 0 0 0.125em rgba(55,90,127,0.25)}html.theme--documenter-dark .button.is-primary:active,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:active,html.theme--documenter-dark .button.is-primary.is-active,html.theme--documenter-dark .docstring>section>a.button.is-active.docs-sourcelink{background-color:#2f4d6d;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-primary[disabled],html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-primary,fieldset[disabled] html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink{background-color:#375a7f;border-color:#375a7f;box-shadow:none}html.theme--documenter-dark .button.is-primary.is-inverted,html.theme--documenter-dark .docstring>section>a.button.is-inverted.docs-sourcelink{background-color:#fff;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-inverted:hover,html.theme--documenter-dark .docstring>section>a.button.is-inverted.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-inverted.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-hovered.docs-sourcelink{background-color:#f2f2f2}html.theme--documenter-dark .button.is-primary.is-inverted[disabled],html.theme--documenter-dark .docstring>section>a.button.is-inverted.docs-sourcelink[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-primary.is-inverted,fieldset[disabled] html.theme--documenter-dark .docstring>section>a.button.is-inverted.docs-sourcelink{background-color:#fff;border-color:transparent;box-shadow:none;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-loading::after,html.theme--documenter-dark .docstring>section>a.button.is-loading.docs-sourcelink::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-primary.is-outlined,html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink{background-color:transparent;border-color:#375a7f;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-outlined:hover,html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-outlined.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-hovered.docs-sourcelink,html.theme--documenter-dark .button.is-primary.is-outlined:focus,html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink:focus,html.theme--documenter-dark .button.is-primary.is-outlined.is-focused,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-focused.docs-sourcelink{background-color:#375a7f;border-color:#375a7f;color:#fff}html.theme--documenter-dark .button.is-primary.is-outlined.is-loading::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.docs-sourcelink::after{border-color:transparent transparent #375a7f #375a7f !important}html.theme--documenter-dark .button.is-primary.is-outlined.is-loading:hover::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.docs-sourcelink:hover::after,html.theme--documenter-dark .button.is-primary.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.is-hovered.docs-sourcelink::after,html.theme--documenter-dark .button.is-primary.is-outlined.is-loading:focus::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.docs-sourcelink:focus::after,html.theme--documenter-dark .button.is-primary.is-outlined.is-loading.is-focused::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.is-focused.docs-sourcelink::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-primary.is-outlined[disabled],html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-primary.is-outlined,fieldset[disabled] html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink{background-color:transparent;border-color:#375a7f;box-shadow:none;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined:hover,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-hovered.docs-sourcelink,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined:focus,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink:focus,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-focused,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-focused.docs-sourcelink{background-color:#fff;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-loading.docs-sourcelink:hover::after,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-loading.is-hovered.docs-sourcelink::after,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-loading.docs-sourcelink:focus::after,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-loading.is-focused::after,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-loading.is-focused.docs-sourcelink::after{border-color:transparent transparent #375a7f #375a7f !important}html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined[disabled],html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined,fieldset[disabled] html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-primary.is-light,html.theme--documenter-dark .docstring>section>a.button.is-light.docs-sourcelink{background-color:#f1f5f9;color:#4d7eb2}html.theme--documenter-dark .button.is-primary.is-light:hover,html.theme--documenter-dark .docstring>section>a.button.is-light.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-light.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-light.is-hovered.docs-sourcelink{background-color:#e8eef5;border-color:transparent;color:#4d7eb2}html.theme--documenter-dark .button.is-primary.is-light:active,html.theme--documenter-dark .docstring>section>a.button.is-light.docs-sourcelink:active,html.theme--documenter-dark .button.is-primary.is-light.is-active,html.theme--documenter-dark .docstring>section>a.button.is-light.is-active.docs-sourcelink{background-color:#dfe8f1;border-color:transparent;color:#4d7eb2}html.theme--documenter-dark .button.is-link{background-color:#1abc9c;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-link:hover,html.theme--documenter-dark .button.is-link.is-hovered{background-color:#18b193;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-link:focus,html.theme--documenter-dark .button.is-link.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-link:focus:not(:active),html.theme--documenter-dark .button.is-link.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .button.is-link:active,html.theme--documenter-dark .button.is-link.is-active{background-color:#17a689;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-link[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-link{background-color:#1abc9c;border-color:#1abc9c;box-shadow:none}html.theme--documenter-dark .button.is-link.is-inverted{background-color:#fff;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-inverted:hover,html.theme--documenter-dark .button.is-link.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-link.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-link.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-link.is-outlined{background-color:transparent;border-color:#1abc9c;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-outlined:hover,html.theme--documenter-dark .button.is-link.is-outlined.is-hovered,html.theme--documenter-dark .button.is-link.is-outlined:focus,html.theme--documenter-dark .button.is-link.is-outlined.is-focused{background-color:#1abc9c;border-color:#1abc9c;color:#fff}html.theme--documenter-dark .button.is-link.is-outlined.is-loading::after{border-color:transparent transparent #1abc9c #1abc9c !important}html.theme--documenter-dark .button.is-link.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-link.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-link.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-link.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-link.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-link.is-outlined{background-color:transparent;border-color:#1abc9c;box-shadow:none;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-link.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-focused{background-color:#fff;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #1abc9c #1abc9c !important}html.theme--documenter-dark .button.is-link.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-link.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-link.is-light{background-color:#edfdf9;color:#15987e}html.theme--documenter-dark .button.is-link.is-light:hover,html.theme--documenter-dark .button.is-link.is-light.is-hovered{background-color:#e2fbf6;border-color:transparent;color:#15987e}html.theme--documenter-dark .button.is-link.is-light:active,html.theme--documenter-dark .button.is-link.is-light.is-active{background-color:#d7f9f3;border-color:transparent;color:#15987e}html.theme--documenter-dark .button.is-info{background-color:#024c7d;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-info:hover,html.theme--documenter-dark .button.is-info.is-hovered{background-color:#024470;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-info:focus,html.theme--documenter-dark .button.is-info.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-info:focus:not(:active),html.theme--documenter-dark .button.is-info.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(2,76,125,0.25)}html.theme--documenter-dark .button.is-info:active,html.theme--documenter-dark .button.is-info.is-active{background-color:#023d64;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-info[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-info{background-color:#024c7d;border-color:#024c7d;box-shadow:none}html.theme--documenter-dark .button.is-info.is-inverted{background-color:#fff;color:#024c7d}html.theme--documenter-dark .button.is-info.is-inverted:hover,html.theme--documenter-dark .button.is-info.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-info.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-info.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#024c7d}html.theme--documenter-dark .button.is-info.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-info.is-outlined{background-color:transparent;border-color:#024c7d;color:#024c7d}html.theme--documenter-dark .button.is-info.is-outlined:hover,html.theme--documenter-dark .button.is-info.is-outlined.is-hovered,html.theme--documenter-dark .button.is-info.is-outlined:focus,html.theme--documenter-dark .button.is-info.is-outlined.is-focused{background-color:#024c7d;border-color:#024c7d;color:#fff}html.theme--documenter-dark .button.is-info.is-outlined.is-loading::after{border-color:transparent transparent #024c7d #024c7d !important}html.theme--documenter-dark .button.is-info.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-info.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-info.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-info.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-info.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-info.is-outlined{background-color:transparent;border-color:#024c7d;box-shadow:none;color:#024c7d}html.theme--documenter-dark .button.is-info.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-info.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-focused{background-color:#fff;color:#024c7d}html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #024c7d #024c7d !important}html.theme--documenter-dark .button.is-info.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-info.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-info.is-light{background-color:#ebf7ff;color:#0e9dfb}html.theme--documenter-dark .button.is-info.is-light:hover,html.theme--documenter-dark .button.is-info.is-light.is-hovered{background-color:#def2fe;border-color:transparent;color:#0e9dfb}html.theme--documenter-dark .button.is-info.is-light:active,html.theme--documenter-dark .button.is-info.is-light.is-active{background-color:#d2edfe;border-color:transparent;color:#0e9dfb}html.theme--documenter-dark .button.is-success{background-color:#008438;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-success:hover,html.theme--documenter-dark .button.is-success.is-hovered{background-color:#073;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-success:focus,html.theme--documenter-dark .button.is-success.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-success:focus:not(:active),html.theme--documenter-dark .button.is-success.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(0,132,56,0.25)}html.theme--documenter-dark .button.is-success:active,html.theme--documenter-dark .button.is-success.is-active{background-color:#006b2d;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-success[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-success{background-color:#008438;border-color:#008438;box-shadow:none}html.theme--documenter-dark .button.is-success.is-inverted{background-color:#fff;color:#008438}html.theme--documenter-dark .button.is-success.is-inverted:hover,html.theme--documenter-dark .button.is-success.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-success.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-success.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#008438}html.theme--documenter-dark .button.is-success.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-success.is-outlined{background-color:transparent;border-color:#008438;color:#008438}html.theme--documenter-dark .button.is-success.is-outlined:hover,html.theme--documenter-dark .button.is-success.is-outlined.is-hovered,html.theme--documenter-dark .button.is-success.is-outlined:focus,html.theme--documenter-dark .button.is-success.is-outlined.is-focused{background-color:#008438;border-color:#008438;color:#fff}html.theme--documenter-dark .button.is-success.is-outlined.is-loading::after{border-color:transparent transparent #008438 #008438 !important}html.theme--documenter-dark .button.is-success.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-success.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-success.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-success.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-success.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-success.is-outlined{background-color:transparent;border-color:#008438;box-shadow:none;color:#008438}html.theme--documenter-dark .button.is-success.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-success.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-focused{background-color:#fff;color:#008438}html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #008438 #008438 !important}html.theme--documenter-dark .button.is-success.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-success.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-success.is-light{background-color:#ebfff3;color:#00eb64}html.theme--documenter-dark .button.is-success.is-light:hover,html.theme--documenter-dark .button.is-success.is-light.is-hovered{background-color:#deffec;border-color:transparent;color:#00eb64}html.theme--documenter-dark .button.is-success.is-light:active,html.theme--documenter-dark .button.is-success.is-light.is-active{background-color:#d1ffe5;border-color:transparent;color:#00eb64}html.theme--documenter-dark .button.is-warning{background-color:#ad8100;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-warning:hover,html.theme--documenter-dark .button.is-warning.is-hovered{background-color:#a07700;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-warning:focus,html.theme--documenter-dark .button.is-warning.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-warning:focus:not(:active),html.theme--documenter-dark .button.is-warning.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(173,129,0,0.25)}html.theme--documenter-dark .button.is-warning:active,html.theme--documenter-dark .button.is-warning.is-active{background-color:#946e00;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-warning[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-warning{background-color:#ad8100;border-color:#ad8100;box-shadow:none}html.theme--documenter-dark .button.is-warning.is-inverted{background-color:#fff;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-inverted:hover,html.theme--documenter-dark .button.is-warning.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-warning.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-warning.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-warning.is-outlined{background-color:transparent;border-color:#ad8100;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-outlined:hover,html.theme--documenter-dark .button.is-warning.is-outlined.is-hovered,html.theme--documenter-dark .button.is-warning.is-outlined:focus,html.theme--documenter-dark .button.is-warning.is-outlined.is-focused{background-color:#ad8100;border-color:#ad8100;color:#fff}html.theme--documenter-dark .button.is-warning.is-outlined.is-loading::after{border-color:transparent transparent #ad8100 #ad8100 !important}html.theme--documenter-dark .button.is-warning.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-warning.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-warning.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-warning.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-warning.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-warning.is-outlined{background-color:transparent;border-color:#ad8100;box-shadow:none;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-focused{background-color:#fff;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #ad8100 #ad8100 !important}html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-warning.is-light{background-color:#fffaeb;color:#d19c00}html.theme--documenter-dark .button.is-warning.is-light:hover,html.theme--documenter-dark .button.is-warning.is-light.is-hovered{background-color:#fff7de;border-color:transparent;color:#d19c00}html.theme--documenter-dark .button.is-warning.is-light:active,html.theme--documenter-dark .button.is-warning.is-light.is-active{background-color:#fff3d1;border-color:transparent;color:#d19c00}html.theme--documenter-dark .button.is-danger{background-color:#9e1b0d;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-danger:hover,html.theme--documenter-dark .button.is-danger.is-hovered{background-color:#92190c;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-danger:focus,html.theme--documenter-dark .button.is-danger.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-danger:focus:not(:active),html.theme--documenter-dark .button.is-danger.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(158,27,13,0.25)}html.theme--documenter-dark .button.is-danger:active,html.theme--documenter-dark .button.is-danger.is-active{background-color:#86170b;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-danger[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-danger{background-color:#9e1b0d;border-color:#9e1b0d;box-shadow:none}html.theme--documenter-dark .button.is-danger.is-inverted{background-color:#fff;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-inverted:hover,html.theme--documenter-dark .button.is-danger.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-danger.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-danger.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-danger.is-outlined{background-color:transparent;border-color:#9e1b0d;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-outlined:hover,html.theme--documenter-dark .button.is-danger.is-outlined.is-hovered,html.theme--documenter-dark .button.is-danger.is-outlined:focus,html.theme--documenter-dark .button.is-danger.is-outlined.is-focused{background-color:#9e1b0d;border-color:#9e1b0d;color:#fff}html.theme--documenter-dark .button.is-danger.is-outlined.is-loading::after{border-color:transparent transparent #9e1b0d #9e1b0d !important}html.theme--documenter-dark .button.is-danger.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-danger.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-danger.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-danger.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-danger.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-danger.is-outlined{background-color:transparent;border-color:#9e1b0d;box-shadow:none;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-focused{background-color:#fff;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #9e1b0d #9e1b0d !important}html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-danger.is-light{background-color:#fdeeec;color:#ec311d}html.theme--documenter-dark .button.is-danger.is-light:hover,html.theme--documenter-dark .button.is-danger.is-light.is-hovered{background-color:#fce3e0;border-color:transparent;color:#ec311d}html.theme--documenter-dark .button.is-danger.is-light:active,html.theme--documenter-dark .button.is-danger.is-light.is-active{background-color:#fcd8d5;border-color:transparent;color:#ec311d}html.theme--documenter-dark .button.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.button{font-size:.75rem}html.theme--documenter-dark .button.is-small:not(.is-rounded),html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.button:not(.is-rounded){border-radius:3px}html.theme--documenter-dark .button.is-normal{font-size:1rem}html.theme--documenter-dark .button.is-medium{font-size:1.25rem}html.theme--documenter-dark .button.is-large{font-size:1.5rem}html.theme--documenter-dark .button[disabled],fieldset[disabled] html.theme--documenter-dark .button{background-color:#8c9b9d;border-color:#5e6d6f;box-shadow:none;opacity:.5}html.theme--documenter-dark .button.is-fullwidth{display:flex;width:100%}html.theme--documenter-dark .button.is-loading{color:transparent !important;pointer-events:none}html.theme--documenter-dark .button.is-loading::after{position:absolute;left:calc(50% - (1em * 0.5));top:calc(50% - (1em * 0.5));position:absolute !important}html.theme--documenter-dark .button.is-static{background-color:#282f2f;border-color:#5e6d6f;color:#dbdee0;box-shadow:none;pointer-events:none}html.theme--documenter-dark .button.is-rounded,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.button{border-radius:9999px;padding-left:calc(1em + 0.25em);padding-right:calc(1em + 0.25em)}html.theme--documenter-dark .buttons{align-items:center;display:flex;flex-wrap:wrap;justify-content:flex-start}html.theme--documenter-dark .buttons .button{margin-bottom:0.5rem}html.theme--documenter-dark .buttons .button:not(:last-child):not(.is-fullwidth){margin-right:.5rem}html.theme--documenter-dark .buttons:last-child{margin-bottom:-0.5rem}html.theme--documenter-dark .buttons:not(:last-child){margin-bottom:1rem}html.theme--documenter-dark .buttons.are-small .button:not(.is-normal):not(.is-medium):not(.is-large){font-size:.75rem}html.theme--documenter-dark .buttons.are-small .button:not(.is-normal):not(.is-medium):not(.is-large):not(.is-rounded){border-radius:3px}html.theme--documenter-dark .buttons.are-medium .button:not(.is-small):not(.is-normal):not(.is-large){font-size:1.25rem}html.theme--documenter-dark .buttons.are-large .button:not(.is-small):not(.is-normal):not(.is-medium){font-size:1.5rem}html.theme--documenter-dark .buttons.has-addons .button:not(:first-child){border-bottom-left-radius:0;border-top-left-radius:0}html.theme--documenter-dark .buttons.has-addons .button:not(:last-child){border-bottom-right-radius:0;border-top-right-radius:0;margin-right:-1px}html.theme--documenter-dark .buttons.has-addons .button:last-child{margin-right:0}html.theme--documenter-dark .buttons.has-addons .button:hover,html.theme--documenter-dark .buttons.has-addons .button.is-hovered{z-index:2}html.theme--documenter-dark .buttons.has-addons .button:focus,html.theme--documenter-dark .buttons.has-addons .button.is-focused,html.theme--documenter-dark .buttons.has-addons .button:active,html.theme--documenter-dark .buttons.has-addons .button.is-active,html.theme--documenter-dark .buttons.has-addons .button.is-selected{z-index:3}html.theme--documenter-dark .buttons.has-addons .button:focus:hover,html.theme--documenter-dark .buttons.has-addons .button.is-focused:hover,html.theme--documenter-dark .buttons.has-addons .button:active:hover,html.theme--documenter-dark .buttons.has-addons .button.is-active:hover,html.theme--documenter-dark .buttons.has-addons .button.is-selected:hover{z-index:4}html.theme--documenter-dark .buttons.has-addons .button.is-expanded{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .buttons.is-centered{justify-content:center}html.theme--documenter-dark .buttons.is-centered:not(.has-addons) .button:not(.is-fullwidth){margin-left:0.25rem;margin-right:0.25rem}html.theme--documenter-dark .buttons.is-right{justify-content:flex-end}html.theme--documenter-dark .buttons.is-right:not(.has-addons) .button:not(.is-fullwidth){margin-left:0.25rem;margin-right:0.25rem}@media screen and (max-width: 768px){html.theme--documenter-dark .button.is-responsive.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-responsive{font-size:.5625rem}html.theme--documenter-dark .button.is-responsive,html.theme--documenter-dark .button.is-responsive.is-normal{font-size:.65625rem}html.theme--documenter-dark .button.is-responsive.is-medium{font-size:.75rem}html.theme--documenter-dark .button.is-responsive.is-large{font-size:1rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .button.is-responsive.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-responsive{font-size:.65625rem}html.theme--documenter-dark .button.is-responsive,html.theme--documenter-dark .button.is-responsive.is-normal{font-size:.75rem}html.theme--documenter-dark .button.is-responsive.is-medium{font-size:1rem}html.theme--documenter-dark .button.is-responsive.is-large{font-size:1.25rem}}html.theme--documenter-dark .container{flex-grow:1;margin:0 auto;position:relative;width:auto}html.theme--documenter-dark .container.is-fluid{max-width:none !important;padding-left:32px;padding-right:32px;width:100%}@media screen and (min-width: 1056px){html.theme--documenter-dark .container{max-width:992px}}@media screen and (max-width: 1215px){html.theme--documenter-dark .container.is-widescreen:not(.is-max-desktop){max-width:1152px}}@media screen and (max-width: 1407px){html.theme--documenter-dark .container.is-fullhd:not(.is-max-desktop):not(.is-max-widescreen){max-width:1344px}}@media screen and (min-width: 1216px){html.theme--documenter-dark .container:not(.is-max-desktop){max-width:1152px}}@media screen and (min-width: 1408px){html.theme--documenter-dark .container:not(.is-max-desktop):not(.is-max-widescreen){max-width:1344px}}html.theme--documenter-dark .content li+li{margin-top:0.25em}html.theme--documenter-dark .content p:not(:last-child),html.theme--documenter-dark .content dl:not(:last-child),html.theme--documenter-dark .content ol:not(:last-child),html.theme--documenter-dark .content ul:not(:last-child),html.theme--documenter-dark .content blockquote:not(:last-child),html.theme--documenter-dark .content pre:not(:last-child),html.theme--documenter-dark .content table:not(:last-child){margin-bottom:1em}html.theme--documenter-dark .content h1,html.theme--documenter-dark .content h2,html.theme--documenter-dark .content h3,html.theme--documenter-dark .content h4,html.theme--documenter-dark .content h5,html.theme--documenter-dark .content h6{color:#f2f2f2;font-weight:600;line-height:1.125}html.theme--documenter-dark .content h1{font-size:2em;margin-bottom:0.5em}html.theme--documenter-dark .content h1:not(:first-child){margin-top:1em}html.theme--documenter-dark .content h2{font-size:1.75em;margin-bottom:0.5714em}html.theme--documenter-dark .content h2:not(:first-child){margin-top:1.1428em}html.theme--documenter-dark .content h3{font-size:1.5em;margin-bottom:0.6666em}html.theme--documenter-dark .content h3:not(:first-child){margin-top:1.3333em}html.theme--documenter-dark .content h4{font-size:1.25em;margin-bottom:0.8em}html.theme--documenter-dark .content h5{font-size:1.125em;margin-bottom:0.8888em}html.theme--documenter-dark .content h6{font-size:1em;margin-bottom:1em}html.theme--documenter-dark .content blockquote{background-color:#282f2f;border-left:5px solid #5e6d6f;padding:1.25em 1.5em}html.theme--documenter-dark .content ol{list-style-position:outside;margin-left:2em;margin-top:1em}html.theme--documenter-dark .content ol:not([type]){list-style-type:decimal}html.theme--documenter-dark .content ol.is-lower-alpha:not([type]){list-style-type:lower-alpha}html.theme--documenter-dark .content ol.is-lower-roman:not([type]){list-style-type:lower-roman}html.theme--documenter-dark .content ol.is-upper-alpha:not([type]){list-style-type:upper-alpha}html.theme--documenter-dark .content ol.is-upper-roman:not([type]){list-style-type:upper-roman}html.theme--documenter-dark .content ul{list-style:disc outside;margin-left:2em;margin-top:1em}html.theme--documenter-dark .content ul ul{list-style-type:circle;margin-top:0.5em}html.theme--documenter-dark .content ul ul ul{list-style-type:square}html.theme--documenter-dark .content dd{margin-left:2em}html.theme--documenter-dark .content figure{margin-left:2em;margin-right:2em;text-align:center}html.theme--documenter-dark .content figure:not(:first-child){margin-top:2em}html.theme--documenter-dark .content figure:not(:last-child){margin-bottom:2em}html.theme--documenter-dark .content figure img{display:inline-block}html.theme--documenter-dark .content figure figcaption{font-style:italic}html.theme--documenter-dark .content pre{-webkit-overflow-scrolling:touch;overflow-x:auto;padding:0;white-space:pre;word-wrap:normal}html.theme--documenter-dark .content sup,html.theme--documenter-dark .content sub{font-size:75%}html.theme--documenter-dark .content table{width:100%}html.theme--documenter-dark .content table td,html.theme--documenter-dark .content table th{border:1px solid #5e6d6f;border-width:0 0 1px;padding:0.5em 0.75em;vertical-align:top}html.theme--documenter-dark .content table th{color:#f2f2f2}html.theme--documenter-dark .content table th:not([align]){text-align:inherit}html.theme--documenter-dark .content table thead td,html.theme--documenter-dark .content table thead th{border-width:0 0 2px;color:#f2f2f2}html.theme--documenter-dark .content table tfoot td,html.theme--documenter-dark .content table tfoot th{border-width:2px 0 0;color:#f2f2f2}html.theme--documenter-dark .content table tbody tr:last-child td,html.theme--documenter-dark .content table tbody tr:last-child th{border-bottom-width:0}html.theme--documenter-dark .content .tabs li+li{margin-top:0}html.theme--documenter-dark .content.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.content{font-size:.75rem}html.theme--documenter-dark .content.is-normal{font-size:1rem}html.theme--documenter-dark .content.is-medium{font-size:1.25rem}html.theme--documenter-dark .content.is-large{font-size:1.5rem}html.theme--documenter-dark .icon{align-items:center;display:inline-flex;justify-content:center;height:1.5rem;width:1.5rem}html.theme--documenter-dark .icon.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.icon{height:1rem;width:1rem}html.theme--documenter-dark .icon.is-medium{height:2rem;width:2rem}html.theme--documenter-dark .icon.is-large{height:3rem;width:3rem}html.theme--documenter-dark .icon-text{align-items:flex-start;color:inherit;display:inline-flex;flex-wrap:wrap;line-height:1.5rem;vertical-align:top}html.theme--documenter-dark .icon-text .icon{flex-grow:0;flex-shrink:0}html.theme--documenter-dark .icon-text .icon:not(:last-child){margin-right:.25em}html.theme--documenter-dark .icon-text .icon:not(:first-child){margin-left:.25em}html.theme--documenter-dark div.icon-text{display:flex}html.theme--documenter-dark .image,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img{display:block;position:relative}html.theme--documenter-dark .image img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img img{display:block;height:auto;width:100%}html.theme--documenter-dark .image img.is-rounded,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img img.is-rounded{border-radius:9999px}html.theme--documenter-dark .image.is-fullwidth,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-fullwidth{width:100%}html.theme--documenter-dark .image.is-square img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square img,html.theme--documenter-dark .image.is-square .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square .has-ratio,html.theme--documenter-dark .image.is-1by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1 img,html.theme--documenter-dark .image.is-1by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1 .has-ratio,html.theme--documenter-dark .image.is-5by4 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4 img,html.theme--documenter-dark .image.is-5by4 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4 .has-ratio,html.theme--documenter-dark .image.is-4by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3 img,html.theme--documenter-dark .image.is-4by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3 .has-ratio,html.theme--documenter-dark .image.is-3by2 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2 img,html.theme--documenter-dark .image.is-3by2 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2 .has-ratio,html.theme--documenter-dark .image.is-5by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3 img,html.theme--documenter-dark .image.is-5by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3 .has-ratio,html.theme--documenter-dark .image.is-16by9 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9 img,html.theme--documenter-dark .image.is-16by9 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9 .has-ratio,html.theme--documenter-dark .image.is-2by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1 img,html.theme--documenter-dark .image.is-2by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1 .has-ratio,html.theme--documenter-dark .image.is-3by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1 img,html.theme--documenter-dark .image.is-3by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1 .has-ratio,html.theme--documenter-dark .image.is-4by5 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5 img,html.theme--documenter-dark .image.is-4by5 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5 .has-ratio,html.theme--documenter-dark .image.is-3by4 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4 img,html.theme--documenter-dark .image.is-3by4 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4 .has-ratio,html.theme--documenter-dark .image.is-2by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3 img,html.theme--documenter-dark .image.is-2by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3 .has-ratio,html.theme--documenter-dark .image.is-3by5 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5 img,html.theme--documenter-dark .image.is-3by5 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5 .has-ratio,html.theme--documenter-dark .image.is-9by16 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16 img,html.theme--documenter-dark .image.is-9by16 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16 .has-ratio,html.theme--documenter-dark .image.is-1by2 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2 img,html.theme--documenter-dark .image.is-1by2 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2 .has-ratio,html.theme--documenter-dark .image.is-1by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3 img,html.theme--documenter-dark .image.is-1by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3 .has-ratio{height:100%;width:100%}html.theme--documenter-dark .image.is-square,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square,html.theme--documenter-dark .image.is-1by1,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1{padding-top:100%}html.theme--documenter-dark .image.is-5by4,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4{padding-top:80%}html.theme--documenter-dark .image.is-4by3,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3{padding-top:75%}html.theme--documenter-dark .image.is-3by2,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2{padding-top:66.6666%}html.theme--documenter-dark .image.is-5by3,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3{padding-top:60%}html.theme--documenter-dark .image.is-16by9,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9{padding-top:56.25%}html.theme--documenter-dark .image.is-2by1,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1{padding-top:50%}html.theme--documenter-dark .image.is-3by1,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1{padding-top:33.3333%}html.theme--documenter-dark .image.is-4by5,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5{padding-top:125%}html.theme--documenter-dark .image.is-3by4,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4{padding-top:133.3333%}html.theme--documenter-dark .image.is-2by3,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3{padding-top:150%}html.theme--documenter-dark .image.is-3by5,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5{padding-top:166.6666%}html.theme--documenter-dark .image.is-9by16,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16{padding-top:177.7777%}html.theme--documenter-dark .image.is-1by2,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2{padding-top:200%}html.theme--documenter-dark .image.is-1by3,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3{padding-top:300%}html.theme--documenter-dark .image.is-16x16,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16x16{height:16px;width:16px}html.theme--documenter-dark .image.is-24x24,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-24x24{height:24px;width:24px}html.theme--documenter-dark .image.is-32x32,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-32x32{height:32px;width:32px}html.theme--documenter-dark .image.is-48x48,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-48x48{height:48px;width:48px}html.theme--documenter-dark .image.is-64x64,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-64x64{height:64px;width:64px}html.theme--documenter-dark .image.is-96x96,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-96x96{height:96px;width:96px}html.theme--documenter-dark .image.is-128x128,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-128x128{height:128px;width:128px}html.theme--documenter-dark .notification{background-color:#282f2f;border-radius:.4em;position:relative;padding:1.25rem 2.5rem 1.25rem 1.5rem}html.theme--documenter-dark .notification a:not(.button):not(.dropdown-item){color:currentColor;text-decoration:underline}html.theme--documenter-dark .notification strong{color:currentColor}html.theme--documenter-dark .notification code,html.theme--documenter-dark .notification pre{background:#fff}html.theme--documenter-dark .notification pre code{background:transparent}html.theme--documenter-dark .notification>.delete{right:.5rem;position:absolute;top:0.5rem}html.theme--documenter-dark .notification .title,html.theme--documenter-dark .notification .subtitle,html.theme--documenter-dark .notification .content{color:currentColor}html.theme--documenter-dark .notification.is-white{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .notification.is-black{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .notification.is-light{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .notification.is-dark,html.theme--documenter-dark .content kbd.notification{background-color:#282f2f;color:#fff}html.theme--documenter-dark .notification.is-primary,html.theme--documenter-dark .docstring>section>a.notification.docs-sourcelink{background-color:#375a7f;color:#fff}html.theme--documenter-dark .notification.is-primary.is-light,html.theme--documenter-dark .docstring>section>a.notification.is-light.docs-sourcelink{background-color:#f1f5f9;color:#4d7eb2}html.theme--documenter-dark .notification.is-link{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .notification.is-link.is-light{background-color:#edfdf9;color:#15987e}html.theme--documenter-dark .notification.is-info{background-color:#024c7d;color:#fff}html.theme--documenter-dark .notification.is-info.is-light{background-color:#ebf7ff;color:#0e9dfb}html.theme--documenter-dark .notification.is-success{background-color:#008438;color:#fff}html.theme--documenter-dark .notification.is-success.is-light{background-color:#ebfff3;color:#00eb64}html.theme--documenter-dark .notification.is-warning{background-color:#ad8100;color:#fff}html.theme--documenter-dark .notification.is-warning.is-light{background-color:#fffaeb;color:#d19c00}html.theme--documenter-dark .notification.is-danger{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .notification.is-danger.is-light{background-color:#fdeeec;color:#ec311d}html.theme--documenter-dark .progress{-moz-appearance:none;-webkit-appearance:none;border:none;border-radius:9999px;display:block;height:1rem;overflow:hidden;padding:0;width:100%}html.theme--documenter-dark .progress::-webkit-progress-bar{background-color:#343c3d}html.theme--documenter-dark .progress::-webkit-progress-value{background-color:#dbdee0}html.theme--documenter-dark .progress::-moz-progress-bar{background-color:#dbdee0}html.theme--documenter-dark .progress::-ms-fill{background-color:#dbdee0;border:none}html.theme--documenter-dark .progress.is-white::-webkit-progress-value{background-color:#fff}html.theme--documenter-dark .progress.is-white::-moz-progress-bar{background-color:#fff}html.theme--documenter-dark .progress.is-white::-ms-fill{background-color:#fff}html.theme--documenter-dark .progress.is-white:indeterminate{background-image:linear-gradient(to right, #fff 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-black::-webkit-progress-value{background-color:#0a0a0a}html.theme--documenter-dark .progress.is-black::-moz-progress-bar{background-color:#0a0a0a}html.theme--documenter-dark .progress.is-black::-ms-fill{background-color:#0a0a0a}html.theme--documenter-dark .progress.is-black:indeterminate{background-image:linear-gradient(to right, #0a0a0a 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-light::-webkit-progress-value{background-color:#ecf0f1}html.theme--documenter-dark .progress.is-light::-moz-progress-bar{background-color:#ecf0f1}html.theme--documenter-dark .progress.is-light::-ms-fill{background-color:#ecf0f1}html.theme--documenter-dark .progress.is-light:indeterminate{background-image:linear-gradient(to right, #ecf0f1 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-dark::-webkit-progress-value,html.theme--documenter-dark .content kbd.progress::-webkit-progress-value{background-color:#282f2f}html.theme--documenter-dark .progress.is-dark::-moz-progress-bar,html.theme--documenter-dark .content kbd.progress::-moz-progress-bar{background-color:#282f2f}html.theme--documenter-dark .progress.is-dark::-ms-fill,html.theme--documenter-dark .content kbd.progress::-ms-fill{background-color:#282f2f}html.theme--documenter-dark .progress.is-dark:indeterminate,html.theme--documenter-dark .content kbd.progress:indeterminate{background-image:linear-gradient(to right, #282f2f 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-primary::-webkit-progress-value,html.theme--documenter-dark .docstring>section>a.progress.docs-sourcelink::-webkit-progress-value{background-color:#375a7f}html.theme--documenter-dark .progress.is-primary::-moz-progress-bar,html.theme--documenter-dark .docstring>section>a.progress.docs-sourcelink::-moz-progress-bar{background-color:#375a7f}html.theme--documenter-dark .progress.is-primary::-ms-fill,html.theme--documenter-dark .docstring>section>a.progress.docs-sourcelink::-ms-fill{background-color:#375a7f}html.theme--documenter-dark .progress.is-primary:indeterminate,html.theme--documenter-dark .docstring>section>a.progress.docs-sourcelink:indeterminate{background-image:linear-gradient(to right, #375a7f 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-link::-webkit-progress-value{background-color:#1abc9c}html.theme--documenter-dark .progress.is-link::-moz-progress-bar{background-color:#1abc9c}html.theme--documenter-dark .progress.is-link::-ms-fill{background-color:#1abc9c}html.theme--documenter-dark .progress.is-link:indeterminate{background-image:linear-gradient(to right, #1abc9c 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-info::-webkit-progress-value{background-color:#024c7d}html.theme--documenter-dark .progress.is-info::-moz-progress-bar{background-color:#024c7d}html.theme--documenter-dark .progress.is-info::-ms-fill{background-color:#024c7d}html.theme--documenter-dark .progress.is-info:indeterminate{background-image:linear-gradient(to right, #024c7d 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-success::-webkit-progress-value{background-color:#008438}html.theme--documenter-dark .progress.is-success::-moz-progress-bar{background-color:#008438}html.theme--documenter-dark .progress.is-success::-ms-fill{background-color:#008438}html.theme--documenter-dark .progress.is-success:indeterminate{background-image:linear-gradient(to right, #008438 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-warning::-webkit-progress-value{background-color:#ad8100}html.theme--documenter-dark .progress.is-warning::-moz-progress-bar{background-color:#ad8100}html.theme--documenter-dark .progress.is-warning::-ms-fill{background-color:#ad8100}html.theme--documenter-dark .progress.is-warning:indeterminate{background-image:linear-gradient(to right, #ad8100 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-danger::-webkit-progress-value{background-color:#9e1b0d}html.theme--documenter-dark .progress.is-danger::-moz-progress-bar{background-color:#9e1b0d}html.theme--documenter-dark .progress.is-danger::-ms-fill{background-color:#9e1b0d}html.theme--documenter-dark .progress.is-danger:indeterminate{background-image:linear-gradient(to right, #9e1b0d 30%, #343c3d 30%)}html.theme--documenter-dark .progress:indeterminate{animation-duration:1.5s;animation-iteration-count:infinite;animation-name:moveIndeterminate;animation-timing-function:linear;background-color:#343c3d;background-image:linear-gradient(to right, #fff 30%, #343c3d 30%);background-position:top left;background-repeat:no-repeat;background-size:150% 150%}html.theme--documenter-dark .progress:indeterminate::-webkit-progress-bar{background-color:transparent}html.theme--documenter-dark .progress:indeterminate::-moz-progress-bar{background-color:transparent}html.theme--documenter-dark .progress:indeterminate::-ms-fill{animation-name:none}html.theme--documenter-dark .progress.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.progress{height:.75rem}html.theme--documenter-dark .progress.is-medium{height:1.25rem}html.theme--documenter-dark .progress.is-large{height:1.5rem}@keyframes moveIndeterminate{from{background-position:200% 0}to{background-position:-200% 0}}html.theme--documenter-dark .table{background-color:#343c3d;color:#fff}html.theme--documenter-dark .table td,html.theme--documenter-dark .table th{border:1px solid #5e6d6f;border-width:0 0 1px;padding:0.5em 0.75em;vertical-align:top}html.theme--documenter-dark .table td.is-white,html.theme--documenter-dark .table th.is-white{background-color:#fff;border-color:#fff;color:#0a0a0a}html.theme--documenter-dark .table td.is-black,html.theme--documenter-dark .table th.is-black{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}html.theme--documenter-dark .table td.is-light,html.theme--documenter-dark .table th.is-light{background-color:#ecf0f1;border-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .table td.is-dark,html.theme--documenter-dark .table th.is-dark{background-color:#282f2f;border-color:#282f2f;color:#fff}html.theme--documenter-dark .table td.is-primary,html.theme--documenter-dark .table th.is-primary{background-color:#375a7f;border-color:#375a7f;color:#fff}html.theme--documenter-dark .table td.is-link,html.theme--documenter-dark .table th.is-link{background-color:#1abc9c;border-color:#1abc9c;color:#fff}html.theme--documenter-dark .table td.is-info,html.theme--documenter-dark .table th.is-info{background-color:#024c7d;border-color:#024c7d;color:#fff}html.theme--documenter-dark .table td.is-success,html.theme--documenter-dark .table th.is-success{background-color:#008438;border-color:#008438;color:#fff}html.theme--documenter-dark .table td.is-warning,html.theme--documenter-dark .table th.is-warning{background-color:#ad8100;border-color:#ad8100;color:#fff}html.theme--documenter-dark .table td.is-danger,html.theme--documenter-dark .table th.is-danger{background-color:#9e1b0d;border-color:#9e1b0d;color:#fff}html.theme--documenter-dark .table td.is-narrow,html.theme--documenter-dark .table th.is-narrow{white-space:nowrap;width:1%}html.theme--documenter-dark .table td.is-selected,html.theme--documenter-dark .table th.is-selected{background-color:#375a7f;color:#fff}html.theme--documenter-dark .table td.is-selected a,html.theme--documenter-dark .table td.is-selected strong,html.theme--documenter-dark .table th.is-selected a,html.theme--documenter-dark .table th.is-selected strong{color:currentColor}html.theme--documenter-dark .table td.is-vcentered,html.theme--documenter-dark .table th.is-vcentered{vertical-align:middle}html.theme--documenter-dark .table th{color:#f2f2f2}html.theme--documenter-dark .table th:not([align]){text-align:left}html.theme--documenter-dark .table tr.is-selected{background-color:#375a7f;color:#fff}html.theme--documenter-dark .table tr.is-selected a,html.theme--documenter-dark .table tr.is-selected strong{color:currentColor}html.theme--documenter-dark .table tr.is-selected td,html.theme--documenter-dark .table tr.is-selected th{border-color:#fff;color:currentColor}html.theme--documenter-dark .table thead{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .table thead td,html.theme--documenter-dark .table thead th{border-width:0 0 2px;color:#f2f2f2}html.theme--documenter-dark .table tfoot{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .table tfoot td,html.theme--documenter-dark .table tfoot th{border-width:2px 0 0;color:#f2f2f2}html.theme--documenter-dark .table tbody{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .table tbody tr:last-child td,html.theme--documenter-dark .table tbody tr:last-child th{border-bottom-width:0}html.theme--documenter-dark .table.is-bordered td,html.theme--documenter-dark .table.is-bordered th{border-width:1px}html.theme--documenter-dark .table.is-bordered tr:last-child td,html.theme--documenter-dark .table.is-bordered tr:last-child th{border-bottom-width:1px}html.theme--documenter-dark .table.is-fullwidth{width:100%}html.theme--documenter-dark .table.is-hoverable tbody tr:not(.is-selected):hover{background-color:#282f2f}html.theme--documenter-dark .table.is-hoverable.is-striped tbody tr:not(.is-selected):hover{background-color:#282f2f}html.theme--documenter-dark .table.is-hoverable.is-striped tbody tr:not(.is-selected):hover:nth-child(even){background-color:#2d3435}html.theme--documenter-dark .table.is-narrow td,html.theme--documenter-dark .table.is-narrow th{padding:0.25em 0.5em}html.theme--documenter-dark .table.is-striped tbody tr:not(.is-selected):nth-child(even){background-color:#282f2f}html.theme--documenter-dark .table-container{-webkit-overflow-scrolling:touch;overflow:auto;overflow-y:hidden;max-width:100%}html.theme--documenter-dark .tags{align-items:center;display:flex;flex-wrap:wrap;justify-content:flex-start}html.theme--documenter-dark .tags .tag,html.theme--documenter-dark .tags .content kbd,html.theme--documenter-dark .content .tags kbd,html.theme--documenter-dark .tags .docstring>section>a.docs-sourcelink{margin-bottom:0.5rem}html.theme--documenter-dark .tags .tag:not(:last-child),html.theme--documenter-dark .tags .content kbd:not(:last-child),html.theme--documenter-dark .content .tags kbd:not(:last-child),html.theme--documenter-dark .tags .docstring>section>a.docs-sourcelink:not(:last-child){margin-right:.5rem}html.theme--documenter-dark .tags:last-child{margin-bottom:-0.5rem}html.theme--documenter-dark .tags:not(:last-child){margin-bottom:1rem}html.theme--documenter-dark .tags.are-medium .tag:not(.is-normal):not(.is-large),html.theme--documenter-dark .tags.are-medium .content kbd:not(.is-normal):not(.is-large),html.theme--documenter-dark .content .tags.are-medium kbd:not(.is-normal):not(.is-large),html.theme--documenter-dark .tags.are-medium .docstring>section>a.docs-sourcelink:not(.is-normal):not(.is-large){font-size:1rem}html.theme--documenter-dark .tags.are-large .tag:not(.is-normal):not(.is-medium),html.theme--documenter-dark .tags.are-large .content kbd:not(.is-normal):not(.is-medium),html.theme--documenter-dark .content .tags.are-large kbd:not(.is-normal):not(.is-medium),html.theme--documenter-dark .tags.are-large .docstring>section>a.docs-sourcelink:not(.is-normal):not(.is-medium){font-size:1.25rem}html.theme--documenter-dark .tags.is-centered{justify-content:center}html.theme--documenter-dark .tags.is-centered .tag,html.theme--documenter-dark .tags.is-centered .content kbd,html.theme--documenter-dark .content .tags.is-centered kbd,html.theme--documenter-dark .tags.is-centered .docstring>section>a.docs-sourcelink{margin-right:0.25rem;margin-left:0.25rem}html.theme--documenter-dark .tags.is-right{justify-content:flex-end}html.theme--documenter-dark .tags.is-right .tag:not(:first-child),html.theme--documenter-dark .tags.is-right .content kbd:not(:first-child),html.theme--documenter-dark .content .tags.is-right kbd:not(:first-child),html.theme--documenter-dark .tags.is-right .docstring>section>a.docs-sourcelink:not(:first-child){margin-left:0.5rem}html.theme--documenter-dark .tags.is-right .tag:not(:last-child),html.theme--documenter-dark .tags.is-right .content kbd:not(:last-child),html.theme--documenter-dark .content .tags.is-right kbd:not(:last-child),html.theme--documenter-dark .tags.is-right .docstring>section>a.docs-sourcelink:not(:last-child){margin-right:0}html.theme--documenter-dark .tags.has-addons .tag,html.theme--documenter-dark .tags.has-addons .content kbd,html.theme--documenter-dark .content .tags.has-addons kbd,html.theme--documenter-dark .tags.has-addons .docstring>section>a.docs-sourcelink{margin-right:0}html.theme--documenter-dark .tags.has-addons .tag:not(:first-child),html.theme--documenter-dark .tags.has-addons .content kbd:not(:first-child),html.theme--documenter-dark .content .tags.has-addons kbd:not(:first-child),html.theme--documenter-dark .tags.has-addons .docstring>section>a.docs-sourcelink:not(:first-child){margin-left:0;border-top-left-radius:0;border-bottom-left-radius:0}html.theme--documenter-dark .tags.has-addons .tag:not(:last-child),html.theme--documenter-dark .tags.has-addons .content kbd:not(:last-child),html.theme--documenter-dark .content .tags.has-addons kbd:not(:last-child),html.theme--documenter-dark .tags.has-addons .docstring>section>a.docs-sourcelink:not(:last-child){border-top-right-radius:0;border-bottom-right-radius:0}html.theme--documenter-dark .tag:not(body),html.theme--documenter-dark .content kbd:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body){align-items:center;background-color:#282f2f;border-radius:.4em;color:#fff;display:inline-flex;font-size:.75rem;height:2em;justify-content:center;line-height:1.5;padding-left:0.75em;padding-right:0.75em;white-space:nowrap}html.theme--documenter-dark .tag:not(body) .delete,html.theme--documenter-dark .content kbd:not(body) .delete,html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body) .delete{margin-left:.25rem;margin-right:-.375rem}html.theme--documenter-dark .tag.is-white:not(body),html.theme--documenter-dark .content kbd.is-white:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-white:not(body){background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .tag.is-black:not(body),html.theme--documenter-dark .content kbd.is-black:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-black:not(body){background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .tag.is-light:not(body),html.theme--documenter-dark .content kbd.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-light:not(body){background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .tag.is-dark:not(body),html.theme--documenter-dark .content kbd:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-dark:not(body),html.theme--documenter-dark .content .docstring>section>kbd:not(body){background-color:#282f2f;color:#fff}html.theme--documenter-dark .tag.is-primary:not(body),html.theme--documenter-dark .content kbd.is-primary:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body){background-color:#375a7f;color:#fff}html.theme--documenter-dark .tag.is-primary.is-light:not(body),html.theme--documenter-dark .content kbd.is-primary.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-light:not(body){background-color:#f1f5f9;color:#4d7eb2}html.theme--documenter-dark .tag.is-link:not(body),html.theme--documenter-dark .content kbd.is-link:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-link:not(body){background-color:#1abc9c;color:#fff}html.theme--documenter-dark .tag.is-link.is-light:not(body),html.theme--documenter-dark .content kbd.is-link.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-link.is-light:not(body){background-color:#edfdf9;color:#15987e}html.theme--documenter-dark .tag.is-info:not(body),html.theme--documenter-dark .content kbd.is-info:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-info:not(body){background-color:#024c7d;color:#fff}html.theme--documenter-dark .tag.is-info.is-light:not(body),html.theme--documenter-dark .content kbd.is-info.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-info.is-light:not(body){background-color:#ebf7ff;color:#0e9dfb}html.theme--documenter-dark .tag.is-success:not(body),html.theme--documenter-dark .content kbd.is-success:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-success:not(body){background-color:#008438;color:#fff}html.theme--documenter-dark .tag.is-success.is-light:not(body),html.theme--documenter-dark .content kbd.is-success.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-success.is-light:not(body){background-color:#ebfff3;color:#00eb64}html.theme--documenter-dark .tag.is-warning:not(body),html.theme--documenter-dark .content kbd.is-warning:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-warning:not(body){background-color:#ad8100;color:#fff}html.theme--documenter-dark .tag.is-warning.is-light:not(body),html.theme--documenter-dark .content kbd.is-warning.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-warning.is-light:not(body){background-color:#fffaeb;color:#d19c00}html.theme--documenter-dark .tag.is-danger:not(body),html.theme--documenter-dark .content kbd.is-danger:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-danger:not(body){background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .tag.is-danger.is-light:not(body),html.theme--documenter-dark .content kbd.is-danger.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-danger.is-light:not(body){background-color:#fdeeec;color:#ec311d}html.theme--documenter-dark .tag.is-normal:not(body),html.theme--documenter-dark .content kbd.is-normal:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-normal:not(body){font-size:.75rem}html.theme--documenter-dark .tag.is-medium:not(body),html.theme--documenter-dark .content kbd.is-medium:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-medium:not(body){font-size:1rem}html.theme--documenter-dark .tag.is-large:not(body),html.theme--documenter-dark .content kbd.is-large:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-large:not(body){font-size:1.25rem}html.theme--documenter-dark .tag:not(body) .icon:first-child:not(:last-child),html.theme--documenter-dark .content kbd:not(body) .icon:first-child:not(:last-child),html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body) .icon:first-child:not(:last-child){margin-left:-.375em;margin-right:.1875em}html.theme--documenter-dark .tag:not(body) .icon:last-child:not(:first-child),html.theme--documenter-dark .content kbd:not(body) .icon:last-child:not(:first-child),html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body) .icon:last-child:not(:first-child){margin-left:.1875em;margin-right:-.375em}html.theme--documenter-dark .tag:not(body) .icon:first-child:last-child,html.theme--documenter-dark .content kbd:not(body) .icon:first-child:last-child,html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body) .icon:first-child:last-child{margin-left:-.375em;margin-right:-.375em}html.theme--documenter-dark .tag.is-delete:not(body),html.theme--documenter-dark .content kbd.is-delete:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body){margin-left:1px;padding:0;position:relative;width:2em}html.theme--documenter-dark .tag.is-delete:not(body)::before,html.theme--documenter-dark .content kbd.is-delete:not(body)::before,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body)::before,html.theme--documenter-dark .tag.is-delete:not(body)::after,html.theme--documenter-dark .content kbd.is-delete:not(body)::after,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body)::after{background-color:currentColor;content:"";display:block;left:50%;position:absolute;top:50%;transform:translateX(-50%) translateY(-50%) rotate(45deg);transform-origin:center center}html.theme--documenter-dark .tag.is-delete:not(body)::before,html.theme--documenter-dark .content kbd.is-delete:not(body)::before,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body)::before{height:1px;width:50%}html.theme--documenter-dark .tag.is-delete:not(body)::after,html.theme--documenter-dark .content kbd.is-delete:not(body)::after,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body)::after{height:50%;width:1px}html.theme--documenter-dark .tag.is-delete:not(body):hover,html.theme--documenter-dark .content kbd.is-delete:not(body):hover,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body):hover,html.theme--documenter-dark .tag.is-delete:not(body):focus,html.theme--documenter-dark .content kbd.is-delete:not(body):focus,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body):focus{background-color:#1d2122}html.theme--documenter-dark .tag.is-delete:not(body):active,html.theme--documenter-dark .content kbd.is-delete:not(body):active,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body):active{background-color:#111414}html.theme--documenter-dark .tag.is-rounded:not(body),html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:not(body),html.theme--documenter-dark .content kbd.is-rounded:not(body),html.theme--documenter-dark #documenter .docs-sidebar .content form.docs-search>input:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-rounded:not(body){border-radius:9999px}html.theme--documenter-dark a.tag:hover,html.theme--documenter-dark .docstring>section>a.docs-sourcelink:hover{text-decoration:underline}html.theme--documenter-dark .title,html.theme--documenter-dark .subtitle{word-break:break-word}html.theme--documenter-dark .title em,html.theme--documenter-dark .title span,html.theme--documenter-dark .subtitle em,html.theme--documenter-dark .subtitle span{font-weight:inherit}html.theme--documenter-dark .title sub,html.theme--documenter-dark .subtitle sub{font-size:.75em}html.theme--documenter-dark .title sup,html.theme--documenter-dark .subtitle sup{font-size:.75em}html.theme--documenter-dark .title .tag,html.theme--documenter-dark .title .content kbd,html.theme--documenter-dark .content .title kbd,html.theme--documenter-dark .title .docstring>section>a.docs-sourcelink,html.theme--documenter-dark .subtitle .tag,html.theme--documenter-dark .subtitle .content kbd,html.theme--documenter-dark .content .subtitle kbd,html.theme--documenter-dark .subtitle .docstring>section>a.docs-sourcelink{vertical-align:middle}html.theme--documenter-dark .title{color:#fff;font-size:2rem;font-weight:500;line-height:1.125}html.theme--documenter-dark .title strong{color:inherit;font-weight:inherit}html.theme--documenter-dark .title:not(.is-spaced)+.subtitle{margin-top:-1.25rem}html.theme--documenter-dark .title.is-1{font-size:3rem}html.theme--documenter-dark .title.is-2{font-size:2.5rem}html.theme--documenter-dark .title.is-3{font-size:2rem}html.theme--documenter-dark .title.is-4{font-size:1.5rem}html.theme--documenter-dark .title.is-5{font-size:1.25rem}html.theme--documenter-dark .title.is-6{font-size:1rem}html.theme--documenter-dark .title.is-7{font-size:.75rem}html.theme--documenter-dark .subtitle{color:#8c9b9d;font-size:1.25rem;font-weight:400;line-height:1.25}html.theme--documenter-dark .subtitle strong{color:#8c9b9d;font-weight:600}html.theme--documenter-dark .subtitle:not(.is-spaced)+.title{margin-top:-1.25rem}html.theme--documenter-dark .subtitle.is-1{font-size:3rem}html.theme--documenter-dark .subtitle.is-2{font-size:2.5rem}html.theme--documenter-dark .subtitle.is-3{font-size:2rem}html.theme--documenter-dark .subtitle.is-4{font-size:1.5rem}html.theme--documenter-dark .subtitle.is-5{font-size:1.25rem}html.theme--documenter-dark .subtitle.is-6{font-size:1rem}html.theme--documenter-dark .subtitle.is-7{font-size:.75rem}html.theme--documenter-dark .heading{display:block;font-size:11px;letter-spacing:1px;margin-bottom:5px;text-transform:uppercase}html.theme--documenter-dark .number{align-items:center;background-color:#282f2f;border-radius:9999px;display:inline-flex;font-size:1.25rem;height:2em;justify-content:center;margin-right:1.5rem;min-width:2.5em;padding:0.25rem 0.5rem;text-align:center;vertical-align:top}html.theme--documenter-dark .select select,html.theme--documenter-dark .textarea,html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{background-color:#1f2424;border-color:#5e6d6f;border-radius:.4em;color:#dbdee0}html.theme--documenter-dark .select select::-moz-placeholder,html.theme--documenter-dark .textarea::-moz-placeholder,html.theme--documenter-dark .input::-moz-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input::-moz-placeholder{color:#868c98}html.theme--documenter-dark .select select::-webkit-input-placeholder,html.theme--documenter-dark .textarea::-webkit-input-placeholder,html.theme--documenter-dark .input::-webkit-input-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input::-webkit-input-placeholder{color:#868c98}html.theme--documenter-dark .select select:-moz-placeholder,html.theme--documenter-dark .textarea:-moz-placeholder,html.theme--documenter-dark .input:-moz-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:-moz-placeholder{color:#868c98}html.theme--documenter-dark .select select:-ms-input-placeholder,html.theme--documenter-dark .textarea:-ms-input-placeholder,html.theme--documenter-dark .input:-ms-input-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:-ms-input-placeholder{color:#868c98}html.theme--documenter-dark .select select:hover,html.theme--documenter-dark .textarea:hover,html.theme--documenter-dark .input:hover,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:hover,html.theme--documenter-dark .select select.is-hovered,html.theme--documenter-dark .is-hovered.textarea,html.theme--documenter-dark .is-hovered.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-hovered{border-color:#8c9b9d}html.theme--documenter-dark .select select:focus,html.theme--documenter-dark .textarea:focus,html.theme--documenter-dark .input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:focus,html.theme--documenter-dark .select select.is-focused,html.theme--documenter-dark .is-focused.textarea,html.theme--documenter-dark .is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .select select:active,html.theme--documenter-dark .textarea:active,html.theme--documenter-dark .input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:active,html.theme--documenter-dark .select select.is-active,html.theme--documenter-dark .is-active.textarea,html.theme--documenter-dark .is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{border-color:#1abc9c;box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .select select[disabled],html.theme--documenter-dark .textarea[disabled],html.theme--documenter-dark .input[disabled],html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled],fieldset[disabled] html.theme--documenter-dark .select select,fieldset[disabled] html.theme--documenter-dark .textarea,fieldset[disabled] html.theme--documenter-dark .input,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{background-color:#8c9b9d;border-color:#282f2f;box-shadow:none;color:#fff}html.theme--documenter-dark .select select[disabled]::-moz-placeholder,html.theme--documenter-dark .textarea[disabled]::-moz-placeholder,html.theme--documenter-dark .input[disabled]::-moz-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled]::-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .select select::-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .textarea::-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .input::-moz-placeholder,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input::-moz-placeholder{color:rgba(255,255,255,0.3)}html.theme--documenter-dark .select select[disabled]::-webkit-input-placeholder,html.theme--documenter-dark .textarea[disabled]::-webkit-input-placeholder,html.theme--documenter-dark .input[disabled]::-webkit-input-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled]::-webkit-input-placeholder,fieldset[disabled] html.theme--documenter-dark .select select::-webkit-input-placeholder,fieldset[disabled] html.theme--documenter-dark .textarea::-webkit-input-placeholder,fieldset[disabled] html.theme--documenter-dark .input::-webkit-input-placeholder,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input::-webkit-input-placeholder{color:rgba(255,255,255,0.3)}html.theme--documenter-dark .select select[disabled]:-moz-placeholder,html.theme--documenter-dark .textarea[disabled]:-moz-placeholder,html.theme--documenter-dark .input[disabled]:-moz-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled]:-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .select select:-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .textarea:-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .input:-moz-placeholder,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:-moz-placeholder{color:rgba(255,255,255,0.3)}html.theme--documenter-dark .select select[disabled]:-ms-input-placeholder,html.theme--documenter-dark .textarea[disabled]:-ms-input-placeholder,html.theme--documenter-dark .input[disabled]:-ms-input-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled]:-ms-input-placeholder,fieldset[disabled] html.theme--documenter-dark .select select:-ms-input-placeholder,fieldset[disabled] html.theme--documenter-dark .textarea:-ms-input-placeholder,fieldset[disabled] html.theme--documenter-dark .input:-ms-input-placeholder,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:-ms-input-placeholder{color:rgba(255,255,255,0.3)}html.theme--documenter-dark .textarea,html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{box-shadow:inset 0 0.0625em 0.125em rgba(10,10,10,0.05);max-width:100%;width:100%}html.theme--documenter-dark .textarea[readonly],html.theme--documenter-dark .input[readonly],html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[readonly]{box-shadow:none}html.theme--documenter-dark .is-white.textarea,html.theme--documenter-dark .is-white.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-white{border-color:#fff}html.theme--documenter-dark .is-white.textarea:focus,html.theme--documenter-dark .is-white.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-white:focus,html.theme--documenter-dark .is-white.is-focused.textarea,html.theme--documenter-dark .is-white.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-white.textarea:active,html.theme--documenter-dark .is-white.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-white:active,html.theme--documenter-dark .is-white.is-active.textarea,html.theme--documenter-dark .is-white.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}html.theme--documenter-dark .is-black.textarea,html.theme--documenter-dark .is-black.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-black{border-color:#0a0a0a}html.theme--documenter-dark .is-black.textarea:focus,html.theme--documenter-dark .is-black.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-black:focus,html.theme--documenter-dark .is-black.is-focused.textarea,html.theme--documenter-dark .is-black.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-black.textarea:active,html.theme--documenter-dark .is-black.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-black:active,html.theme--documenter-dark .is-black.is-active.textarea,html.theme--documenter-dark .is-black.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}html.theme--documenter-dark .is-light.textarea,html.theme--documenter-dark .is-light.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-light{border-color:#ecf0f1}html.theme--documenter-dark .is-light.textarea:focus,html.theme--documenter-dark .is-light.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-light:focus,html.theme--documenter-dark .is-light.is-focused.textarea,html.theme--documenter-dark .is-light.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-light.textarea:active,html.theme--documenter-dark .is-light.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-light:active,html.theme--documenter-dark .is-light.is-active.textarea,html.theme--documenter-dark .is-light.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(236,240,241,0.25)}html.theme--documenter-dark .is-dark.textarea,html.theme--documenter-dark .content kbd.textarea,html.theme--documenter-dark .is-dark.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-dark,html.theme--documenter-dark .content kbd.input{border-color:#282f2f}html.theme--documenter-dark .is-dark.textarea:focus,html.theme--documenter-dark .content kbd.textarea:focus,html.theme--documenter-dark .is-dark.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-dark:focus,html.theme--documenter-dark .content kbd.input:focus,html.theme--documenter-dark .is-dark.is-focused.textarea,html.theme--documenter-dark .content kbd.is-focused.textarea,html.theme--documenter-dark .is-dark.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .content kbd.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar .content form.docs-search>input.is-focused,html.theme--documenter-dark .is-dark.textarea:active,html.theme--documenter-dark .content kbd.textarea:active,html.theme--documenter-dark .is-dark.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-dark:active,html.theme--documenter-dark .content kbd.input:active,html.theme--documenter-dark .is-dark.is-active.textarea,html.theme--documenter-dark .content kbd.is-active.textarea,html.theme--documenter-dark .is-dark.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active,html.theme--documenter-dark .content kbd.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar .content form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(40,47,47,0.25)}html.theme--documenter-dark .is-primary.textarea,html.theme--documenter-dark .docstring>section>a.textarea.docs-sourcelink,html.theme--documenter-dark .is-primary.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-primary,html.theme--documenter-dark .docstring>section>a.input.docs-sourcelink{border-color:#375a7f}html.theme--documenter-dark .is-primary.textarea:focus,html.theme--documenter-dark .docstring>section>a.textarea.docs-sourcelink:focus,html.theme--documenter-dark .is-primary.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-primary:focus,html.theme--documenter-dark .docstring>section>a.input.docs-sourcelink:focus,html.theme--documenter-dark .is-primary.is-focused.textarea,html.theme--documenter-dark .docstring>section>a.is-focused.textarea.docs-sourcelink,html.theme--documenter-dark .is-primary.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .docstring>section>a.is-focused.input.docs-sourcelink,html.theme--documenter-dark .is-primary.textarea:active,html.theme--documenter-dark .docstring>section>a.textarea.docs-sourcelink:active,html.theme--documenter-dark .is-primary.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-primary:active,html.theme--documenter-dark .docstring>section>a.input.docs-sourcelink:active,html.theme--documenter-dark .is-primary.is-active.textarea,html.theme--documenter-dark .docstring>section>a.is-active.textarea.docs-sourcelink,html.theme--documenter-dark .is-primary.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active,html.theme--documenter-dark .docstring>section>a.is-active.input.docs-sourcelink{box-shadow:0 0 0 0.125em rgba(55,90,127,0.25)}html.theme--documenter-dark .is-link.textarea,html.theme--documenter-dark .is-link.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-link{border-color:#1abc9c}html.theme--documenter-dark .is-link.textarea:focus,html.theme--documenter-dark .is-link.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-link:focus,html.theme--documenter-dark .is-link.is-focused.textarea,html.theme--documenter-dark .is-link.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-link.textarea:active,html.theme--documenter-dark .is-link.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-link:active,html.theme--documenter-dark .is-link.is-active.textarea,html.theme--documenter-dark .is-link.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .is-info.textarea,html.theme--documenter-dark .is-info.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-info{border-color:#024c7d}html.theme--documenter-dark .is-info.textarea:focus,html.theme--documenter-dark .is-info.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-info:focus,html.theme--documenter-dark .is-info.is-focused.textarea,html.theme--documenter-dark .is-info.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-info.textarea:active,html.theme--documenter-dark .is-info.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-info:active,html.theme--documenter-dark .is-info.is-active.textarea,html.theme--documenter-dark .is-info.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(2,76,125,0.25)}html.theme--documenter-dark .is-success.textarea,html.theme--documenter-dark .is-success.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-success{border-color:#008438}html.theme--documenter-dark .is-success.textarea:focus,html.theme--documenter-dark .is-success.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-success:focus,html.theme--documenter-dark .is-success.is-focused.textarea,html.theme--documenter-dark .is-success.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-success.textarea:active,html.theme--documenter-dark .is-success.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-success:active,html.theme--documenter-dark .is-success.is-active.textarea,html.theme--documenter-dark .is-success.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(0,132,56,0.25)}html.theme--documenter-dark .is-warning.textarea,html.theme--documenter-dark .is-warning.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-warning{border-color:#ad8100}html.theme--documenter-dark .is-warning.textarea:focus,html.theme--documenter-dark .is-warning.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-warning:focus,html.theme--documenter-dark .is-warning.is-focused.textarea,html.theme--documenter-dark .is-warning.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-warning.textarea:active,html.theme--documenter-dark .is-warning.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-warning:active,html.theme--documenter-dark .is-warning.is-active.textarea,html.theme--documenter-dark .is-warning.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(173,129,0,0.25)}html.theme--documenter-dark .is-danger.textarea,html.theme--documenter-dark .is-danger.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-danger{border-color:#9e1b0d}html.theme--documenter-dark .is-danger.textarea:focus,html.theme--documenter-dark .is-danger.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-danger:focus,html.theme--documenter-dark .is-danger.is-focused.textarea,html.theme--documenter-dark .is-danger.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-danger.textarea:active,html.theme--documenter-dark .is-danger.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-danger:active,html.theme--documenter-dark .is-danger.is-active.textarea,html.theme--documenter-dark .is-danger.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(158,27,13,0.25)}html.theme--documenter-dark .is-small.textarea,html.theme--documenter-dark .is-small.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{border-radius:3px;font-size:.75rem}html.theme--documenter-dark .is-medium.textarea,html.theme--documenter-dark .is-medium.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-medium{font-size:1.25rem}html.theme--documenter-dark .is-large.textarea,html.theme--documenter-dark .is-large.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-large{font-size:1.5rem}html.theme--documenter-dark .is-fullwidth.textarea,html.theme--documenter-dark .is-fullwidth.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-fullwidth{display:block;width:100%}html.theme--documenter-dark .is-inline.textarea,html.theme--documenter-dark .is-inline.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-inline{display:inline;width:auto}html.theme--documenter-dark .input.is-rounded,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{border-radius:9999px;padding-left:calc(calc(0.75em - 1px) + 0.375em);padding-right:calc(calc(0.75em - 1px) + 0.375em)}html.theme--documenter-dark .input.is-static,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-static{background-color:transparent;border-color:transparent;box-shadow:none;padding-left:0;padding-right:0}html.theme--documenter-dark .textarea{display:block;max-width:100%;min-width:100%;padding:calc(0.75em - 1px);resize:vertical}html.theme--documenter-dark .textarea:not([rows]){max-height:40em;min-height:8em}html.theme--documenter-dark .textarea[rows]{height:initial}html.theme--documenter-dark .textarea.has-fixed-size{resize:none}html.theme--documenter-dark .radio,html.theme--documenter-dark .checkbox{cursor:pointer;display:inline-block;line-height:1.25;position:relative}html.theme--documenter-dark .radio input,html.theme--documenter-dark .checkbox input{cursor:pointer}html.theme--documenter-dark .radio:hover,html.theme--documenter-dark .checkbox:hover{color:#8c9b9d}html.theme--documenter-dark .radio[disabled],html.theme--documenter-dark .checkbox[disabled],fieldset[disabled] html.theme--documenter-dark .radio,fieldset[disabled] html.theme--documenter-dark .checkbox,html.theme--documenter-dark .radio input[disabled],html.theme--documenter-dark .checkbox input[disabled]{color:#fff;cursor:not-allowed}html.theme--documenter-dark .radio+.radio{margin-left:.5em}html.theme--documenter-dark .select{display:inline-block;max-width:100%;position:relative;vertical-align:top}html.theme--documenter-dark .select:not(.is-multiple){height:2.5em}html.theme--documenter-dark .select:not(.is-multiple):not(.is-loading)::after{border-color:#1abc9c;right:1.125em;z-index:4}html.theme--documenter-dark .select.is-rounded select,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.select select{border-radius:9999px;padding-left:1em}html.theme--documenter-dark .select select{cursor:pointer;display:block;font-size:1em;max-width:100%;outline:none}html.theme--documenter-dark .select select::-ms-expand{display:none}html.theme--documenter-dark .select select[disabled]:hover,fieldset[disabled] html.theme--documenter-dark .select select:hover{border-color:#282f2f}html.theme--documenter-dark .select select:not([multiple]){padding-right:2.5em}html.theme--documenter-dark .select select[multiple]{height:auto;padding:0}html.theme--documenter-dark .select select[multiple] option{padding:0.5em 1em}html.theme--documenter-dark .select:not(.is-multiple):not(.is-loading):hover::after{border-color:#8c9b9d}html.theme--documenter-dark .select.is-white:not(:hover)::after{border-color:#fff}html.theme--documenter-dark .select.is-white select{border-color:#fff}html.theme--documenter-dark .select.is-white select:hover,html.theme--documenter-dark .select.is-white select.is-hovered{border-color:#f2f2f2}html.theme--documenter-dark .select.is-white select:focus,html.theme--documenter-dark .select.is-white select.is-focused,html.theme--documenter-dark .select.is-white select:active,html.theme--documenter-dark .select.is-white select.is-active{box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}html.theme--documenter-dark .select.is-black:not(:hover)::after{border-color:#0a0a0a}html.theme--documenter-dark .select.is-black select{border-color:#0a0a0a}html.theme--documenter-dark .select.is-black select:hover,html.theme--documenter-dark .select.is-black select.is-hovered{border-color:#000}html.theme--documenter-dark .select.is-black select:focus,html.theme--documenter-dark .select.is-black select.is-focused,html.theme--documenter-dark .select.is-black select:active,html.theme--documenter-dark .select.is-black select.is-active{box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}html.theme--documenter-dark .select.is-light:not(:hover)::after{border-color:#ecf0f1}html.theme--documenter-dark .select.is-light select{border-color:#ecf0f1}html.theme--documenter-dark .select.is-light select:hover,html.theme--documenter-dark .select.is-light select.is-hovered{border-color:#dde4e6}html.theme--documenter-dark .select.is-light select:focus,html.theme--documenter-dark .select.is-light select.is-focused,html.theme--documenter-dark .select.is-light select:active,html.theme--documenter-dark .select.is-light select.is-active{box-shadow:0 0 0 0.125em rgba(236,240,241,0.25)}html.theme--documenter-dark .select.is-dark:not(:hover)::after,html.theme--documenter-dark .content kbd.select:not(:hover)::after{border-color:#282f2f}html.theme--documenter-dark .select.is-dark select,html.theme--documenter-dark .content kbd.select select{border-color:#282f2f}html.theme--documenter-dark .select.is-dark select:hover,html.theme--documenter-dark .content kbd.select select:hover,html.theme--documenter-dark .select.is-dark select.is-hovered,html.theme--documenter-dark .content kbd.select select.is-hovered{border-color:#1d2122}html.theme--documenter-dark .select.is-dark select:focus,html.theme--documenter-dark .content kbd.select select:focus,html.theme--documenter-dark .select.is-dark select.is-focused,html.theme--documenter-dark .content kbd.select select.is-focused,html.theme--documenter-dark .select.is-dark select:active,html.theme--documenter-dark .content kbd.select select:active,html.theme--documenter-dark .select.is-dark select.is-active,html.theme--documenter-dark .content kbd.select select.is-active{box-shadow:0 0 0 0.125em rgba(40,47,47,0.25)}html.theme--documenter-dark .select.is-primary:not(:hover)::after,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink:not(:hover)::after{border-color:#375a7f}html.theme--documenter-dark .select.is-primary select,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select{border-color:#375a7f}html.theme--documenter-dark .select.is-primary select:hover,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select:hover,html.theme--documenter-dark .select.is-primary select.is-hovered,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select.is-hovered{border-color:#2f4d6d}html.theme--documenter-dark .select.is-primary select:focus,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select:focus,html.theme--documenter-dark .select.is-primary select.is-focused,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select.is-focused,html.theme--documenter-dark .select.is-primary select:active,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select:active,html.theme--documenter-dark .select.is-primary select.is-active,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select.is-active{box-shadow:0 0 0 0.125em rgba(55,90,127,0.25)}html.theme--documenter-dark .select.is-link:not(:hover)::after{border-color:#1abc9c}html.theme--documenter-dark .select.is-link select{border-color:#1abc9c}html.theme--documenter-dark .select.is-link select:hover,html.theme--documenter-dark .select.is-link select.is-hovered{border-color:#17a689}html.theme--documenter-dark .select.is-link select:focus,html.theme--documenter-dark .select.is-link select.is-focused,html.theme--documenter-dark .select.is-link select:active,html.theme--documenter-dark .select.is-link select.is-active{box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .select.is-info:not(:hover)::after{border-color:#024c7d}html.theme--documenter-dark .select.is-info select{border-color:#024c7d}html.theme--documenter-dark .select.is-info select:hover,html.theme--documenter-dark .select.is-info select.is-hovered{border-color:#023d64}html.theme--documenter-dark .select.is-info select:focus,html.theme--documenter-dark .select.is-info select.is-focused,html.theme--documenter-dark .select.is-info select:active,html.theme--documenter-dark .select.is-info select.is-active{box-shadow:0 0 0 0.125em rgba(2,76,125,0.25)}html.theme--documenter-dark .select.is-success:not(:hover)::after{border-color:#008438}html.theme--documenter-dark .select.is-success select{border-color:#008438}html.theme--documenter-dark .select.is-success select:hover,html.theme--documenter-dark .select.is-success select.is-hovered{border-color:#006b2d}html.theme--documenter-dark .select.is-success select:focus,html.theme--documenter-dark .select.is-success select.is-focused,html.theme--documenter-dark .select.is-success select:active,html.theme--documenter-dark .select.is-success select.is-active{box-shadow:0 0 0 0.125em rgba(0,132,56,0.25)}html.theme--documenter-dark .select.is-warning:not(:hover)::after{border-color:#ad8100}html.theme--documenter-dark .select.is-warning select{border-color:#ad8100}html.theme--documenter-dark .select.is-warning select:hover,html.theme--documenter-dark .select.is-warning select.is-hovered{border-color:#946e00}html.theme--documenter-dark .select.is-warning select:focus,html.theme--documenter-dark .select.is-warning select.is-focused,html.theme--documenter-dark .select.is-warning select:active,html.theme--documenter-dark .select.is-warning select.is-active{box-shadow:0 0 0 0.125em rgba(173,129,0,0.25)}html.theme--documenter-dark .select.is-danger:not(:hover)::after{border-color:#9e1b0d}html.theme--documenter-dark .select.is-danger select{border-color:#9e1b0d}html.theme--documenter-dark .select.is-danger select:hover,html.theme--documenter-dark .select.is-danger select.is-hovered{border-color:#86170b}html.theme--documenter-dark .select.is-danger select:focus,html.theme--documenter-dark .select.is-danger select.is-focused,html.theme--documenter-dark .select.is-danger select:active,html.theme--documenter-dark .select.is-danger select.is-active{box-shadow:0 0 0 0.125em rgba(158,27,13,0.25)}html.theme--documenter-dark .select.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.select{border-radius:3px;font-size:.75rem}html.theme--documenter-dark .select.is-medium{font-size:1.25rem}html.theme--documenter-dark .select.is-large{font-size:1.5rem}html.theme--documenter-dark .select.is-disabled::after{border-color:#fff !important;opacity:0.5}html.theme--documenter-dark .select.is-fullwidth{width:100%}html.theme--documenter-dark .select.is-fullwidth select{width:100%}html.theme--documenter-dark .select.is-loading::after{margin-top:0;position:absolute;right:.625em;top:0.625em;transform:none}html.theme--documenter-dark .select.is-loading.is-small:after,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-loading:after{font-size:.75rem}html.theme--documenter-dark .select.is-loading.is-medium:after{font-size:1.25rem}html.theme--documenter-dark .select.is-loading.is-large:after{font-size:1.5rem}html.theme--documenter-dark .file{align-items:stretch;display:flex;justify-content:flex-start;position:relative}html.theme--documenter-dark .file.is-white .file-cta{background-color:#fff;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .file.is-white:hover .file-cta,html.theme--documenter-dark .file.is-white.is-hovered .file-cta{background-color:#f9f9f9;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .file.is-white:focus .file-cta,html.theme--documenter-dark .file.is-white.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(255,255,255,0.25);color:#0a0a0a}html.theme--documenter-dark .file.is-white:active .file-cta,html.theme--documenter-dark .file.is-white.is-active .file-cta{background-color:#f2f2f2;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .file.is-black .file-cta{background-color:#0a0a0a;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-black:hover .file-cta,html.theme--documenter-dark .file.is-black.is-hovered .file-cta{background-color:#040404;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-black:focus .file-cta,html.theme--documenter-dark .file.is-black.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(10,10,10,0.25);color:#fff}html.theme--documenter-dark .file.is-black:active .file-cta,html.theme--documenter-dark .file.is-black.is-active .file-cta{background-color:#000;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-light .file-cta{background-color:#ecf0f1;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .file.is-light:hover .file-cta,html.theme--documenter-dark .file.is-light.is-hovered .file-cta{background-color:#e5eaec;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .file.is-light:focus .file-cta,html.theme--documenter-dark .file.is-light.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(236,240,241,0.25);color:rgba(0,0,0,0.7)}html.theme--documenter-dark .file.is-light:active .file-cta,html.theme--documenter-dark .file.is-light.is-active .file-cta{background-color:#dde4e6;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .file.is-dark .file-cta,html.theme--documenter-dark .content kbd.file .file-cta{background-color:#282f2f;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-dark:hover .file-cta,html.theme--documenter-dark .content kbd.file:hover .file-cta,html.theme--documenter-dark .file.is-dark.is-hovered .file-cta,html.theme--documenter-dark .content kbd.file.is-hovered .file-cta{background-color:#232829;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-dark:focus .file-cta,html.theme--documenter-dark .content kbd.file:focus .file-cta,html.theme--documenter-dark .file.is-dark.is-focused .file-cta,html.theme--documenter-dark .content kbd.file.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(40,47,47,0.25);color:#fff}html.theme--documenter-dark .file.is-dark:active .file-cta,html.theme--documenter-dark .content kbd.file:active .file-cta,html.theme--documenter-dark .file.is-dark.is-active .file-cta,html.theme--documenter-dark .content kbd.file.is-active .file-cta{background-color:#1d2122;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-primary .file-cta,html.theme--documenter-dark .docstring>section>a.file.docs-sourcelink .file-cta{background-color:#375a7f;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-primary:hover .file-cta,html.theme--documenter-dark .docstring>section>a.file.docs-sourcelink:hover .file-cta,html.theme--documenter-dark .file.is-primary.is-hovered .file-cta,html.theme--documenter-dark .docstring>section>a.file.is-hovered.docs-sourcelink .file-cta{background-color:#335476;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-primary:focus .file-cta,html.theme--documenter-dark .docstring>section>a.file.docs-sourcelink:focus .file-cta,html.theme--documenter-dark .file.is-primary.is-focused .file-cta,html.theme--documenter-dark .docstring>section>a.file.is-focused.docs-sourcelink .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(55,90,127,0.25);color:#fff}html.theme--documenter-dark .file.is-primary:active .file-cta,html.theme--documenter-dark .docstring>section>a.file.docs-sourcelink:active .file-cta,html.theme--documenter-dark .file.is-primary.is-active .file-cta,html.theme--documenter-dark .docstring>section>a.file.is-active.docs-sourcelink .file-cta{background-color:#2f4d6d;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-link .file-cta{background-color:#1abc9c;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-link:hover .file-cta,html.theme--documenter-dark .file.is-link.is-hovered .file-cta{background-color:#18b193;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-link:focus .file-cta,html.theme--documenter-dark .file.is-link.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(26,188,156,0.25);color:#fff}html.theme--documenter-dark .file.is-link:active .file-cta,html.theme--documenter-dark .file.is-link.is-active .file-cta{background-color:#17a689;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-info .file-cta{background-color:#024c7d;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-info:hover .file-cta,html.theme--documenter-dark .file.is-info.is-hovered .file-cta{background-color:#024470;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-info:focus .file-cta,html.theme--documenter-dark .file.is-info.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(2,76,125,0.25);color:#fff}html.theme--documenter-dark .file.is-info:active .file-cta,html.theme--documenter-dark .file.is-info.is-active .file-cta{background-color:#023d64;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-success .file-cta{background-color:#008438;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-success:hover .file-cta,html.theme--documenter-dark .file.is-success.is-hovered .file-cta{background-color:#073;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-success:focus .file-cta,html.theme--documenter-dark .file.is-success.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(0,132,56,0.25);color:#fff}html.theme--documenter-dark .file.is-success:active .file-cta,html.theme--documenter-dark .file.is-success.is-active .file-cta{background-color:#006b2d;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-warning .file-cta{background-color:#ad8100;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-warning:hover .file-cta,html.theme--documenter-dark .file.is-warning.is-hovered .file-cta{background-color:#a07700;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-warning:focus .file-cta,html.theme--documenter-dark .file.is-warning.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(173,129,0,0.25);color:#fff}html.theme--documenter-dark .file.is-warning:active .file-cta,html.theme--documenter-dark .file.is-warning.is-active .file-cta{background-color:#946e00;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-danger .file-cta{background-color:#9e1b0d;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-danger:hover .file-cta,html.theme--documenter-dark .file.is-danger.is-hovered .file-cta{background-color:#92190c;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-danger:focus .file-cta,html.theme--documenter-dark .file.is-danger.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(158,27,13,0.25);color:#fff}html.theme--documenter-dark .file.is-danger:active .file-cta,html.theme--documenter-dark .file.is-danger.is-active .file-cta{background-color:#86170b;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.file{font-size:.75rem}html.theme--documenter-dark .file.is-normal{font-size:1rem}html.theme--documenter-dark .file.is-medium{font-size:1.25rem}html.theme--documenter-dark .file.is-medium .file-icon .fa{font-size:21px}html.theme--documenter-dark .file.is-large{font-size:1.5rem}html.theme--documenter-dark .file.is-large .file-icon .fa{font-size:28px}html.theme--documenter-dark .file.has-name .file-cta{border-bottom-right-radius:0;border-top-right-radius:0}html.theme--documenter-dark .file.has-name .file-name{border-bottom-left-radius:0;border-top-left-radius:0}html.theme--documenter-dark .file.has-name.is-empty .file-cta{border-radius:.4em}html.theme--documenter-dark .file.has-name.is-empty .file-name{display:none}html.theme--documenter-dark .file.is-boxed .file-label{flex-direction:column}html.theme--documenter-dark .file.is-boxed .file-cta{flex-direction:column;height:auto;padding:1em 3em}html.theme--documenter-dark .file.is-boxed .file-name{border-width:0 1px 1px}html.theme--documenter-dark .file.is-boxed .file-icon{height:1.5em;width:1.5em}html.theme--documenter-dark .file.is-boxed .file-icon .fa{font-size:21px}html.theme--documenter-dark .file.is-boxed.is-small .file-icon .fa,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-boxed .file-icon .fa{font-size:14px}html.theme--documenter-dark .file.is-boxed.is-medium .file-icon .fa{font-size:28px}html.theme--documenter-dark .file.is-boxed.is-large .file-icon .fa{font-size:35px}html.theme--documenter-dark .file.is-boxed.has-name .file-cta{border-radius:.4em .4em 0 0}html.theme--documenter-dark .file.is-boxed.has-name .file-name{border-radius:0 0 .4em .4em;border-width:0 1px 1px}html.theme--documenter-dark .file.is-centered{justify-content:center}html.theme--documenter-dark .file.is-fullwidth .file-label{width:100%}html.theme--documenter-dark .file.is-fullwidth .file-name{flex-grow:1;max-width:none}html.theme--documenter-dark .file.is-right{justify-content:flex-end}html.theme--documenter-dark .file.is-right .file-cta{border-radius:0 .4em .4em 0}html.theme--documenter-dark .file.is-right .file-name{border-radius:.4em 0 0 .4em;border-width:1px 0 1px 1px;order:-1}html.theme--documenter-dark .file-label{align-items:stretch;display:flex;cursor:pointer;justify-content:flex-start;overflow:hidden;position:relative}html.theme--documenter-dark .file-label:hover .file-cta{background-color:#232829;color:#f2f2f2}html.theme--documenter-dark .file-label:hover .file-name{border-color:#596668}html.theme--documenter-dark .file-label:active .file-cta{background-color:#1d2122;color:#f2f2f2}html.theme--documenter-dark .file-label:active .file-name{border-color:#535f61}html.theme--documenter-dark .file-input{height:100%;left:0;opacity:0;outline:none;position:absolute;top:0;width:100%}html.theme--documenter-dark .file-cta,html.theme--documenter-dark .file-name{border-color:#5e6d6f;border-radius:.4em;font-size:1em;padding-left:1em;padding-right:1em;white-space:nowrap}html.theme--documenter-dark .file-cta{background-color:#282f2f;color:#fff}html.theme--documenter-dark .file-name{border-color:#5e6d6f;border-style:solid;border-width:1px 1px 1px 0;display:block;max-width:16em;overflow:hidden;text-align:inherit;text-overflow:ellipsis}html.theme--documenter-dark .file-icon{align-items:center;display:flex;height:1em;justify-content:center;margin-right:.5em;width:1em}html.theme--documenter-dark .file-icon .fa{font-size:14px}html.theme--documenter-dark .label{color:#f2f2f2;display:block;font-size:1rem;font-weight:700}html.theme--documenter-dark .label:not(:last-child){margin-bottom:0.5em}html.theme--documenter-dark .label.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.label{font-size:.75rem}html.theme--documenter-dark .label.is-medium{font-size:1.25rem}html.theme--documenter-dark .label.is-large{font-size:1.5rem}html.theme--documenter-dark .help{display:block;font-size:.75rem;margin-top:0.25rem}html.theme--documenter-dark .help.is-white{color:#fff}html.theme--documenter-dark .help.is-black{color:#0a0a0a}html.theme--documenter-dark .help.is-light{color:#ecf0f1}html.theme--documenter-dark .help.is-dark,html.theme--documenter-dark .content kbd.help{color:#282f2f}html.theme--documenter-dark .help.is-primary,html.theme--documenter-dark .docstring>section>a.help.docs-sourcelink{color:#375a7f}html.theme--documenter-dark .help.is-link{color:#1abc9c}html.theme--documenter-dark .help.is-info{color:#024c7d}html.theme--documenter-dark .help.is-success{color:#008438}html.theme--documenter-dark .help.is-warning{color:#ad8100}html.theme--documenter-dark .help.is-danger{color:#9e1b0d}html.theme--documenter-dark .field:not(:last-child){margin-bottom:0.75rem}html.theme--documenter-dark .field.has-addons{display:flex;justify-content:flex-start}html.theme--documenter-dark .field.has-addons .control:not(:last-child){margin-right:-1px}html.theme--documenter-dark .field.has-addons .control:not(:first-child):not(:last-child) .button,html.theme--documenter-dark .field.has-addons .control:not(:first-child):not(:last-child) .input,html.theme--documenter-dark .field.has-addons .control:not(:first-child):not(:last-child) #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control:not(:first-child):not(:last-child) form.docs-search>input,html.theme--documenter-dark .field.has-addons .control:not(:first-child):not(:last-child) .select select{border-radius:0}html.theme--documenter-dark .field.has-addons .control:first-child:not(:only-child) .button,html.theme--documenter-dark .field.has-addons .control:first-child:not(:only-child) .input,html.theme--documenter-dark .field.has-addons .control:first-child:not(:only-child) #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control:first-child:not(:only-child) form.docs-search>input,html.theme--documenter-dark .field.has-addons .control:first-child:not(:only-child) .select select{border-bottom-right-radius:0;border-top-right-radius:0}html.theme--documenter-dark .field.has-addons .control:last-child:not(:only-child) .button,html.theme--documenter-dark .field.has-addons .control:last-child:not(:only-child) .input,html.theme--documenter-dark .field.has-addons .control:last-child:not(:only-child) #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control:last-child:not(:only-child) form.docs-search>input,html.theme--documenter-dark .field.has-addons .control:last-child:not(:only-child) .select select{border-bottom-left-radius:0;border-top-left-radius:0}html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .button.is-hovered:not([disabled]),html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .input.is-hovered:not([disabled]),html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-hovered:not([disabled]),html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-hovered:not([disabled]),html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .select select.is-hovered:not([disabled]){z-index:2}html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):focus,html.theme--documenter-dark .field.has-addons .control .button.is-focused:not([disabled]),html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):active,html.theme--documenter-dark .field.has-addons .control .button.is-active:not([disabled]),html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):focus,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):focus,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):focus,html.theme--documenter-dark .field.has-addons .control .input.is-focused:not([disabled]),html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-focused:not([disabled]),html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-focused:not([disabled]),html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):active,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):active,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):active,html.theme--documenter-dark .field.has-addons .control .input.is-active:not([disabled]),html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-active:not([disabled]),html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-active:not([disabled]),html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):focus,html.theme--documenter-dark .field.has-addons .control .select select.is-focused:not([disabled]),html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):active,html.theme--documenter-dark .field.has-addons .control .select select.is-active:not([disabled]){z-index:3}html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):focus:hover,html.theme--documenter-dark .field.has-addons .control .button.is-focused:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):active:hover,html.theme--documenter-dark .field.has-addons .control .button.is-active:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):focus:hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):focus:hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):focus:hover,html.theme--documenter-dark .field.has-addons .control .input.is-focused:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-focused:not([disabled]):hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-focused:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):active:hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):active:hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):active:hover,html.theme--documenter-dark .field.has-addons .control .input.is-active:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-active:not([disabled]):hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-active:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):focus:hover,html.theme--documenter-dark .field.has-addons .control .select select.is-focused:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):active:hover,html.theme--documenter-dark .field.has-addons .control .select select.is-active:not([disabled]):hover{z-index:4}html.theme--documenter-dark .field.has-addons .control.is-expanded{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .field.has-addons.has-addons-centered{justify-content:center}html.theme--documenter-dark .field.has-addons.has-addons-right{justify-content:flex-end}html.theme--documenter-dark .field.has-addons.has-addons-fullwidth .control{flex-grow:1;flex-shrink:0}html.theme--documenter-dark .field.is-grouped{display:flex;justify-content:flex-start}html.theme--documenter-dark .field.is-grouped>.control{flex-shrink:0}html.theme--documenter-dark .field.is-grouped>.control:not(:last-child){margin-bottom:0;margin-right:.75rem}html.theme--documenter-dark .field.is-grouped>.control.is-expanded{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .field.is-grouped.is-grouped-centered{justify-content:center}html.theme--documenter-dark .field.is-grouped.is-grouped-right{justify-content:flex-end}html.theme--documenter-dark .field.is-grouped.is-grouped-multiline{flex-wrap:wrap}html.theme--documenter-dark .field.is-grouped.is-grouped-multiline>.control:last-child,html.theme--documenter-dark .field.is-grouped.is-grouped-multiline>.control:not(:last-child){margin-bottom:0.75rem}html.theme--documenter-dark .field.is-grouped.is-grouped-multiline:last-child{margin-bottom:-0.75rem}html.theme--documenter-dark .field.is-grouped.is-grouped-multiline:not(:last-child){margin-bottom:0}@media screen and (min-width: 769px),print{html.theme--documenter-dark .field.is-horizontal{display:flex}}html.theme--documenter-dark .field-label .label{font-size:inherit}@media screen and (max-width: 768px){html.theme--documenter-dark .field-label{margin-bottom:0.5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .field-label{flex-basis:0;flex-grow:1;flex-shrink:0;margin-right:1.5rem;text-align:right}html.theme--documenter-dark .field-label.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.field-label{font-size:.75rem;padding-top:0.375em}html.theme--documenter-dark .field-label.is-normal{padding-top:0.375em}html.theme--documenter-dark .field-label.is-medium{font-size:1.25rem;padding-top:0.375em}html.theme--documenter-dark .field-label.is-large{font-size:1.5rem;padding-top:0.375em}}html.theme--documenter-dark .field-body .field .field{margin-bottom:0}@media screen and (min-width: 769px),print{html.theme--documenter-dark .field-body{display:flex;flex-basis:0;flex-grow:5;flex-shrink:1}html.theme--documenter-dark .field-body .field{margin-bottom:0}html.theme--documenter-dark .field-body>.field{flex-shrink:1}html.theme--documenter-dark .field-body>.field:not(.is-narrow){flex-grow:1}html.theme--documenter-dark .field-body>.field:not(:last-child){margin-right:.75rem}}html.theme--documenter-dark .control{box-sizing:border-box;clear:both;font-size:1rem;position:relative;text-align:inherit}html.theme--documenter-dark .control.has-icons-left .input:focus~.icon,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input:focus~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input:focus~.icon,html.theme--documenter-dark .control.has-icons-left .select:focus~.icon,html.theme--documenter-dark .control.has-icons-right .input:focus~.icon,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input:focus~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input:focus~.icon,html.theme--documenter-dark .control.has-icons-right .select:focus~.icon{color:#282f2f}html.theme--documenter-dark .control.has-icons-left .input.is-small~.icon,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input~.icon,html.theme--documenter-dark .control.has-icons-left .select.is-small~.icon,html.theme--documenter-dark .control.has-icons-right .input.is-small~.icon,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input~.icon,html.theme--documenter-dark .control.has-icons-right .select.is-small~.icon{font-size:.75rem}html.theme--documenter-dark .control.has-icons-left .input.is-medium~.icon,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input.is-medium~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input.is-medium~.icon,html.theme--documenter-dark .control.has-icons-left .select.is-medium~.icon,html.theme--documenter-dark .control.has-icons-right .input.is-medium~.icon,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input.is-medium~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input.is-medium~.icon,html.theme--documenter-dark .control.has-icons-right .select.is-medium~.icon{font-size:1.25rem}html.theme--documenter-dark .control.has-icons-left .input.is-large~.icon,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input.is-large~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input.is-large~.icon,html.theme--documenter-dark .control.has-icons-left .select.is-large~.icon,html.theme--documenter-dark .control.has-icons-right .input.is-large~.icon,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input.is-large~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input.is-large~.icon,html.theme--documenter-dark .control.has-icons-right .select.is-large~.icon{font-size:1.5rem}html.theme--documenter-dark .control.has-icons-left .icon,html.theme--documenter-dark .control.has-icons-right .icon{color:#5e6d6f;height:2.5em;pointer-events:none;position:absolute;top:0;width:2.5em;z-index:4}html.theme--documenter-dark .control.has-icons-left .input,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input,html.theme--documenter-dark .control.has-icons-left .select select{padding-left:2.5em}html.theme--documenter-dark .control.has-icons-left .icon.is-left{left:0}html.theme--documenter-dark .control.has-icons-right .input,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input,html.theme--documenter-dark .control.has-icons-right .select select{padding-right:2.5em}html.theme--documenter-dark .control.has-icons-right .icon.is-right{right:0}html.theme--documenter-dark .control.is-loading::after{position:absolute !important;right:.625em;top:0.625em;z-index:4}html.theme--documenter-dark .control.is-loading.is-small:after,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-loading:after{font-size:.75rem}html.theme--documenter-dark .control.is-loading.is-medium:after{font-size:1.25rem}html.theme--documenter-dark .control.is-loading.is-large:after{font-size:1.5rem}html.theme--documenter-dark .breadcrumb{font-size:1rem;white-space:nowrap}html.theme--documenter-dark .breadcrumb a{align-items:center;color:#1abc9c;display:flex;justify-content:center;padding:0 .75em}html.theme--documenter-dark .breadcrumb a:hover{color:#1dd2af}html.theme--documenter-dark .breadcrumb li{align-items:center;display:flex}html.theme--documenter-dark .breadcrumb li:first-child a{padding-left:0}html.theme--documenter-dark .breadcrumb li.is-active a{color:#f2f2f2;cursor:default;pointer-events:none}html.theme--documenter-dark .breadcrumb li+li::before{color:#8c9b9d;content:"\0002f"}html.theme--documenter-dark .breadcrumb ul,html.theme--documenter-dark .breadcrumb ol{align-items:flex-start;display:flex;flex-wrap:wrap;justify-content:flex-start}html.theme--documenter-dark .breadcrumb .icon:first-child{margin-right:.5em}html.theme--documenter-dark .breadcrumb .icon:last-child{margin-left:.5em}html.theme--documenter-dark .breadcrumb.is-centered ol,html.theme--documenter-dark .breadcrumb.is-centered ul{justify-content:center}html.theme--documenter-dark .breadcrumb.is-right ol,html.theme--documenter-dark .breadcrumb.is-right ul{justify-content:flex-end}html.theme--documenter-dark .breadcrumb.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.breadcrumb{font-size:.75rem}html.theme--documenter-dark .breadcrumb.is-medium{font-size:1.25rem}html.theme--documenter-dark .breadcrumb.is-large{font-size:1.5rem}html.theme--documenter-dark .breadcrumb.has-arrow-separator li+li::before{content:"\02192"}html.theme--documenter-dark .breadcrumb.has-bullet-separator li+li::before{content:"\02022"}html.theme--documenter-dark .breadcrumb.has-dot-separator li+li::before{content:"\000b7"}html.theme--documenter-dark .breadcrumb.has-succeeds-separator li+li::before{content:"\0227B"}html.theme--documenter-dark .card{background-color:#fff;border-radius:.25rem;box-shadow:#171717;color:#fff;max-width:100%;position:relative}html.theme--documenter-dark .card-footer:first-child,html.theme--documenter-dark .card-content:first-child,html.theme--documenter-dark .card-header:first-child{border-top-left-radius:.25rem;border-top-right-radius:.25rem}html.theme--documenter-dark .card-footer:last-child,html.theme--documenter-dark .card-content:last-child,html.theme--documenter-dark .card-header:last-child{border-bottom-left-radius:.25rem;border-bottom-right-radius:.25rem}html.theme--documenter-dark .card-header{background-color:rgba(0,0,0,0);align-items:stretch;box-shadow:0 0.125em 0.25em rgba(10,10,10,0.1);display:flex}html.theme--documenter-dark .card-header-title{align-items:center;color:#f2f2f2;display:flex;flex-grow:1;font-weight:700;padding:0.75rem 1rem}html.theme--documenter-dark .card-header-title.is-centered{justify-content:center}html.theme--documenter-dark .card-header-icon{-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;color:currentColor;font-family:inherit;font-size:1em;margin:0;padding:0;align-items:center;cursor:pointer;display:flex;justify-content:center;padding:0.75rem 1rem}html.theme--documenter-dark .card-image{display:block;position:relative}html.theme--documenter-dark .card-image:first-child img{border-top-left-radius:.25rem;border-top-right-radius:.25rem}html.theme--documenter-dark .card-image:last-child img{border-bottom-left-radius:.25rem;border-bottom-right-radius:.25rem}html.theme--documenter-dark .card-content{background-color:rgba(0,0,0,0);padding:1.5rem}html.theme--documenter-dark .card-footer{background-color:rgba(0,0,0,0);border-top:1px solid #ededed;align-items:stretch;display:flex}html.theme--documenter-dark .card-footer-item{align-items:center;display:flex;flex-basis:0;flex-grow:1;flex-shrink:0;justify-content:center;padding:.75rem}html.theme--documenter-dark .card-footer-item:not(:last-child){border-right:1px solid #ededed}html.theme--documenter-dark .card .media:not(:last-child){margin-bottom:1.5rem}html.theme--documenter-dark .dropdown{display:inline-flex;position:relative;vertical-align:top}html.theme--documenter-dark .dropdown.is-active .dropdown-menu,html.theme--documenter-dark .dropdown.is-hoverable:hover .dropdown-menu{display:block}html.theme--documenter-dark .dropdown.is-right .dropdown-menu{left:auto;right:0}html.theme--documenter-dark .dropdown.is-up .dropdown-menu{bottom:100%;padding-bottom:4px;padding-top:initial;top:auto}html.theme--documenter-dark .dropdown-menu{display:none;left:0;min-width:12rem;padding-top:4px;position:absolute;top:100%;z-index:20}html.theme--documenter-dark .dropdown-content{background-color:#282f2f;border-radius:.4em;box-shadow:#171717;padding-bottom:.5rem;padding-top:.5rem}html.theme--documenter-dark .dropdown-item{color:#fff;display:block;font-size:0.875rem;line-height:1.5;padding:0.375rem 1rem;position:relative}html.theme--documenter-dark a.dropdown-item,html.theme--documenter-dark button.dropdown-item{padding-right:3rem;text-align:inherit;white-space:nowrap;width:100%}html.theme--documenter-dark a.dropdown-item:hover,html.theme--documenter-dark button.dropdown-item:hover{background-color:#282f2f;color:#0a0a0a}html.theme--documenter-dark a.dropdown-item.is-active,html.theme--documenter-dark button.dropdown-item.is-active{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .dropdown-divider{background-color:#ededed;border:none;display:block;height:1px;margin:0.5rem 0}html.theme--documenter-dark .level{align-items:center;justify-content:space-between}html.theme--documenter-dark .level code{border-radius:.4em}html.theme--documenter-dark .level img{display:inline-block;vertical-align:top}html.theme--documenter-dark .level.is-mobile{display:flex}html.theme--documenter-dark .level.is-mobile .level-left,html.theme--documenter-dark .level.is-mobile .level-right{display:flex}html.theme--documenter-dark .level.is-mobile .level-left+.level-right{margin-top:0}html.theme--documenter-dark .level.is-mobile .level-item:not(:last-child){margin-bottom:0;margin-right:.75rem}html.theme--documenter-dark .level.is-mobile .level-item:not(.is-narrow){flex-grow:1}@media screen and (min-width: 769px),print{html.theme--documenter-dark .level{display:flex}html.theme--documenter-dark .level>.level-item:not(.is-narrow){flex-grow:1}}html.theme--documenter-dark .level-item{align-items:center;display:flex;flex-basis:auto;flex-grow:0;flex-shrink:0;justify-content:center}html.theme--documenter-dark .level-item .title,html.theme--documenter-dark .level-item .subtitle{margin-bottom:0}@media screen and (max-width: 768px){html.theme--documenter-dark .level-item:not(:last-child){margin-bottom:.75rem}}html.theme--documenter-dark .level-left,html.theme--documenter-dark .level-right{flex-basis:auto;flex-grow:0;flex-shrink:0}html.theme--documenter-dark .level-left .level-item.is-flexible,html.theme--documenter-dark .level-right .level-item.is-flexible{flex-grow:1}@media screen and (min-width: 769px),print{html.theme--documenter-dark .level-left .level-item:not(:last-child),html.theme--documenter-dark .level-right .level-item:not(:last-child){margin-right:.75rem}}html.theme--documenter-dark .level-left{align-items:center;justify-content:flex-start}@media screen and (max-width: 768px){html.theme--documenter-dark .level-left+.level-right{margin-top:1.5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .level-left{display:flex}}html.theme--documenter-dark .level-right{align-items:center;justify-content:flex-end}@media screen and (min-width: 769px),print{html.theme--documenter-dark .level-right{display:flex}}html.theme--documenter-dark .media{align-items:flex-start;display:flex;text-align:inherit}html.theme--documenter-dark .media .content:not(:last-child){margin-bottom:.75rem}html.theme--documenter-dark .media .media{border-top:1px solid rgba(94,109,111,0.5);display:flex;padding-top:.75rem}html.theme--documenter-dark .media .media .content:not(:last-child),html.theme--documenter-dark .media .media .control:not(:last-child){margin-bottom:.5rem}html.theme--documenter-dark .media .media .media{padding-top:.5rem}html.theme--documenter-dark .media .media .media+.media{margin-top:.5rem}html.theme--documenter-dark .media+.media{border-top:1px solid rgba(94,109,111,0.5);margin-top:1rem;padding-top:1rem}html.theme--documenter-dark .media.is-large+.media{margin-top:1.5rem;padding-top:1.5rem}html.theme--documenter-dark .media-left,html.theme--documenter-dark .media-right{flex-basis:auto;flex-grow:0;flex-shrink:0}html.theme--documenter-dark .media-left{margin-right:1rem}html.theme--documenter-dark .media-right{margin-left:1rem}html.theme--documenter-dark .media-content{flex-basis:auto;flex-grow:1;flex-shrink:1;text-align:inherit}@media screen and (max-width: 768px){html.theme--documenter-dark .media-content{overflow-x:auto}}html.theme--documenter-dark .menu{font-size:1rem}html.theme--documenter-dark .menu.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.menu{font-size:.75rem}html.theme--documenter-dark .menu.is-medium{font-size:1.25rem}html.theme--documenter-dark .menu.is-large{font-size:1.5rem}html.theme--documenter-dark .menu-list{line-height:1.25}html.theme--documenter-dark .menu-list a{border-radius:3px;color:#fff;display:block;padding:0.5em 0.75em}html.theme--documenter-dark .menu-list a:hover{background-color:#282f2f;color:#f2f2f2}html.theme--documenter-dark .menu-list a.is-active{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .menu-list li ul{border-left:1px solid #5e6d6f;margin:.75em;padding-left:.75em}html.theme--documenter-dark .menu-label{color:#fff;font-size:.75em;letter-spacing:.1em;text-transform:uppercase}html.theme--documenter-dark .menu-label:not(:first-child){margin-top:1em}html.theme--documenter-dark .menu-label:not(:last-child){margin-bottom:1em}html.theme--documenter-dark .message{background-color:#282f2f;border-radius:.4em;font-size:1rem}html.theme--documenter-dark .message strong{color:currentColor}html.theme--documenter-dark .message a:not(.button):not(.tag):not(.dropdown-item){color:currentColor;text-decoration:underline}html.theme--documenter-dark .message.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.message{font-size:.75rem}html.theme--documenter-dark .message.is-medium{font-size:1.25rem}html.theme--documenter-dark .message.is-large{font-size:1.5rem}html.theme--documenter-dark .message.is-white{background-color:#fff}html.theme--documenter-dark .message.is-white .message-header{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .message.is-white .message-body{border-color:#fff}html.theme--documenter-dark .message.is-black{background-color:#fafafa}html.theme--documenter-dark .message.is-black .message-header{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .message.is-black .message-body{border-color:#0a0a0a}html.theme--documenter-dark .message.is-light{background-color:#f9fafb}html.theme--documenter-dark .message.is-light .message-header{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .message.is-light .message-body{border-color:#ecf0f1}html.theme--documenter-dark .message.is-dark,html.theme--documenter-dark .content kbd.message{background-color:#f9fafa}html.theme--documenter-dark .message.is-dark .message-header,html.theme--documenter-dark .content kbd.message .message-header{background-color:#282f2f;color:#fff}html.theme--documenter-dark .message.is-dark .message-body,html.theme--documenter-dark .content kbd.message .message-body{border-color:#282f2f}html.theme--documenter-dark .message.is-primary,html.theme--documenter-dark .docstring>section>a.message.docs-sourcelink{background-color:#f1f5f9}html.theme--documenter-dark .message.is-primary .message-header,html.theme--documenter-dark .docstring>section>a.message.docs-sourcelink .message-header{background-color:#375a7f;color:#fff}html.theme--documenter-dark .message.is-primary .message-body,html.theme--documenter-dark .docstring>section>a.message.docs-sourcelink .message-body{border-color:#375a7f;color:#4d7eb2}html.theme--documenter-dark .message.is-link{background-color:#edfdf9}html.theme--documenter-dark .message.is-link .message-header{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .message.is-link .message-body{border-color:#1abc9c;color:#15987e}html.theme--documenter-dark .message.is-info{background-color:#ebf7ff}html.theme--documenter-dark .message.is-info .message-header{background-color:#024c7d;color:#fff}html.theme--documenter-dark .message.is-info .message-body{border-color:#024c7d;color:#0e9dfb}html.theme--documenter-dark .message.is-success{background-color:#ebfff3}html.theme--documenter-dark .message.is-success .message-header{background-color:#008438;color:#fff}html.theme--documenter-dark .message.is-success .message-body{border-color:#008438;color:#00eb64}html.theme--documenter-dark .message.is-warning{background-color:#fffaeb}html.theme--documenter-dark .message.is-warning .message-header{background-color:#ad8100;color:#fff}html.theme--documenter-dark .message.is-warning .message-body{border-color:#ad8100;color:#d19c00}html.theme--documenter-dark .message.is-danger{background-color:#fdeeec}html.theme--documenter-dark .message.is-danger .message-header{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .message.is-danger .message-body{border-color:#9e1b0d;color:#ec311d}html.theme--documenter-dark .message-header{align-items:center;background-color:#fff;border-radius:.4em .4em 0 0;color:rgba(0,0,0,0.7);display:flex;font-weight:700;justify-content:space-between;line-height:1.25;padding:0.75em 1em;position:relative}html.theme--documenter-dark .message-header .delete{flex-grow:0;flex-shrink:0;margin-left:.75em}html.theme--documenter-dark .message-header+.message-body{border-width:0;border-top-left-radius:0;border-top-right-radius:0}html.theme--documenter-dark .message-body{border-color:#5e6d6f;border-radius:.4em;border-style:solid;border-width:0 0 0 4px;color:#fff;padding:1.25em 1.5em}html.theme--documenter-dark .message-body code,html.theme--documenter-dark .message-body pre{background-color:#fff}html.theme--documenter-dark .message-body pre code{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .modal{align-items:center;display:none;flex-direction:column;justify-content:center;overflow:hidden;position:fixed;z-index:40}html.theme--documenter-dark .modal.is-active{display:flex}html.theme--documenter-dark .modal-background{background-color:rgba(10,10,10,0.86)}html.theme--documenter-dark .modal-content,html.theme--documenter-dark .modal-card{margin:0 20px;max-height:calc(100vh - 160px);overflow:auto;position:relative;width:100%}@media screen and (min-width: 769px){html.theme--documenter-dark .modal-content,html.theme--documenter-dark .modal-card{margin:0 auto;max-height:calc(100vh - 40px);width:640px}}html.theme--documenter-dark .modal-close{background:none;height:40px;position:fixed;right:20px;top:20px;width:40px}html.theme--documenter-dark .modal-card{display:flex;flex-direction:column;max-height:calc(100vh - 40px);overflow:hidden;-ms-overflow-y:visible}html.theme--documenter-dark .modal-card-head,html.theme--documenter-dark .modal-card-foot{align-items:center;background-color:#282f2f;display:flex;flex-shrink:0;justify-content:flex-start;padding:20px;position:relative}html.theme--documenter-dark .modal-card-head{border-bottom:1px solid #5e6d6f;border-top-left-radius:8px;border-top-right-radius:8px}html.theme--documenter-dark .modal-card-title{color:#f2f2f2;flex-grow:1;flex-shrink:0;font-size:1.5rem;line-height:1}html.theme--documenter-dark .modal-card-foot{border-bottom-left-radius:8px;border-bottom-right-radius:8px;border-top:1px solid #5e6d6f}html.theme--documenter-dark .modal-card-foot .button:not(:last-child){margin-right:.5em}html.theme--documenter-dark .modal-card-body{-webkit-overflow-scrolling:touch;background-color:#fff;flex-grow:1;flex-shrink:1;overflow:auto;padding:20px}html.theme--documenter-dark .navbar{background-color:#375a7f;min-height:4rem;position:relative;z-index:30}html.theme--documenter-dark .navbar.is-white{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link{color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-white .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-white .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link::after{border-color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-burger{color:#0a0a0a}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-white .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-white .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link{color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-white .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-white .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-white .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-white .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-white .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link::after{border-color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-white .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-white .navbar-item.has-dropdown.is-active .navbar-link{background-color:#f2f2f2;color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-dropdown a.navbar-item.is-active{background-color:#fff;color:#0a0a0a}}html.theme--documenter-dark .navbar.is-black{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-black .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-black .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link.is-active{background-color:#000;color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-black .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-black .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-black .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-black .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-black .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-black .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-black .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link.is-active{background-color:#000;color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-black .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-black .navbar-item.has-dropdown.is-active .navbar-link{background-color:#000;color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-dropdown a.navbar-item.is-active{background-color:#0a0a0a;color:#fff}}html.theme--documenter-dark .navbar.is-light{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-light .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-light .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link.is-active{background-color:#dde4e6;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link::after{border-color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-burger{color:rgba(0,0,0,0.7)}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-light .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-light .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-light .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-light .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-light .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-light .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-light .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link.is-active{background-color:#dde4e6;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link::after{border-color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-light .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-light .navbar-item.has-dropdown.is-active .navbar-link{background-color:#dde4e6;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-dropdown a.navbar-item.is-active{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}}html.theme--documenter-dark .navbar.is-dark,html.theme--documenter-dark .content kbd.navbar{background-color:#282f2f;color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-brand>.navbar-item,html.theme--documenter-dark .content kbd.navbar .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .content kbd.navbar .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-dark .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .content kbd.navbar .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-dark .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link:focus,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link:hover,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link.is-active{background-color:#1d2122;color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link::after,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-burger,html.theme--documenter-dark .content kbd.navbar .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-dark .navbar-start>.navbar-item,html.theme--documenter-dark .content kbd.navbar .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-dark .navbar-end>.navbar-item,html.theme--documenter-dark .content kbd.navbar .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .content kbd.navbar .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-dark .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .content kbd.navbar .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-dark .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link:focus,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link:hover,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-dark .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .content kbd.navbar .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-dark .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .content kbd.navbar .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-dark .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link:focus,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link:hover,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link.is-active{background-color:#1d2122;color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link::after,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link::after,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-dark .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-dark .navbar-item.has-dropdown.is-active .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-item.has-dropdown.is-active .navbar-link{background-color:#1d2122;color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-dropdown a.navbar-item.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-dropdown a.navbar-item.is-active{background-color:#282f2f;color:#fff}}html.theme--documenter-dark .navbar.is-primary,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink{background-color:#375a7f;color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-brand>.navbar-item,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-primary .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-primary .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link.is-active{background-color:#2f4d6d;color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link::after,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-burger,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-primary .navbar-start>.navbar-item,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-primary .navbar-end>.navbar-item,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-primary .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-primary .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-primary .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-primary .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-primary .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link.is-active{background-color:#2f4d6d;color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link::after,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link::after,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-primary .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-primary .navbar-item.has-dropdown.is-active .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown.is-active .navbar-link{background-color:#2f4d6d;color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-dropdown a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-dropdown a.navbar-item.is-active{background-color:#375a7f;color:#fff}}html.theme--documenter-dark .navbar.is-link{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-link .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-link .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link.is-active{background-color:#17a689;color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-link .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-link .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-link .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-link .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-link .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-link .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-link .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link.is-active{background-color:#17a689;color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-link .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-link .navbar-item.has-dropdown.is-active .navbar-link{background-color:#17a689;color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-dropdown a.navbar-item.is-active{background-color:#1abc9c;color:#fff}}html.theme--documenter-dark .navbar.is-info{background-color:#024c7d;color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-info .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-info .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link.is-active{background-color:#023d64;color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-info .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-info .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-info .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-info .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-info .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-info .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-info .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link.is-active{background-color:#023d64;color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-info .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-info .navbar-item.has-dropdown.is-active .navbar-link{background-color:#023d64;color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-dropdown a.navbar-item.is-active{background-color:#024c7d;color:#fff}}html.theme--documenter-dark .navbar.is-success{background-color:#008438;color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-success .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-success .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link.is-active{background-color:#006b2d;color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-success .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-success .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-success .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-success .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-success .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-success .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-success .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link.is-active{background-color:#006b2d;color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-success .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-success .navbar-item.has-dropdown.is-active .navbar-link{background-color:#006b2d;color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-dropdown a.navbar-item.is-active{background-color:#008438;color:#fff}}html.theme--documenter-dark .navbar.is-warning{background-color:#ad8100;color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-warning .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-warning .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link.is-active{background-color:#946e00;color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-warning .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-warning .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-warning .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-warning .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-warning .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-warning .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-warning .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link.is-active{background-color:#946e00;color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-warning .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-warning .navbar-item.has-dropdown.is-active .navbar-link{background-color:#946e00;color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-dropdown a.navbar-item.is-active{background-color:#ad8100;color:#fff}}html.theme--documenter-dark .navbar.is-danger{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-danger .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-danger .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link.is-active{background-color:#86170b;color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-danger .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-danger .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-danger .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-danger .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-danger .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-danger .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-danger .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link.is-active{background-color:#86170b;color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-danger .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-danger .navbar-item.has-dropdown.is-active .navbar-link{background-color:#86170b;color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-dropdown a.navbar-item.is-active{background-color:#9e1b0d;color:#fff}}html.theme--documenter-dark .navbar>.container{align-items:stretch;display:flex;min-height:4rem;width:100%}html.theme--documenter-dark .navbar.has-shadow{box-shadow:0 2px 0 0 #282f2f}html.theme--documenter-dark .navbar.is-fixed-bottom,html.theme--documenter-dark .navbar.is-fixed-top{left:0;position:fixed;right:0;z-index:30}html.theme--documenter-dark .navbar.is-fixed-bottom{bottom:0}html.theme--documenter-dark .navbar.is-fixed-bottom.has-shadow{box-shadow:0 -2px 0 0 #282f2f}html.theme--documenter-dark .navbar.is-fixed-top{top:0}html.theme--documenter-dark html.has-navbar-fixed-top,html.theme--documenter-dark body.has-navbar-fixed-top{padding-top:4rem}html.theme--documenter-dark html.has-navbar-fixed-bottom,html.theme--documenter-dark body.has-navbar-fixed-bottom{padding-bottom:4rem}html.theme--documenter-dark .navbar-brand,html.theme--documenter-dark .navbar-tabs{align-items:stretch;display:flex;flex-shrink:0;min-height:4rem}html.theme--documenter-dark .navbar-brand a.navbar-item:focus,html.theme--documenter-dark .navbar-brand a.navbar-item:hover{background-color:transparent}html.theme--documenter-dark .navbar-tabs{-webkit-overflow-scrolling:touch;max-width:100vw;overflow-x:auto;overflow-y:hidden}html.theme--documenter-dark .navbar-burger{color:#fff;-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;cursor:pointer;display:block;height:4rem;position:relative;width:4rem;margin-left:auto}html.theme--documenter-dark .navbar-burger span{background-color:currentColor;display:block;height:1px;left:calc(50% - 8px);position:absolute;transform-origin:center;transition-duration:86ms;transition-property:background-color, opacity, transform;transition-timing-function:ease-out;width:16px}html.theme--documenter-dark .navbar-burger span:nth-child(1){top:calc(50% - 6px)}html.theme--documenter-dark .navbar-burger span:nth-child(2){top:calc(50% - 1px)}html.theme--documenter-dark .navbar-burger span:nth-child(3){top:calc(50% + 4px)}html.theme--documenter-dark .navbar-burger:hover{background-color:rgba(0,0,0,0.05)}html.theme--documenter-dark .navbar-burger.is-active span:nth-child(1){transform:translateY(5px) rotate(45deg)}html.theme--documenter-dark .navbar-burger.is-active span:nth-child(2){opacity:0}html.theme--documenter-dark .navbar-burger.is-active span:nth-child(3){transform:translateY(-5px) rotate(-45deg)}html.theme--documenter-dark .navbar-menu{display:none}html.theme--documenter-dark .navbar-item,html.theme--documenter-dark .navbar-link{color:#fff;display:block;line-height:1.5;padding:0.5rem 0.75rem;position:relative}html.theme--documenter-dark .navbar-item .icon:only-child,html.theme--documenter-dark .navbar-link .icon:only-child{margin-left:-0.25rem;margin-right:-0.25rem}html.theme--documenter-dark a.navbar-item,html.theme--documenter-dark .navbar-link{cursor:pointer}html.theme--documenter-dark a.navbar-item:focus,html.theme--documenter-dark a.navbar-item:focus-within,html.theme--documenter-dark a.navbar-item:hover,html.theme--documenter-dark a.navbar-item.is-active,html.theme--documenter-dark .navbar-link:focus,html.theme--documenter-dark .navbar-link:focus-within,html.theme--documenter-dark .navbar-link:hover,html.theme--documenter-dark .navbar-link.is-active{background-color:rgba(0,0,0,0);color:#1abc9c}html.theme--documenter-dark .navbar-item{flex-grow:0;flex-shrink:0}html.theme--documenter-dark .navbar-item img{max-height:1.75rem}html.theme--documenter-dark .navbar-item.has-dropdown{padding:0}html.theme--documenter-dark .navbar-item.is-expanded{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .navbar-item.is-tab{border-bottom:1px solid transparent;min-height:4rem;padding-bottom:calc(0.5rem - 1px)}html.theme--documenter-dark .navbar-item.is-tab:focus,html.theme--documenter-dark .navbar-item.is-tab:hover{background-color:rgba(0,0,0,0);border-bottom-color:#1abc9c}html.theme--documenter-dark .navbar-item.is-tab.is-active{background-color:rgba(0,0,0,0);border-bottom-color:#1abc9c;border-bottom-style:solid;border-bottom-width:3px;color:#1abc9c;padding-bottom:calc(0.5rem - 3px)}html.theme--documenter-dark .navbar-content{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .navbar-link:not(.is-arrowless){padding-right:2.5em}html.theme--documenter-dark .navbar-link:not(.is-arrowless)::after{border-color:#fff;margin-top:-0.375em;right:1.125em}html.theme--documenter-dark .navbar-dropdown{font-size:0.875rem;padding-bottom:0.5rem;padding-top:0.5rem}html.theme--documenter-dark .navbar-dropdown .navbar-item{padding-left:1.5rem;padding-right:1.5rem}html.theme--documenter-dark .navbar-divider{background-color:rgba(0,0,0,0.2);border:none;display:none;height:2px;margin:0.5rem 0}@media screen and (max-width: 1055px){html.theme--documenter-dark .navbar>.container{display:block}html.theme--documenter-dark .navbar-brand .navbar-item,html.theme--documenter-dark .navbar-tabs .navbar-item{align-items:center;display:flex}html.theme--documenter-dark .navbar-link::after{display:none}html.theme--documenter-dark .navbar-menu{background-color:#375a7f;box-shadow:0 8px 16px rgba(10,10,10,0.1);padding:0.5rem 0}html.theme--documenter-dark .navbar-menu.is-active{display:block}html.theme--documenter-dark .navbar.is-fixed-bottom-touch,html.theme--documenter-dark .navbar.is-fixed-top-touch{left:0;position:fixed;right:0;z-index:30}html.theme--documenter-dark .navbar.is-fixed-bottom-touch{bottom:0}html.theme--documenter-dark .navbar.is-fixed-bottom-touch.has-shadow{box-shadow:0 -2px 3px rgba(10,10,10,0.1)}html.theme--documenter-dark .navbar.is-fixed-top-touch{top:0}html.theme--documenter-dark .navbar.is-fixed-top .navbar-menu,html.theme--documenter-dark .navbar.is-fixed-top-touch .navbar-menu{-webkit-overflow-scrolling:touch;max-height:calc(100vh - 4rem);overflow:auto}html.theme--documenter-dark html.has-navbar-fixed-top-touch,html.theme--documenter-dark body.has-navbar-fixed-top-touch{padding-top:4rem}html.theme--documenter-dark html.has-navbar-fixed-bottom-touch,html.theme--documenter-dark body.has-navbar-fixed-bottom-touch{padding-bottom:4rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar,html.theme--documenter-dark .navbar-menu,html.theme--documenter-dark .navbar-start,html.theme--documenter-dark .navbar-end{align-items:stretch;display:flex}html.theme--documenter-dark .navbar{min-height:4rem}html.theme--documenter-dark .navbar.is-spaced{padding:1rem 2rem}html.theme--documenter-dark .navbar.is-spaced .navbar-start,html.theme--documenter-dark .navbar.is-spaced .navbar-end{align-items:center}html.theme--documenter-dark .navbar.is-spaced a.navbar-item,html.theme--documenter-dark .navbar.is-spaced .navbar-link{border-radius:.4em}html.theme--documenter-dark .navbar.is-transparent a.navbar-item:focus,html.theme--documenter-dark .navbar.is-transparent a.navbar-item:hover,html.theme--documenter-dark .navbar.is-transparent a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-transparent .navbar-link:focus,html.theme--documenter-dark .navbar.is-transparent .navbar-link:hover,html.theme--documenter-dark .navbar.is-transparent .navbar-link.is-active{background-color:transparent !important}html.theme--documenter-dark .navbar.is-transparent .navbar-item.has-dropdown.is-active .navbar-link,html.theme--documenter-dark .navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:focus .navbar-link,html.theme--documenter-dark .navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:focus-within .navbar-link,html.theme--documenter-dark .navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:hover .navbar-link{background-color:transparent !important}html.theme--documenter-dark .navbar.is-transparent .navbar-dropdown a.navbar-item:focus,html.theme--documenter-dark .navbar.is-transparent .navbar-dropdown a.navbar-item:hover{background-color:rgba(0,0,0,0);color:#dbdee0}html.theme--documenter-dark .navbar.is-transparent .navbar-dropdown a.navbar-item.is-active{background-color:rgba(0,0,0,0);color:#1abc9c}html.theme--documenter-dark .navbar-burger{display:none}html.theme--documenter-dark .navbar-item,html.theme--documenter-dark .navbar-link{align-items:center;display:flex}html.theme--documenter-dark .navbar-item.has-dropdown{align-items:stretch}html.theme--documenter-dark .navbar-item.has-dropdown-up .navbar-link::after{transform:rotate(135deg) translate(0.25em, -0.25em)}html.theme--documenter-dark .navbar-item.has-dropdown-up .navbar-dropdown{border-bottom:1px solid rgba(0,0,0,0.2);border-radius:8px 8px 0 0;border-top:none;bottom:100%;box-shadow:0 -8px 8px rgba(10,10,10,0.1);top:auto}html.theme--documenter-dark .navbar-item.is-active .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:focus .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:focus-within .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:hover .navbar-dropdown{display:block}.navbar.is-spaced html.theme--documenter-dark .navbar-item.is-active .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-active .navbar-dropdown.is-boxed,.navbar.is-spaced html.theme--documenter-dark .navbar-item.is-hoverable:focus .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:focus .navbar-dropdown.is-boxed,.navbar.is-spaced html.theme--documenter-dark .navbar-item.is-hoverable:focus-within .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:focus-within .navbar-dropdown.is-boxed,.navbar.is-spaced html.theme--documenter-dark .navbar-item.is-hoverable:hover .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:hover .navbar-dropdown.is-boxed{opacity:1;pointer-events:auto;transform:translateY(0)}html.theme--documenter-dark .navbar-menu{flex-grow:1;flex-shrink:0}html.theme--documenter-dark .navbar-start{justify-content:flex-start;margin-right:auto}html.theme--documenter-dark .navbar-end{justify-content:flex-end;margin-left:auto}html.theme--documenter-dark .navbar-dropdown{background-color:#375a7f;border-bottom-left-radius:8px;border-bottom-right-radius:8px;border-top:1px solid rgba(0,0,0,0.2);box-shadow:0 8px 8px rgba(10,10,10,0.1);display:none;font-size:0.875rem;left:0;min-width:100%;position:absolute;top:100%;z-index:20}html.theme--documenter-dark .navbar-dropdown .navbar-item{padding:0.375rem 1rem;white-space:nowrap}html.theme--documenter-dark .navbar-dropdown a.navbar-item{padding-right:3rem}html.theme--documenter-dark .navbar-dropdown a.navbar-item:focus,html.theme--documenter-dark .navbar-dropdown a.navbar-item:hover{background-color:rgba(0,0,0,0);color:#dbdee0}html.theme--documenter-dark .navbar-dropdown a.navbar-item.is-active{background-color:rgba(0,0,0,0);color:#1abc9c}.navbar.is-spaced html.theme--documenter-dark .navbar-dropdown,html.theme--documenter-dark .navbar-dropdown.is-boxed{border-radius:8px;border-top:none;box-shadow:0 8px 8px rgba(10,10,10,0.1), 0 0 0 1px rgba(10,10,10,0.1);display:block;opacity:0;pointer-events:none;top:calc(100% + (-4px));transform:translateY(-5px);transition-duration:86ms;transition-property:opacity, transform}html.theme--documenter-dark .navbar-dropdown.is-right{left:auto;right:0}html.theme--documenter-dark .navbar-divider{display:block}html.theme--documenter-dark .navbar>.container .navbar-brand,html.theme--documenter-dark .container>.navbar .navbar-brand{margin-left:-.75rem}html.theme--documenter-dark .navbar>.container .navbar-menu,html.theme--documenter-dark .container>.navbar .navbar-menu{margin-right:-.75rem}html.theme--documenter-dark .navbar.is-fixed-bottom-desktop,html.theme--documenter-dark .navbar.is-fixed-top-desktop{left:0;position:fixed;right:0;z-index:30}html.theme--documenter-dark .navbar.is-fixed-bottom-desktop{bottom:0}html.theme--documenter-dark .navbar.is-fixed-bottom-desktop.has-shadow{box-shadow:0 -2px 3px rgba(10,10,10,0.1)}html.theme--documenter-dark .navbar.is-fixed-top-desktop{top:0}html.theme--documenter-dark html.has-navbar-fixed-top-desktop,html.theme--documenter-dark body.has-navbar-fixed-top-desktop{padding-top:4rem}html.theme--documenter-dark html.has-navbar-fixed-bottom-desktop,html.theme--documenter-dark body.has-navbar-fixed-bottom-desktop{padding-bottom:4rem}html.theme--documenter-dark html.has-spaced-navbar-fixed-top,html.theme--documenter-dark body.has-spaced-navbar-fixed-top{padding-top:6rem}html.theme--documenter-dark html.has-spaced-navbar-fixed-bottom,html.theme--documenter-dark body.has-spaced-navbar-fixed-bottom{padding-bottom:6rem}html.theme--documenter-dark a.navbar-item.is-active,html.theme--documenter-dark .navbar-link.is-active{color:#1abc9c}html.theme--documenter-dark a.navbar-item.is-active:not(:focus):not(:hover),html.theme--documenter-dark .navbar-link.is-active:not(:focus):not(:hover){background-color:rgba(0,0,0,0)}html.theme--documenter-dark .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar-item.has-dropdown.is-active .navbar-link{background-color:rgba(0,0,0,0)}}html.theme--documenter-dark .hero.is-fullheight-with-navbar{min-height:calc(100vh - 4rem)}html.theme--documenter-dark .pagination{font-size:1rem;margin:-.25rem}html.theme--documenter-dark .pagination.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.pagination{font-size:.75rem}html.theme--documenter-dark .pagination.is-medium{font-size:1.25rem}html.theme--documenter-dark .pagination.is-large{font-size:1.5rem}html.theme--documenter-dark .pagination.is-rounded .pagination-previous,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.pagination .pagination-previous,html.theme--documenter-dark .pagination.is-rounded .pagination-next,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.pagination .pagination-next{padding-left:1em;padding-right:1em;border-radius:9999px}html.theme--documenter-dark .pagination.is-rounded .pagination-link,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.pagination .pagination-link{border-radius:9999px}html.theme--documenter-dark .pagination,html.theme--documenter-dark .pagination-list{align-items:center;display:flex;justify-content:center;text-align:center}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-ellipsis{font-size:1em;justify-content:center;margin:.25rem;padding-left:.5em;padding-right:.5em;text-align:center}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link{border-color:#5e6d6f;color:#1abc9c;min-width:2.5em}html.theme--documenter-dark .pagination-previous:hover,html.theme--documenter-dark .pagination-next:hover,html.theme--documenter-dark .pagination-link:hover{border-color:#8c9b9d;color:#1dd2af}html.theme--documenter-dark .pagination-previous:focus,html.theme--documenter-dark .pagination-next:focus,html.theme--documenter-dark .pagination-link:focus{border-color:#8c9b9d}html.theme--documenter-dark .pagination-previous:active,html.theme--documenter-dark .pagination-next:active,html.theme--documenter-dark .pagination-link:active{box-shadow:inset 0 1px 2px rgba(10,10,10,0.2)}html.theme--documenter-dark .pagination-previous[disabled],html.theme--documenter-dark .pagination-previous.is-disabled,html.theme--documenter-dark .pagination-next[disabled],html.theme--documenter-dark .pagination-next.is-disabled,html.theme--documenter-dark .pagination-link[disabled],html.theme--documenter-dark .pagination-link.is-disabled{background-color:#5e6d6f;border-color:#5e6d6f;box-shadow:none;color:#fff;opacity:0.5}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next{padding-left:.75em;padding-right:.75em;white-space:nowrap}html.theme--documenter-dark .pagination-link.is-current{background-color:#1abc9c;border-color:#1abc9c;color:#fff}html.theme--documenter-dark .pagination-ellipsis{color:#8c9b9d;pointer-events:none}html.theme--documenter-dark .pagination-list{flex-wrap:wrap}html.theme--documenter-dark .pagination-list li{list-style:none}@media screen and (max-width: 768px){html.theme--documenter-dark .pagination{flex-wrap:wrap}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .pagination-list li{flex-grow:1;flex-shrink:1}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .pagination-list{flex-grow:1;flex-shrink:1;justify-content:flex-start;order:1}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-ellipsis{margin-bottom:0;margin-top:0}html.theme--documenter-dark .pagination-previous{order:2}html.theme--documenter-dark .pagination-next{order:3}html.theme--documenter-dark .pagination{justify-content:space-between;margin-bottom:0;margin-top:0}html.theme--documenter-dark .pagination.is-centered .pagination-previous{order:1}html.theme--documenter-dark .pagination.is-centered .pagination-list{justify-content:center;order:2}html.theme--documenter-dark .pagination.is-centered .pagination-next{order:3}html.theme--documenter-dark .pagination.is-right .pagination-previous{order:1}html.theme--documenter-dark .pagination.is-right .pagination-next{order:2}html.theme--documenter-dark .pagination.is-right .pagination-list{justify-content:flex-end;order:3}}html.theme--documenter-dark .panel{border-radius:8px;box-shadow:#171717;font-size:1rem}html.theme--documenter-dark .panel:not(:last-child){margin-bottom:1.5rem}html.theme--documenter-dark .panel.is-white .panel-heading{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .panel.is-white .panel-tabs a.is-active{border-bottom-color:#fff}html.theme--documenter-dark .panel.is-white .panel-block.is-active .panel-icon{color:#fff}html.theme--documenter-dark .panel.is-black .panel-heading{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .panel.is-black .panel-tabs a.is-active{border-bottom-color:#0a0a0a}html.theme--documenter-dark .panel.is-black .panel-block.is-active .panel-icon{color:#0a0a0a}html.theme--documenter-dark .panel.is-light .panel-heading{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .panel.is-light .panel-tabs a.is-active{border-bottom-color:#ecf0f1}html.theme--documenter-dark .panel.is-light .panel-block.is-active .panel-icon{color:#ecf0f1}html.theme--documenter-dark .panel.is-dark .panel-heading,html.theme--documenter-dark .content kbd.panel .panel-heading{background-color:#282f2f;color:#fff}html.theme--documenter-dark .panel.is-dark .panel-tabs a.is-active,html.theme--documenter-dark .content kbd.panel .panel-tabs a.is-active{border-bottom-color:#282f2f}html.theme--documenter-dark .panel.is-dark .panel-block.is-active .panel-icon,html.theme--documenter-dark .content kbd.panel .panel-block.is-active .panel-icon{color:#282f2f}html.theme--documenter-dark .panel.is-primary .panel-heading,html.theme--documenter-dark .docstring>section>a.panel.docs-sourcelink .panel-heading{background-color:#375a7f;color:#fff}html.theme--documenter-dark .panel.is-primary .panel-tabs a.is-active,html.theme--documenter-dark .docstring>section>a.panel.docs-sourcelink .panel-tabs a.is-active{border-bottom-color:#375a7f}html.theme--documenter-dark .panel.is-primary .panel-block.is-active .panel-icon,html.theme--documenter-dark .docstring>section>a.panel.docs-sourcelink .panel-block.is-active .panel-icon{color:#375a7f}html.theme--documenter-dark .panel.is-link .panel-heading{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .panel.is-link .panel-tabs a.is-active{border-bottom-color:#1abc9c}html.theme--documenter-dark .panel.is-link .panel-block.is-active .panel-icon{color:#1abc9c}html.theme--documenter-dark .panel.is-info .panel-heading{background-color:#024c7d;color:#fff}html.theme--documenter-dark .panel.is-info .panel-tabs a.is-active{border-bottom-color:#024c7d}html.theme--documenter-dark .panel.is-info .panel-block.is-active .panel-icon{color:#024c7d}html.theme--documenter-dark .panel.is-success .panel-heading{background-color:#008438;color:#fff}html.theme--documenter-dark .panel.is-success .panel-tabs a.is-active{border-bottom-color:#008438}html.theme--documenter-dark .panel.is-success .panel-block.is-active .panel-icon{color:#008438}html.theme--documenter-dark .panel.is-warning .panel-heading{background-color:#ad8100;color:#fff}html.theme--documenter-dark .panel.is-warning .panel-tabs a.is-active{border-bottom-color:#ad8100}html.theme--documenter-dark .panel.is-warning .panel-block.is-active .panel-icon{color:#ad8100}html.theme--documenter-dark .panel.is-danger .panel-heading{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .panel.is-danger .panel-tabs a.is-active{border-bottom-color:#9e1b0d}html.theme--documenter-dark .panel.is-danger .panel-block.is-active .panel-icon{color:#9e1b0d}html.theme--documenter-dark .panel-tabs:not(:last-child),html.theme--documenter-dark .panel-block:not(:last-child){border-bottom:1px solid #ededed}html.theme--documenter-dark .panel-heading{background-color:#343c3d;border-radius:8px 8px 0 0;color:#f2f2f2;font-size:1.25em;font-weight:700;line-height:1.25;padding:0.75em 1em}html.theme--documenter-dark .panel-tabs{align-items:flex-end;display:flex;font-size:.875em;justify-content:center}html.theme--documenter-dark .panel-tabs a{border-bottom:1px solid #5e6d6f;margin-bottom:-1px;padding:0.5em}html.theme--documenter-dark .panel-tabs a.is-active{border-bottom-color:#343c3d;color:#17a689}html.theme--documenter-dark .panel-list a{color:#fff}html.theme--documenter-dark .panel-list a:hover{color:#1abc9c}html.theme--documenter-dark .panel-block{align-items:center;color:#f2f2f2;display:flex;justify-content:flex-start;padding:0.5em 0.75em}html.theme--documenter-dark .panel-block input[type="checkbox"]{margin-right:.75em}html.theme--documenter-dark .panel-block>.control{flex-grow:1;flex-shrink:1;width:100%}html.theme--documenter-dark .panel-block.is-wrapped{flex-wrap:wrap}html.theme--documenter-dark .panel-block.is-active{border-left-color:#1abc9c;color:#17a689}html.theme--documenter-dark .panel-block.is-active .panel-icon{color:#1abc9c}html.theme--documenter-dark .panel-block:last-child{border-bottom-left-radius:8px;border-bottom-right-radius:8px}html.theme--documenter-dark a.panel-block,html.theme--documenter-dark label.panel-block{cursor:pointer}html.theme--documenter-dark a.panel-block:hover,html.theme--documenter-dark label.panel-block:hover{background-color:#282f2f}html.theme--documenter-dark .panel-icon{display:inline-block;font-size:14px;height:1em;line-height:1em;text-align:center;vertical-align:top;width:1em;color:#fff;margin-right:.75em}html.theme--documenter-dark .panel-icon .fa{font-size:inherit;line-height:inherit}html.theme--documenter-dark .tabs{-webkit-overflow-scrolling:touch;align-items:stretch;display:flex;font-size:1rem;justify-content:space-between;overflow:hidden;overflow-x:auto;white-space:nowrap}html.theme--documenter-dark .tabs a{align-items:center;border-bottom-color:#5e6d6f;border-bottom-style:solid;border-bottom-width:1px;color:#fff;display:flex;justify-content:center;margin-bottom:-1px;padding:0.5em 1em;vertical-align:top}html.theme--documenter-dark .tabs a:hover{border-bottom-color:#f2f2f2;color:#f2f2f2}html.theme--documenter-dark .tabs li{display:block}html.theme--documenter-dark .tabs li.is-active a{border-bottom-color:#1abc9c;color:#1abc9c}html.theme--documenter-dark .tabs ul{align-items:center;border-bottom-color:#5e6d6f;border-bottom-style:solid;border-bottom-width:1px;display:flex;flex-grow:1;flex-shrink:0;justify-content:flex-start}html.theme--documenter-dark .tabs ul.is-left{padding-right:0.75em}html.theme--documenter-dark .tabs ul.is-center{flex:none;justify-content:center;padding-left:0.75em;padding-right:0.75em}html.theme--documenter-dark .tabs ul.is-right{justify-content:flex-end;padding-left:0.75em}html.theme--documenter-dark .tabs .icon:first-child{margin-right:.5em}html.theme--documenter-dark .tabs .icon:last-child{margin-left:.5em}html.theme--documenter-dark .tabs.is-centered ul{justify-content:center}html.theme--documenter-dark .tabs.is-right ul{justify-content:flex-end}html.theme--documenter-dark .tabs.is-boxed a{border:1px solid transparent;border-radius:.4em .4em 0 0}html.theme--documenter-dark .tabs.is-boxed a:hover{background-color:#282f2f;border-bottom-color:#5e6d6f}html.theme--documenter-dark .tabs.is-boxed li.is-active a{background-color:#fff;border-color:#5e6d6f;border-bottom-color:rgba(0,0,0,0) !important}html.theme--documenter-dark .tabs.is-fullwidth li{flex-grow:1;flex-shrink:0}html.theme--documenter-dark .tabs.is-toggle a{border-color:#5e6d6f;border-style:solid;border-width:1px;margin-bottom:0;position:relative}html.theme--documenter-dark .tabs.is-toggle a:hover{background-color:#282f2f;border-color:#8c9b9d;z-index:2}html.theme--documenter-dark .tabs.is-toggle li+li{margin-left:-1px}html.theme--documenter-dark .tabs.is-toggle li:first-child a{border-top-left-radius:.4em;border-bottom-left-radius:.4em}html.theme--documenter-dark .tabs.is-toggle li:last-child a{border-top-right-radius:.4em;border-bottom-right-radius:.4em}html.theme--documenter-dark .tabs.is-toggle li.is-active a{background-color:#1abc9c;border-color:#1abc9c;color:#fff;z-index:1}html.theme--documenter-dark .tabs.is-toggle ul{border-bottom:none}html.theme--documenter-dark .tabs.is-toggle.is-toggle-rounded li:first-child a{border-bottom-left-radius:9999px;border-top-left-radius:9999px;padding-left:1.25em}html.theme--documenter-dark .tabs.is-toggle.is-toggle-rounded li:last-child a{border-bottom-right-radius:9999px;border-top-right-radius:9999px;padding-right:1.25em}html.theme--documenter-dark .tabs.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.tabs{font-size:.75rem}html.theme--documenter-dark .tabs.is-medium{font-size:1.25rem}html.theme--documenter-dark .tabs.is-large{font-size:1.5rem}html.theme--documenter-dark .column{display:block;flex-basis:0;flex-grow:1;flex-shrink:1;padding:.75rem}.columns.is-mobile>html.theme--documenter-dark .column.is-narrow{flex:none;width:unset}.columns.is-mobile>html.theme--documenter-dark .column.is-full{flex:none;width:100%}.columns.is-mobile>html.theme--documenter-dark .column.is-three-quarters{flex:none;width:75%}.columns.is-mobile>html.theme--documenter-dark .column.is-two-thirds{flex:none;width:66.6666%}.columns.is-mobile>html.theme--documenter-dark .column.is-half{flex:none;width:50%}.columns.is-mobile>html.theme--documenter-dark .column.is-one-third{flex:none;width:33.3333%}.columns.is-mobile>html.theme--documenter-dark .column.is-one-quarter{flex:none;width:25%}.columns.is-mobile>html.theme--documenter-dark .column.is-one-fifth{flex:none;width:20%}.columns.is-mobile>html.theme--documenter-dark .column.is-two-fifths{flex:none;width:40%}.columns.is-mobile>html.theme--documenter-dark .column.is-three-fifths{flex:none;width:60%}.columns.is-mobile>html.theme--documenter-dark .column.is-four-fifths{flex:none;width:80%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-three-quarters{margin-left:75%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-two-thirds{margin-left:66.6666%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-half{margin-left:50%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-one-third{margin-left:33.3333%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-one-quarter{margin-left:25%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-one-fifth{margin-left:20%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-two-fifths{margin-left:40%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-three-fifths{margin-left:60%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-four-fifths{margin-left:80%}.columns.is-mobile>html.theme--documenter-dark .column.is-0{flex:none;width:0%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-0{margin-left:0%}.columns.is-mobile>html.theme--documenter-dark .column.is-1{flex:none;width:8.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-1{margin-left:8.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-2{flex:none;width:16.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-2{margin-left:16.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-3{flex:none;width:25%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-3{margin-left:25%}.columns.is-mobile>html.theme--documenter-dark .column.is-4{flex:none;width:33.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-4{margin-left:33.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-5{flex:none;width:41.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-5{margin-left:41.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-6{flex:none;width:50%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-6{margin-left:50%}.columns.is-mobile>html.theme--documenter-dark .column.is-7{flex:none;width:58.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-7{margin-left:58.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-8{flex:none;width:66.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-8{margin-left:66.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-9{flex:none;width:75%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-9{margin-left:75%}.columns.is-mobile>html.theme--documenter-dark .column.is-10{flex:none;width:83.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-10{margin-left:83.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-11{flex:none;width:91.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-11{margin-left:91.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-12{flex:none;width:100%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-12{margin-left:100%}@media screen and (max-width: 768px){html.theme--documenter-dark .column.is-narrow-mobile{flex:none;width:unset}html.theme--documenter-dark .column.is-full-mobile{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-mobile{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-mobile{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-mobile{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-mobile{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-mobile{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-mobile{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-mobile{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-mobile{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-mobile{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-mobile{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-mobile{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-mobile{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-mobile{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-mobile{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-mobile{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-mobile{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-mobile{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-mobile{margin-left:80%}html.theme--documenter-dark .column.is-0-mobile{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-mobile{margin-left:0%}html.theme--documenter-dark .column.is-1-mobile{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-mobile{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-mobile{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-mobile{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-mobile{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-mobile{margin-left:25%}html.theme--documenter-dark .column.is-4-mobile{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-mobile{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-mobile{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-mobile{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-mobile{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-mobile{margin-left:50%}html.theme--documenter-dark .column.is-7-mobile{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-mobile{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-mobile{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-mobile{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-mobile{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-mobile{margin-left:75%}html.theme--documenter-dark .column.is-10-mobile{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-mobile{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-mobile{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-mobile{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-mobile{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-mobile{margin-left:100%}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .column.is-narrow,html.theme--documenter-dark .column.is-narrow-tablet{flex:none;width:unset}html.theme--documenter-dark .column.is-full,html.theme--documenter-dark .column.is-full-tablet{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters,html.theme--documenter-dark .column.is-three-quarters-tablet{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds,html.theme--documenter-dark .column.is-two-thirds-tablet{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half,html.theme--documenter-dark .column.is-half-tablet{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third,html.theme--documenter-dark .column.is-one-third-tablet{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter,html.theme--documenter-dark .column.is-one-quarter-tablet{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth,html.theme--documenter-dark .column.is-one-fifth-tablet{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths,html.theme--documenter-dark .column.is-two-fifths-tablet{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths,html.theme--documenter-dark .column.is-three-fifths-tablet{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths,html.theme--documenter-dark .column.is-four-fifths-tablet{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters,html.theme--documenter-dark .column.is-offset-three-quarters-tablet{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds,html.theme--documenter-dark .column.is-offset-two-thirds-tablet{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half,html.theme--documenter-dark .column.is-offset-half-tablet{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third,html.theme--documenter-dark .column.is-offset-one-third-tablet{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter,html.theme--documenter-dark .column.is-offset-one-quarter-tablet{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth,html.theme--documenter-dark .column.is-offset-one-fifth-tablet{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths,html.theme--documenter-dark .column.is-offset-two-fifths-tablet{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths,html.theme--documenter-dark .column.is-offset-three-fifths-tablet{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths,html.theme--documenter-dark .column.is-offset-four-fifths-tablet{margin-left:80%}html.theme--documenter-dark .column.is-0,html.theme--documenter-dark .column.is-0-tablet{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0,html.theme--documenter-dark .column.is-offset-0-tablet{margin-left:0%}html.theme--documenter-dark .column.is-1,html.theme--documenter-dark .column.is-1-tablet{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1,html.theme--documenter-dark .column.is-offset-1-tablet{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2,html.theme--documenter-dark .column.is-2-tablet{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2,html.theme--documenter-dark .column.is-offset-2-tablet{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3,html.theme--documenter-dark .column.is-3-tablet{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3,html.theme--documenter-dark .column.is-offset-3-tablet{margin-left:25%}html.theme--documenter-dark .column.is-4,html.theme--documenter-dark .column.is-4-tablet{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4,html.theme--documenter-dark .column.is-offset-4-tablet{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5,html.theme--documenter-dark .column.is-5-tablet{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5,html.theme--documenter-dark .column.is-offset-5-tablet{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6,html.theme--documenter-dark .column.is-6-tablet{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6,html.theme--documenter-dark .column.is-offset-6-tablet{margin-left:50%}html.theme--documenter-dark .column.is-7,html.theme--documenter-dark .column.is-7-tablet{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7,html.theme--documenter-dark .column.is-offset-7-tablet{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8,html.theme--documenter-dark .column.is-8-tablet{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8,html.theme--documenter-dark .column.is-offset-8-tablet{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9,html.theme--documenter-dark .column.is-9-tablet{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9,html.theme--documenter-dark .column.is-offset-9-tablet{margin-left:75%}html.theme--documenter-dark .column.is-10,html.theme--documenter-dark .column.is-10-tablet{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10,html.theme--documenter-dark .column.is-offset-10-tablet{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11,html.theme--documenter-dark .column.is-11-tablet{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11,html.theme--documenter-dark .column.is-offset-11-tablet{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12,html.theme--documenter-dark .column.is-12-tablet{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12,html.theme--documenter-dark .column.is-offset-12-tablet{margin-left:100%}}@media screen and (max-width: 1055px){html.theme--documenter-dark .column.is-narrow-touch{flex:none;width:unset}html.theme--documenter-dark .column.is-full-touch{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-touch{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-touch{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-touch{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-touch{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-touch{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-touch{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-touch{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-touch{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-touch{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-touch{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-touch{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-touch{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-touch{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-touch{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-touch{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-touch{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-touch{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-touch{margin-left:80%}html.theme--documenter-dark .column.is-0-touch{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-touch{margin-left:0%}html.theme--documenter-dark .column.is-1-touch{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-touch{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-touch{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-touch{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-touch{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-touch{margin-left:25%}html.theme--documenter-dark .column.is-4-touch{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-touch{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-touch{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-touch{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-touch{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-touch{margin-left:50%}html.theme--documenter-dark .column.is-7-touch{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-touch{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-touch{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-touch{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-touch{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-touch{margin-left:75%}html.theme--documenter-dark .column.is-10-touch{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-touch{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-touch{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-touch{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-touch{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-touch{margin-left:100%}}@media screen and (min-width: 1056px){html.theme--documenter-dark .column.is-narrow-desktop{flex:none;width:unset}html.theme--documenter-dark .column.is-full-desktop{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-desktop{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-desktop{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-desktop{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-desktop{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-desktop{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-desktop{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-desktop{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-desktop{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-desktop{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-desktop{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-desktop{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-desktop{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-desktop{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-desktop{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-desktop{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-desktop{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-desktop{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-desktop{margin-left:80%}html.theme--documenter-dark .column.is-0-desktop{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-desktop{margin-left:0%}html.theme--documenter-dark .column.is-1-desktop{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-desktop{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-desktop{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-desktop{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-desktop{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-desktop{margin-left:25%}html.theme--documenter-dark .column.is-4-desktop{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-desktop{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-desktop{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-desktop{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-desktop{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-desktop{margin-left:50%}html.theme--documenter-dark .column.is-7-desktop{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-desktop{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-desktop{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-desktop{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-desktop{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-desktop{margin-left:75%}html.theme--documenter-dark .column.is-10-desktop{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-desktop{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-desktop{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-desktop{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-desktop{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-desktop{margin-left:100%}}@media screen and (min-width: 1216px){html.theme--documenter-dark .column.is-narrow-widescreen{flex:none;width:unset}html.theme--documenter-dark .column.is-full-widescreen{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-widescreen{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-widescreen{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-widescreen{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-widescreen{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-widescreen{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-widescreen{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-widescreen{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-widescreen{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-widescreen{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-widescreen{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-widescreen{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-widescreen{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-widescreen{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-widescreen{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-widescreen{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-widescreen{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-widescreen{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-widescreen{margin-left:80%}html.theme--documenter-dark .column.is-0-widescreen{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-widescreen{margin-left:0%}html.theme--documenter-dark .column.is-1-widescreen{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-widescreen{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-widescreen{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-widescreen{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-widescreen{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-widescreen{margin-left:25%}html.theme--documenter-dark .column.is-4-widescreen{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-widescreen{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-widescreen{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-widescreen{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-widescreen{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-widescreen{margin-left:50%}html.theme--documenter-dark .column.is-7-widescreen{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-widescreen{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-widescreen{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-widescreen{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-widescreen{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-widescreen{margin-left:75%}html.theme--documenter-dark .column.is-10-widescreen{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-widescreen{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-widescreen{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-widescreen{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-widescreen{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-widescreen{margin-left:100%}}@media screen and (min-width: 1408px){html.theme--documenter-dark .column.is-narrow-fullhd{flex:none;width:unset}html.theme--documenter-dark .column.is-full-fullhd{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-fullhd{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-fullhd{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-fullhd{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-fullhd{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-fullhd{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-fullhd{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-fullhd{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-fullhd{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-fullhd{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-fullhd{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-fullhd{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-fullhd{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-fullhd{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-fullhd{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-fullhd{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-fullhd{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-fullhd{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-fullhd{margin-left:80%}html.theme--documenter-dark .column.is-0-fullhd{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-fullhd{margin-left:0%}html.theme--documenter-dark .column.is-1-fullhd{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-fullhd{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-fullhd{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-fullhd{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-fullhd{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-fullhd{margin-left:25%}html.theme--documenter-dark .column.is-4-fullhd{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-fullhd{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-fullhd{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-fullhd{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-fullhd{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-fullhd{margin-left:50%}html.theme--documenter-dark .column.is-7-fullhd{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-fullhd{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-fullhd{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-fullhd{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-fullhd{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-fullhd{margin-left:75%}html.theme--documenter-dark .column.is-10-fullhd{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-fullhd{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-fullhd{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-fullhd{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-fullhd{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-fullhd{margin-left:100%}}html.theme--documenter-dark .columns{margin-left:-.75rem;margin-right:-.75rem;margin-top:-.75rem}html.theme--documenter-dark .columns:last-child{margin-bottom:-.75rem}html.theme--documenter-dark .columns:not(:last-child){margin-bottom:calc(1.5rem - .75rem)}html.theme--documenter-dark .columns.is-centered{justify-content:center}html.theme--documenter-dark .columns.is-gapless{margin-left:0;margin-right:0;margin-top:0}html.theme--documenter-dark .columns.is-gapless>.column{margin:0;padding:0 !important}html.theme--documenter-dark .columns.is-gapless:not(:last-child){margin-bottom:1.5rem}html.theme--documenter-dark .columns.is-gapless:last-child{margin-bottom:0}html.theme--documenter-dark .columns.is-mobile{display:flex}html.theme--documenter-dark .columns.is-multiline{flex-wrap:wrap}html.theme--documenter-dark .columns.is-vcentered{align-items:center}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns:not(.is-desktop){display:flex}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-desktop{display:flex}}html.theme--documenter-dark .columns.is-variable{--columnGap: 0.75rem;margin-left:calc(-1 * var(--columnGap));margin-right:calc(-1 * var(--columnGap))}html.theme--documenter-dark .columns.is-variable>.column{padding-left:var(--columnGap);padding-right:var(--columnGap)}html.theme--documenter-dark .columns.is-variable.is-0{--columnGap: 0rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-0-mobile{--columnGap: 0rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-0-tablet{--columnGap: 0rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-0-tablet-only{--columnGap: 0rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-0-touch{--columnGap: 0rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-0-desktop{--columnGap: 0rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-0-desktop-only{--columnGap: 0rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-0-widescreen{--columnGap: 0rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-0-widescreen-only{--columnGap: 0rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-0-fullhd{--columnGap: 0rem}}html.theme--documenter-dark .columns.is-variable.is-1{--columnGap: .25rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-1-mobile{--columnGap: .25rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-1-tablet{--columnGap: .25rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-1-tablet-only{--columnGap: .25rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-1-touch{--columnGap: .25rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-1-desktop{--columnGap: .25rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-1-desktop-only{--columnGap: .25rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-1-widescreen{--columnGap: .25rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-1-widescreen-only{--columnGap: .25rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-1-fullhd{--columnGap: .25rem}}html.theme--documenter-dark .columns.is-variable.is-2{--columnGap: .5rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-2-mobile{--columnGap: .5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-2-tablet{--columnGap: .5rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-2-tablet-only{--columnGap: .5rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-2-touch{--columnGap: .5rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-2-desktop{--columnGap: .5rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-2-desktop-only{--columnGap: .5rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-2-widescreen{--columnGap: .5rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-2-widescreen-only{--columnGap: .5rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-2-fullhd{--columnGap: .5rem}}html.theme--documenter-dark .columns.is-variable.is-3{--columnGap: .75rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-3-mobile{--columnGap: .75rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-3-tablet{--columnGap: .75rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-3-tablet-only{--columnGap: .75rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-3-touch{--columnGap: .75rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-3-desktop{--columnGap: .75rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-3-desktop-only{--columnGap: .75rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-3-widescreen{--columnGap: .75rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-3-widescreen-only{--columnGap: .75rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-3-fullhd{--columnGap: .75rem}}html.theme--documenter-dark .columns.is-variable.is-4{--columnGap: 1rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-4-mobile{--columnGap: 1rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-4-tablet{--columnGap: 1rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-4-tablet-only{--columnGap: 1rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-4-touch{--columnGap: 1rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-4-desktop{--columnGap: 1rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-4-desktop-only{--columnGap: 1rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-4-widescreen{--columnGap: 1rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-4-widescreen-only{--columnGap: 1rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-4-fullhd{--columnGap: 1rem}}html.theme--documenter-dark .columns.is-variable.is-5{--columnGap: 1.25rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-5-mobile{--columnGap: 1.25rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-5-tablet{--columnGap: 1.25rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-5-tablet-only{--columnGap: 1.25rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-5-touch{--columnGap: 1.25rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-5-desktop{--columnGap: 1.25rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-5-desktop-only{--columnGap: 1.25rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-5-widescreen{--columnGap: 1.25rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-5-widescreen-only{--columnGap: 1.25rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-5-fullhd{--columnGap: 1.25rem}}html.theme--documenter-dark .columns.is-variable.is-6{--columnGap: 1.5rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-6-mobile{--columnGap: 1.5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-6-tablet{--columnGap: 1.5rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-6-tablet-only{--columnGap: 1.5rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-6-touch{--columnGap: 1.5rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-6-desktop{--columnGap: 1.5rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-6-desktop-only{--columnGap: 1.5rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-6-widescreen{--columnGap: 1.5rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-6-widescreen-only{--columnGap: 1.5rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-6-fullhd{--columnGap: 1.5rem}}html.theme--documenter-dark .columns.is-variable.is-7{--columnGap: 1.75rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-7-mobile{--columnGap: 1.75rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-7-tablet{--columnGap: 1.75rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-7-tablet-only{--columnGap: 1.75rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-7-touch{--columnGap: 1.75rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-7-desktop{--columnGap: 1.75rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-7-desktop-only{--columnGap: 1.75rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-7-widescreen{--columnGap: 1.75rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-7-widescreen-only{--columnGap: 1.75rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-7-fullhd{--columnGap: 1.75rem}}html.theme--documenter-dark .columns.is-variable.is-8{--columnGap: 2rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-8-mobile{--columnGap: 2rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-8-tablet{--columnGap: 2rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-8-tablet-only{--columnGap: 2rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-8-touch{--columnGap: 2rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-8-desktop{--columnGap: 2rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-8-desktop-only{--columnGap: 2rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-8-widescreen{--columnGap: 2rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-8-widescreen-only{--columnGap: 2rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-8-fullhd{--columnGap: 2rem}}html.theme--documenter-dark .tile{align-items:stretch;display:block;flex-basis:0;flex-grow:1;flex-shrink:1;min-height:min-content}html.theme--documenter-dark .tile.is-ancestor{margin-left:-.75rem;margin-right:-.75rem;margin-top:-.75rem}html.theme--documenter-dark .tile.is-ancestor:last-child{margin-bottom:-.75rem}html.theme--documenter-dark .tile.is-ancestor:not(:last-child){margin-bottom:.75rem}html.theme--documenter-dark .tile.is-child{margin:0 !important}html.theme--documenter-dark .tile.is-parent{padding:.75rem}html.theme--documenter-dark .tile.is-vertical{flex-direction:column}html.theme--documenter-dark .tile.is-vertical>.tile.is-child:not(:last-child){margin-bottom:1.5rem !important}@media screen and (min-width: 769px),print{html.theme--documenter-dark .tile:not(.is-child){display:flex}html.theme--documenter-dark .tile.is-1{flex:none;width:8.33333337%}html.theme--documenter-dark .tile.is-2{flex:none;width:16.66666674%}html.theme--documenter-dark .tile.is-3{flex:none;width:25%}html.theme--documenter-dark .tile.is-4{flex:none;width:33.33333337%}html.theme--documenter-dark .tile.is-5{flex:none;width:41.66666674%}html.theme--documenter-dark .tile.is-6{flex:none;width:50%}html.theme--documenter-dark .tile.is-7{flex:none;width:58.33333337%}html.theme--documenter-dark .tile.is-8{flex:none;width:66.66666674%}html.theme--documenter-dark .tile.is-9{flex:none;width:75%}html.theme--documenter-dark .tile.is-10{flex:none;width:83.33333337%}html.theme--documenter-dark .tile.is-11{flex:none;width:91.66666674%}html.theme--documenter-dark .tile.is-12{flex:none;width:100%}}html.theme--documenter-dark .hero{align-items:stretch;display:flex;flex-direction:column;justify-content:space-between}html.theme--documenter-dark .hero .navbar{background:none}html.theme--documenter-dark .hero .tabs ul{border-bottom:none}html.theme--documenter-dark .hero.is-white{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .hero.is-white a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-white strong{color:inherit}html.theme--documenter-dark .hero.is-white .title{color:#0a0a0a}html.theme--documenter-dark .hero.is-white .subtitle{color:rgba(10,10,10,0.9)}html.theme--documenter-dark .hero.is-white .subtitle a:not(.button),html.theme--documenter-dark .hero.is-white .subtitle strong{color:#0a0a0a}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-white .navbar-menu{background-color:#fff}}html.theme--documenter-dark .hero.is-white .navbar-item,html.theme--documenter-dark .hero.is-white .navbar-link{color:rgba(10,10,10,0.7)}html.theme--documenter-dark .hero.is-white a.navbar-item:hover,html.theme--documenter-dark .hero.is-white a.navbar-item.is-active,html.theme--documenter-dark .hero.is-white .navbar-link:hover,html.theme--documenter-dark .hero.is-white .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}html.theme--documenter-dark .hero.is-white .tabs a{color:#0a0a0a;opacity:0.9}html.theme--documenter-dark .hero.is-white .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-white .tabs li.is-active a{color:#fff !important;opacity:1}html.theme--documenter-dark .hero.is-white .tabs.is-boxed a,html.theme--documenter-dark .hero.is-white .tabs.is-toggle a{color:#0a0a0a}html.theme--documenter-dark .hero.is-white .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-white .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-white .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-white .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-white .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-white .tabs.is-toggle li.is-active a:hover{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}html.theme--documenter-dark .hero.is-white.is-bold{background-image:linear-gradient(141deg, #e8e3e4 0%, #fff 71%, #fff 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-white.is-bold .navbar-menu{background-image:linear-gradient(141deg, #e8e3e4 0%, #fff 71%, #fff 100%)}}html.theme--documenter-dark .hero.is-black{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .hero.is-black a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-black strong{color:inherit}html.theme--documenter-dark .hero.is-black .title{color:#fff}html.theme--documenter-dark .hero.is-black .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-black .subtitle a:not(.button),html.theme--documenter-dark .hero.is-black .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-black .navbar-menu{background-color:#0a0a0a}}html.theme--documenter-dark .hero.is-black .navbar-item,html.theme--documenter-dark .hero.is-black .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-black a.navbar-item:hover,html.theme--documenter-dark .hero.is-black a.navbar-item.is-active,html.theme--documenter-dark .hero.is-black .navbar-link:hover,html.theme--documenter-dark .hero.is-black .navbar-link.is-active{background-color:#000;color:#fff}html.theme--documenter-dark .hero.is-black .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-black .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-black .tabs li.is-active a{color:#0a0a0a !important;opacity:1}html.theme--documenter-dark .hero.is-black .tabs.is-boxed a,html.theme--documenter-dark .hero.is-black .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-black .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-black .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-black .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-black .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-black .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-black .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#0a0a0a}html.theme--documenter-dark .hero.is-black.is-bold{background-image:linear-gradient(141deg, #000 0%, #0a0a0a 71%, #181616 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-black.is-bold .navbar-menu{background-image:linear-gradient(141deg, #000 0%, #0a0a0a 71%, #181616 100%)}}html.theme--documenter-dark .hero.is-light{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-light strong{color:inherit}html.theme--documenter-dark .hero.is-light .title{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light .subtitle{color:rgba(0,0,0,0.9)}html.theme--documenter-dark .hero.is-light .subtitle a:not(.button),html.theme--documenter-dark .hero.is-light .subtitle strong{color:rgba(0,0,0,0.7)}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-light .navbar-menu{background-color:#ecf0f1}}html.theme--documenter-dark .hero.is-light .navbar-item,html.theme--documenter-dark .hero.is-light .navbar-link{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light a.navbar-item:hover,html.theme--documenter-dark .hero.is-light a.navbar-item.is-active,html.theme--documenter-dark .hero.is-light .navbar-link:hover,html.theme--documenter-dark .hero.is-light .navbar-link.is-active{background-color:#dde4e6;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light .tabs a{color:rgba(0,0,0,0.7);opacity:0.9}html.theme--documenter-dark .hero.is-light .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-light .tabs li.is-active a{color:#ecf0f1 !important;opacity:1}html.theme--documenter-dark .hero.is-light .tabs.is-boxed a,html.theme--documenter-dark .hero.is-light .tabs.is-toggle a{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-light .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-light .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-light .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-light .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-light .tabs.is-toggle li.is-active a:hover{background-color:rgba(0,0,0,0.7);border-color:rgba(0,0,0,0.7);color:#ecf0f1}html.theme--documenter-dark .hero.is-light.is-bold{background-image:linear-gradient(141deg, #cadfe0 0%, #ecf0f1 71%, #fafbfc 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-light.is-bold .navbar-menu{background-image:linear-gradient(141deg, #cadfe0 0%, #ecf0f1 71%, #fafbfc 100%)}}html.theme--documenter-dark .hero.is-dark,html.theme--documenter-dark .content kbd.hero{background-color:#282f2f;color:#fff}html.theme--documenter-dark .hero.is-dark a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .content kbd.hero a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-dark strong,html.theme--documenter-dark .content kbd.hero strong{color:inherit}html.theme--documenter-dark .hero.is-dark .title,html.theme--documenter-dark .content kbd.hero .title{color:#fff}html.theme--documenter-dark .hero.is-dark .subtitle,html.theme--documenter-dark .content kbd.hero .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-dark .subtitle a:not(.button),html.theme--documenter-dark .content kbd.hero .subtitle a:not(.button),html.theme--documenter-dark .hero.is-dark .subtitle strong,html.theme--documenter-dark .content kbd.hero .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-dark .navbar-menu,html.theme--documenter-dark .content kbd.hero .navbar-menu{background-color:#282f2f}}html.theme--documenter-dark .hero.is-dark .navbar-item,html.theme--documenter-dark .content kbd.hero .navbar-item,html.theme--documenter-dark .hero.is-dark .navbar-link,html.theme--documenter-dark .content kbd.hero .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-dark a.navbar-item:hover,html.theme--documenter-dark .content kbd.hero a.navbar-item:hover,html.theme--documenter-dark .hero.is-dark a.navbar-item.is-active,html.theme--documenter-dark .content kbd.hero a.navbar-item.is-active,html.theme--documenter-dark .hero.is-dark .navbar-link:hover,html.theme--documenter-dark .content kbd.hero .navbar-link:hover,html.theme--documenter-dark .hero.is-dark .navbar-link.is-active,html.theme--documenter-dark .content kbd.hero .navbar-link.is-active{background-color:#1d2122;color:#fff}html.theme--documenter-dark .hero.is-dark .tabs a,html.theme--documenter-dark .content kbd.hero .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-dark .tabs a:hover,html.theme--documenter-dark .content kbd.hero .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-dark .tabs li.is-active a,html.theme--documenter-dark .content kbd.hero .tabs li.is-active a{color:#282f2f !important;opacity:1}html.theme--documenter-dark .hero.is-dark .tabs.is-boxed a,html.theme--documenter-dark .content kbd.hero .tabs.is-boxed a,html.theme--documenter-dark .hero.is-dark .tabs.is-toggle a,html.theme--documenter-dark .content kbd.hero .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-dark .tabs.is-boxed a:hover,html.theme--documenter-dark .content kbd.hero .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-dark .tabs.is-toggle a:hover,html.theme--documenter-dark .content kbd.hero .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-dark .tabs.is-boxed li.is-active a,html.theme--documenter-dark .content kbd.hero .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-dark .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-dark .tabs.is-toggle li.is-active a,html.theme--documenter-dark .content kbd.hero .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-dark .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#282f2f}html.theme--documenter-dark .hero.is-dark.is-bold,html.theme--documenter-dark .content kbd.hero.is-bold{background-image:linear-gradient(141deg, #0f1615 0%, #282f2f 71%, #313c40 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-dark.is-bold .navbar-menu,html.theme--documenter-dark .content kbd.hero.is-bold .navbar-menu{background-image:linear-gradient(141deg, #0f1615 0%, #282f2f 71%, #313c40 100%)}}html.theme--documenter-dark .hero.is-primary,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink{background-color:#375a7f;color:#fff}html.theme--documenter-dark .hero.is-primary a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-primary strong,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink strong{color:inherit}html.theme--documenter-dark .hero.is-primary .title,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .title{color:#fff}html.theme--documenter-dark .hero.is-primary .subtitle,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-primary .subtitle a:not(.button),html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .subtitle a:not(.button),html.theme--documenter-dark .hero.is-primary .subtitle strong,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-primary .navbar-menu,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-menu{background-color:#375a7f}}html.theme--documenter-dark .hero.is-primary .navbar-item,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-item,html.theme--documenter-dark .hero.is-primary .navbar-link,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-primary a.navbar-item:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink a.navbar-item:hover,html.theme--documenter-dark .hero.is-primary a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink a.navbar-item.is-active,html.theme--documenter-dark .hero.is-primary .navbar-link:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-link:hover,html.theme--documenter-dark .hero.is-primary .navbar-link.is-active,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-link.is-active{background-color:#2f4d6d;color:#fff}html.theme--documenter-dark .hero.is-primary .tabs a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-primary .tabs a:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-primary .tabs li.is-active a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs li.is-active a{color:#375a7f !important;opacity:1}html.theme--documenter-dark .hero.is-primary .tabs.is-boxed a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-boxed a,html.theme--documenter-dark .hero.is-primary .tabs.is-toggle a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-primary .tabs.is-boxed a:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-primary .tabs.is-toggle a:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-primary .tabs.is-boxed li.is-active a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-primary .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-primary .tabs.is-toggle li.is-active a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-primary .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#375a7f}html.theme--documenter-dark .hero.is-primary.is-bold,html.theme--documenter-dark .docstring>section>a.hero.is-bold.docs-sourcelink{background-image:linear-gradient(141deg, #214b62 0%, #375a7f 71%, #3a5796 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-primary.is-bold .navbar-menu,html.theme--documenter-dark .docstring>section>a.hero.is-bold.docs-sourcelink .navbar-menu{background-image:linear-gradient(141deg, #214b62 0%, #375a7f 71%, #3a5796 100%)}}html.theme--documenter-dark .hero.is-link{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .hero.is-link a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-link strong{color:inherit}html.theme--documenter-dark .hero.is-link .title{color:#fff}html.theme--documenter-dark .hero.is-link .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-link .subtitle a:not(.button),html.theme--documenter-dark .hero.is-link .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-link .navbar-menu{background-color:#1abc9c}}html.theme--documenter-dark .hero.is-link .navbar-item,html.theme--documenter-dark .hero.is-link .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-link a.navbar-item:hover,html.theme--documenter-dark .hero.is-link a.navbar-item.is-active,html.theme--documenter-dark .hero.is-link .navbar-link:hover,html.theme--documenter-dark .hero.is-link .navbar-link.is-active{background-color:#17a689;color:#fff}html.theme--documenter-dark .hero.is-link .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-link .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-link .tabs li.is-active a{color:#1abc9c !important;opacity:1}html.theme--documenter-dark .hero.is-link .tabs.is-boxed a,html.theme--documenter-dark .hero.is-link .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-link .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-link .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-link .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-link .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-link .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-link .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#1abc9c}html.theme--documenter-dark .hero.is-link.is-bold{background-image:linear-gradient(141deg, #0c9764 0%, #1abc9c 71%, #17d8d2 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-link.is-bold .navbar-menu{background-image:linear-gradient(141deg, #0c9764 0%, #1abc9c 71%, #17d8d2 100%)}}html.theme--documenter-dark .hero.is-info{background-color:#024c7d;color:#fff}html.theme--documenter-dark .hero.is-info a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-info strong{color:inherit}html.theme--documenter-dark .hero.is-info .title{color:#fff}html.theme--documenter-dark .hero.is-info .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-info .subtitle a:not(.button),html.theme--documenter-dark .hero.is-info .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-info .navbar-menu{background-color:#024c7d}}html.theme--documenter-dark .hero.is-info .navbar-item,html.theme--documenter-dark .hero.is-info .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-info a.navbar-item:hover,html.theme--documenter-dark .hero.is-info a.navbar-item.is-active,html.theme--documenter-dark .hero.is-info .navbar-link:hover,html.theme--documenter-dark .hero.is-info .navbar-link.is-active{background-color:#023d64;color:#fff}html.theme--documenter-dark .hero.is-info .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-info .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-info .tabs li.is-active a{color:#024c7d !important;opacity:1}html.theme--documenter-dark .hero.is-info .tabs.is-boxed a,html.theme--documenter-dark .hero.is-info .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-info .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-info .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-info .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-info .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-info .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-info .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#024c7d}html.theme--documenter-dark .hero.is-info.is-bold{background-image:linear-gradient(141deg, #003a4c 0%, #024c7d 71%, #004299 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-info.is-bold .navbar-menu{background-image:linear-gradient(141deg, #003a4c 0%, #024c7d 71%, #004299 100%)}}html.theme--documenter-dark .hero.is-success{background-color:#008438;color:#fff}html.theme--documenter-dark .hero.is-success a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-success strong{color:inherit}html.theme--documenter-dark .hero.is-success .title{color:#fff}html.theme--documenter-dark .hero.is-success .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-success .subtitle a:not(.button),html.theme--documenter-dark .hero.is-success .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-success .navbar-menu{background-color:#008438}}html.theme--documenter-dark .hero.is-success .navbar-item,html.theme--documenter-dark .hero.is-success .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-success a.navbar-item:hover,html.theme--documenter-dark .hero.is-success a.navbar-item.is-active,html.theme--documenter-dark .hero.is-success .navbar-link:hover,html.theme--documenter-dark .hero.is-success .navbar-link.is-active{background-color:#006b2d;color:#fff}html.theme--documenter-dark .hero.is-success .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-success .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-success .tabs li.is-active a{color:#008438 !important;opacity:1}html.theme--documenter-dark .hero.is-success .tabs.is-boxed a,html.theme--documenter-dark .hero.is-success .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-success .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-success .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-success .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-success .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-success .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-success .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#008438}html.theme--documenter-dark .hero.is-success.is-bold{background-image:linear-gradient(141deg, #005115 0%, #008438 71%, #009e5d 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-success.is-bold .navbar-menu{background-image:linear-gradient(141deg, #005115 0%, #008438 71%, #009e5d 100%)}}html.theme--documenter-dark .hero.is-warning{background-color:#ad8100;color:#fff}html.theme--documenter-dark .hero.is-warning a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-warning strong{color:inherit}html.theme--documenter-dark .hero.is-warning .title{color:#fff}html.theme--documenter-dark .hero.is-warning .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-warning .subtitle a:not(.button),html.theme--documenter-dark .hero.is-warning .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-warning .navbar-menu{background-color:#ad8100}}html.theme--documenter-dark .hero.is-warning .navbar-item,html.theme--documenter-dark .hero.is-warning .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-warning a.navbar-item:hover,html.theme--documenter-dark .hero.is-warning a.navbar-item.is-active,html.theme--documenter-dark .hero.is-warning .navbar-link:hover,html.theme--documenter-dark .hero.is-warning .navbar-link.is-active{background-color:#946e00;color:#fff}html.theme--documenter-dark .hero.is-warning .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-warning .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-warning .tabs li.is-active a{color:#ad8100 !important;opacity:1}html.theme--documenter-dark .hero.is-warning .tabs.is-boxed a,html.theme--documenter-dark .hero.is-warning .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-warning .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-warning .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-warning .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-warning .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-warning .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-warning .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#ad8100}html.theme--documenter-dark .hero.is-warning.is-bold{background-image:linear-gradient(141deg, #7a4700 0%, #ad8100 71%, #c7b500 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-warning.is-bold .navbar-menu{background-image:linear-gradient(141deg, #7a4700 0%, #ad8100 71%, #c7b500 100%)}}html.theme--documenter-dark .hero.is-danger{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .hero.is-danger a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-danger strong{color:inherit}html.theme--documenter-dark .hero.is-danger .title{color:#fff}html.theme--documenter-dark .hero.is-danger .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-danger .subtitle a:not(.button),html.theme--documenter-dark .hero.is-danger .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-danger .navbar-menu{background-color:#9e1b0d}}html.theme--documenter-dark .hero.is-danger .navbar-item,html.theme--documenter-dark .hero.is-danger .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-danger a.navbar-item:hover,html.theme--documenter-dark .hero.is-danger a.navbar-item.is-active,html.theme--documenter-dark .hero.is-danger .navbar-link:hover,html.theme--documenter-dark .hero.is-danger .navbar-link.is-active{background-color:#86170b;color:#fff}html.theme--documenter-dark .hero.is-danger .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-danger .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-danger .tabs li.is-active a{color:#9e1b0d !important;opacity:1}html.theme--documenter-dark .hero.is-danger .tabs.is-boxed a,html.theme--documenter-dark .hero.is-danger .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-danger .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-danger .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-danger .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-danger .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-danger .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-danger .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#9e1b0d}html.theme--documenter-dark .hero.is-danger.is-bold{background-image:linear-gradient(141deg, #75030b 0%, #9e1b0d 71%, #ba380a 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-danger.is-bold .navbar-menu{background-image:linear-gradient(141deg, #75030b 0%, #9e1b0d 71%, #ba380a 100%)}}html.theme--documenter-dark .hero.is-small .hero-body,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.hero .hero-body{padding:1.5rem}@media screen and (min-width: 769px),print{html.theme--documenter-dark .hero.is-medium .hero-body{padding:9rem 4.5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .hero.is-large .hero-body{padding:18rem 6rem}}html.theme--documenter-dark .hero.is-halfheight .hero-body,html.theme--documenter-dark .hero.is-fullheight .hero-body,html.theme--documenter-dark .hero.is-fullheight-with-navbar .hero-body{align-items:center;display:flex}html.theme--documenter-dark .hero.is-halfheight .hero-body>.container,html.theme--documenter-dark .hero.is-fullheight .hero-body>.container,html.theme--documenter-dark .hero.is-fullheight-with-navbar .hero-body>.container{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .hero.is-halfheight{min-height:50vh}html.theme--documenter-dark .hero.is-fullheight{min-height:100vh}html.theme--documenter-dark .hero-video{overflow:hidden}html.theme--documenter-dark .hero-video video{left:50%;min-height:100%;min-width:100%;position:absolute;top:50%;transform:translate3d(-50%, -50%, 0)}html.theme--documenter-dark .hero-video.is-transparent{opacity:0.3}@media screen and (max-width: 768px){html.theme--documenter-dark .hero-video{display:none}}html.theme--documenter-dark .hero-buttons{margin-top:1.5rem}@media screen and (max-width: 768px){html.theme--documenter-dark .hero-buttons .button{display:flex}html.theme--documenter-dark .hero-buttons .button:not(:last-child){margin-bottom:0.75rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .hero-buttons{display:flex;justify-content:center}html.theme--documenter-dark .hero-buttons .button:not(:last-child){margin-right:1.5rem}}html.theme--documenter-dark .hero-head,html.theme--documenter-dark .hero-foot{flex-grow:0;flex-shrink:0}html.theme--documenter-dark .hero-body{flex-grow:1;flex-shrink:0;padding:3rem 1.5rem}@media screen and (min-width: 769px),print{html.theme--documenter-dark .hero-body{padding:3rem 3rem}}html.theme--documenter-dark .section{padding:3rem 1.5rem}@media screen and (min-width: 1056px){html.theme--documenter-dark .section{padding:3rem 3rem}html.theme--documenter-dark .section.is-medium{padding:9rem 4.5rem}html.theme--documenter-dark .section.is-large{padding:18rem 6rem}}html.theme--documenter-dark .footer{background-color:#282f2f;padding:3rem 1.5rem 6rem}html.theme--documenter-dark hr{height:1px}html.theme--documenter-dark h6{text-transform:uppercase;letter-spacing:0.5px}html.theme--documenter-dark .hero{background-color:#343c3d}html.theme--documenter-dark a{transition:all 200ms ease}html.theme--documenter-dark .button{transition:all 200ms ease;border-width:1px;color:#fff}html.theme--documenter-dark .button.is-active,html.theme--documenter-dark .button.is-focused,html.theme--documenter-dark .button:active,html.theme--documenter-dark .button:focus{box-shadow:0 0 0 2px rgba(140,155,157,0.5)}html.theme--documenter-dark .button.is-white.is-hovered,html.theme--documenter-dark .button.is-white:hover{background-color:#fff}html.theme--documenter-dark .button.is-white.is-active,html.theme--documenter-dark .button.is-white.is-focused,html.theme--documenter-dark .button.is-white:active,html.theme--documenter-dark .button.is-white:focus{border-color:#fff;box-shadow:0 0 0 2px rgba(255,255,255,0.5)}html.theme--documenter-dark .button.is-black.is-hovered,html.theme--documenter-dark .button.is-black:hover{background-color:#1d1d1d}html.theme--documenter-dark .button.is-black.is-active,html.theme--documenter-dark .button.is-black.is-focused,html.theme--documenter-dark .button.is-black:active,html.theme--documenter-dark .button.is-black:focus{border-color:#0a0a0a;box-shadow:0 0 0 2px rgba(10,10,10,0.5)}html.theme--documenter-dark .button.is-light.is-hovered,html.theme--documenter-dark .button.is-light:hover{background-color:#fff}html.theme--documenter-dark .button.is-light.is-active,html.theme--documenter-dark .button.is-light.is-focused,html.theme--documenter-dark .button.is-light:active,html.theme--documenter-dark .button.is-light:focus{border-color:#ecf0f1;box-shadow:0 0 0 2px rgba(236,240,241,0.5)}html.theme--documenter-dark .button.is-dark.is-hovered,html.theme--documenter-dark .content kbd.button.is-hovered,html.theme--documenter-dark .button.is-dark:hover,html.theme--documenter-dark .content kbd.button:hover{background-color:#3a4344}html.theme--documenter-dark .button.is-dark.is-active,html.theme--documenter-dark .content kbd.button.is-active,html.theme--documenter-dark .button.is-dark.is-focused,html.theme--documenter-dark .content kbd.button.is-focused,html.theme--documenter-dark .button.is-dark:active,html.theme--documenter-dark .content kbd.button:active,html.theme--documenter-dark .button.is-dark:focus,html.theme--documenter-dark .content kbd.button:focus{border-color:#282f2f;box-shadow:0 0 0 2px rgba(40,47,47,0.5)}html.theme--documenter-dark .button.is-primary.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-hovered.docs-sourcelink,html.theme--documenter-dark .button.is-primary:hover,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:hover{background-color:#436d9a}html.theme--documenter-dark .button.is-primary.is-active,html.theme--documenter-dark .docstring>section>a.button.is-active.docs-sourcelink,html.theme--documenter-dark .button.is-primary.is-focused,html.theme--documenter-dark .docstring>section>a.button.is-focused.docs-sourcelink,html.theme--documenter-dark .button.is-primary:active,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:active,html.theme--documenter-dark .button.is-primary:focus,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:focus{border-color:#375a7f;box-shadow:0 0 0 2px rgba(55,90,127,0.5)}html.theme--documenter-dark .button.is-link.is-hovered,html.theme--documenter-dark .button.is-link:hover{background-color:#1fdeb8}html.theme--documenter-dark .button.is-link.is-active,html.theme--documenter-dark .button.is-link.is-focused,html.theme--documenter-dark .button.is-link:active,html.theme--documenter-dark .button.is-link:focus{border-color:#1abc9c;box-shadow:0 0 0 2px rgba(26,188,156,0.5)}html.theme--documenter-dark .button.is-info.is-hovered,html.theme--documenter-dark .button.is-info:hover{background-color:#0363a3}html.theme--documenter-dark .button.is-info.is-active,html.theme--documenter-dark .button.is-info.is-focused,html.theme--documenter-dark .button.is-info:active,html.theme--documenter-dark .button.is-info:focus{border-color:#024c7d;box-shadow:0 0 0 2px rgba(2,76,125,0.5)}html.theme--documenter-dark .button.is-success.is-hovered,html.theme--documenter-dark .button.is-success:hover{background-color:#00aa48}html.theme--documenter-dark .button.is-success.is-active,html.theme--documenter-dark .button.is-success.is-focused,html.theme--documenter-dark .button.is-success:active,html.theme--documenter-dark .button.is-success:focus{border-color:#008438;box-shadow:0 0 0 2px rgba(0,132,56,0.5)}html.theme--documenter-dark .button.is-warning.is-hovered,html.theme--documenter-dark .button.is-warning:hover{background-color:#d39e00}html.theme--documenter-dark .button.is-warning.is-active,html.theme--documenter-dark .button.is-warning.is-focused,html.theme--documenter-dark .button.is-warning:active,html.theme--documenter-dark .button.is-warning:focus{border-color:#ad8100;box-shadow:0 0 0 2px rgba(173,129,0,0.5)}html.theme--documenter-dark .button.is-danger.is-hovered,html.theme--documenter-dark .button.is-danger:hover{background-color:#c12110}html.theme--documenter-dark .button.is-danger.is-active,html.theme--documenter-dark .button.is-danger.is-focused,html.theme--documenter-dark .button.is-danger:active,html.theme--documenter-dark .button.is-danger:focus{border-color:#9e1b0d;box-shadow:0 0 0 2px rgba(158,27,13,0.5)}html.theme--documenter-dark .label{color:#dbdee0}html.theme--documenter-dark .button,html.theme--documenter-dark .control.has-icons-left .icon,html.theme--documenter-dark .control.has-icons-right .icon,html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark .pagination-ellipsis,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .select,html.theme--documenter-dark .select select,html.theme--documenter-dark .textarea{height:2.5em}html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark .textarea{transition:all 200ms ease;box-shadow:none;border-width:1px;padding-left:1em;padding-right:1em}html.theme--documenter-dark .select:after,html.theme--documenter-dark .select select{border-width:1px}html.theme--documenter-dark .control.has-addons .button,html.theme--documenter-dark .control.has-addons .input,html.theme--documenter-dark .control.has-addons #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .control.has-addons form.docs-search>input,html.theme--documenter-dark .control.has-addons .select{margin-right:-1px}html.theme--documenter-dark .notification{background-color:#343c3d}html.theme--documenter-dark .card{box-shadow:none;border:1px solid #343c3d;background-color:#282f2f;border-radius:.4em}html.theme--documenter-dark .card .card-image img{border-radius:.4em .4em 0 0}html.theme--documenter-dark .card .card-header{box-shadow:none;background-color:rgba(18,18,18,0.2);border-radius:.4em .4em 0 0}html.theme--documenter-dark .card .card-footer{background-color:rgba(18,18,18,0.2)}html.theme--documenter-dark .card .card-footer,html.theme--documenter-dark .card .card-footer-item{border-width:1px;border-color:#343c3d}html.theme--documenter-dark .notification.is-white a:not(.button){color:#0a0a0a;text-decoration:underline}html.theme--documenter-dark .notification.is-black a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-light a:not(.button){color:rgba(0,0,0,0.7);text-decoration:underline}html.theme--documenter-dark .notification.is-dark a:not(.button),html.theme--documenter-dark .content kbd.notification a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-primary a:not(.button),html.theme--documenter-dark .docstring>section>a.notification.docs-sourcelink a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-link a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-info a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-success a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-warning a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-danger a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .tag,html.theme--documenter-dark .content kbd,html.theme--documenter-dark .docstring>section>a.docs-sourcelink{border-radius:.4em}html.theme--documenter-dark .menu-list a{transition:all 300ms ease}html.theme--documenter-dark .modal-card-body{background-color:#282f2f}html.theme--documenter-dark .modal-card-foot,html.theme--documenter-dark .modal-card-head{border-color:#343c3d}html.theme--documenter-dark .message-header{font-weight:700;background-color:#343c3d;color:#fff}html.theme--documenter-dark .message-body{border-width:1px;border-color:#343c3d}html.theme--documenter-dark .navbar{border-radius:.4em}html.theme--documenter-dark .navbar.is-transparent{background:none}html.theme--documenter-dark .navbar.is-primary .navbar-dropdown a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-dropdown a.navbar-item.is-active{background-color:#1abc9c}@media screen and (max-width: 1055px){html.theme--documenter-dark .navbar .navbar-menu{background-color:#375a7f;border-radius:0 0 .4em .4em}}html.theme--documenter-dark .hero .navbar,html.theme--documenter-dark body>.navbar{border-radius:0}html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-previous{border-width:1px}html.theme--documenter-dark .panel-block,html.theme--documenter-dark .panel-heading,html.theme--documenter-dark .panel-tabs{border-width:1px}html.theme--documenter-dark .panel-block:first-child,html.theme--documenter-dark .panel-heading:first-child,html.theme--documenter-dark .panel-tabs:first-child{border-top-width:1px}html.theme--documenter-dark .panel-heading{font-weight:700}html.theme--documenter-dark .panel-tabs a{border-width:1px;margin-bottom:-1px}html.theme--documenter-dark .panel-tabs a.is-active{border-bottom-color:#17a689}html.theme--documenter-dark .panel-block:hover{color:#1dd2af}html.theme--documenter-dark .panel-block:hover .panel-icon{color:#1dd2af}html.theme--documenter-dark .panel-block.is-active .panel-icon{color:#17a689}html.theme--documenter-dark .tabs a{border-bottom-width:1px;margin-bottom:-1px}html.theme--documenter-dark .tabs ul{border-bottom-width:1px}html.theme--documenter-dark .tabs.is-boxed a{border-width:1px}html.theme--documenter-dark .tabs.is-boxed li.is-active a{background-color:#1f2424}html.theme--documenter-dark .tabs.is-toggle li a{border-width:1px;margin-bottom:0}html.theme--documenter-dark .tabs.is-toggle li+li{margin-left:-1px}html.theme--documenter-dark .hero.is-white .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-black .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-light .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-dark .navbar .navbar-dropdown .navbar-item:hover,html.theme--documenter-dark .content kbd.hero .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-primary .navbar .navbar-dropdown .navbar-item:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-link .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-info .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-success .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-warning .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-danger .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark h1 .docs-heading-anchor,html.theme--documenter-dark h1 .docs-heading-anchor:hover,html.theme--documenter-dark h1 .docs-heading-anchor:visited,html.theme--documenter-dark h2 .docs-heading-anchor,html.theme--documenter-dark h2 .docs-heading-anchor:hover,html.theme--documenter-dark h2 .docs-heading-anchor:visited,html.theme--documenter-dark h3 .docs-heading-anchor,html.theme--documenter-dark h3 .docs-heading-anchor:hover,html.theme--documenter-dark h3 .docs-heading-anchor:visited,html.theme--documenter-dark h4 .docs-heading-anchor,html.theme--documenter-dark h4 .docs-heading-anchor:hover,html.theme--documenter-dark h4 .docs-heading-anchor:visited,html.theme--documenter-dark h5 .docs-heading-anchor,html.theme--documenter-dark h5 .docs-heading-anchor:hover,html.theme--documenter-dark h5 .docs-heading-anchor:visited,html.theme--documenter-dark h6 .docs-heading-anchor,html.theme--documenter-dark h6 .docs-heading-anchor:hover,html.theme--documenter-dark h6 .docs-heading-anchor:visited{color:#f2f2f2}html.theme--documenter-dark h1 .docs-heading-anchor-permalink,html.theme--documenter-dark h2 .docs-heading-anchor-permalink,html.theme--documenter-dark h3 .docs-heading-anchor-permalink,html.theme--documenter-dark h4 .docs-heading-anchor-permalink,html.theme--documenter-dark h5 .docs-heading-anchor-permalink,html.theme--documenter-dark h6 .docs-heading-anchor-permalink{visibility:hidden;vertical-align:middle;margin-left:0.5em;font-size:0.7rem}html.theme--documenter-dark h1 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h2 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h3 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h4 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h5 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h6 .docs-heading-anchor-permalink::before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f0c1"}html.theme--documenter-dark h1:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h2:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h3:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h4:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h5:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h6:hover .docs-heading-anchor-permalink{visibility:visible}html.theme--documenter-dark .docs-light-only{display:none !important}html.theme--documenter-dark pre{position:relative;overflow:hidden}html.theme--documenter-dark pre code,html.theme--documenter-dark pre code.hljs{padding:0 .75rem !important;overflow:auto;display:block}html.theme--documenter-dark pre code:first-of-type,html.theme--documenter-dark pre code.hljs:first-of-type{padding-top:0.5rem !important}html.theme--documenter-dark pre code:last-of-type,html.theme--documenter-dark pre code.hljs:last-of-type{padding-bottom:0.5rem !important}html.theme--documenter-dark pre .copy-button{opacity:0.2;transition:opacity 0.2s;position:absolute;right:0em;top:0em;padding:0.5em;width:2.5em;height:2.5em;background:transparent;border:none;font-family:"Font Awesome 6 Free";color:#fff;cursor:pointer;text-align:center}html.theme--documenter-dark pre .copy-button:focus,html.theme--documenter-dark pre .copy-button:hover{opacity:1;background:rgba(255,255,255,0.1);color:#1abc9c}html.theme--documenter-dark pre .copy-button.success{color:#259a12;opacity:1}html.theme--documenter-dark pre .copy-button.error{color:#cb3c33;opacity:1}html.theme--documenter-dark pre:hover .copy-button{opacity:1}html.theme--documenter-dark .admonition{background-color:#282f2f;border-style:solid;border-width:1px;border-color:#5e6d6f;border-radius:.4em;font-size:1rem}html.theme--documenter-dark .admonition strong{color:currentColor}html.theme--documenter-dark .admonition.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.admonition{font-size:.75rem}html.theme--documenter-dark .admonition.is-medium{font-size:1.25rem}html.theme--documenter-dark .admonition.is-large{font-size:1.5rem}html.theme--documenter-dark .admonition.is-default{background-color:#282f2f;border-color:#5e6d6f}html.theme--documenter-dark .admonition.is-default>.admonition-header{background-color:#5e6d6f;color:#fff}html.theme--documenter-dark .admonition.is-default>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-info{background-color:#282f2f;border-color:#024c7d}html.theme--documenter-dark .admonition.is-info>.admonition-header{background-color:#024c7d;color:#fff}html.theme--documenter-dark .admonition.is-info>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-success{background-color:#282f2f;border-color:#008438}html.theme--documenter-dark .admonition.is-success>.admonition-header{background-color:#008438;color:#fff}html.theme--documenter-dark .admonition.is-success>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-warning{background-color:#282f2f;border-color:#ad8100}html.theme--documenter-dark .admonition.is-warning>.admonition-header{background-color:#ad8100;color:#fff}html.theme--documenter-dark .admonition.is-warning>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-danger{background-color:#282f2f;border-color:#9e1b0d}html.theme--documenter-dark .admonition.is-danger>.admonition-header{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .admonition.is-danger>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-compat{background-color:#282f2f;border-color:#137886}html.theme--documenter-dark .admonition.is-compat>.admonition-header{background-color:#137886;color:#fff}html.theme--documenter-dark .admonition.is-compat>.admonition-body{color:#fff}html.theme--documenter-dark .admonition-header{color:#fff;background-color:#5e6d6f;align-items:center;font-weight:700;justify-content:space-between;line-height:1.25;padding:0.5rem .75rem;position:relative}html.theme--documenter-dark .admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;margin-right:.75rem;content:"\f06a"}html.theme--documenter-dark details.admonition.is-details>.admonition-header{list-style:none}html.theme--documenter-dark details.admonition.is-details>.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f055"}html.theme--documenter-dark details.admonition.is-details[open]>.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f056"}html.theme--documenter-dark .admonition-body{color:#fff;padding:0.5rem .75rem}html.theme--documenter-dark .admonition-body pre{background-color:#282f2f}html.theme--documenter-dark .admonition-body code{background-color:rgba(255,255,255,0.05)}html.theme--documenter-dark .docstring{margin-bottom:1em;background-color:rgba(0,0,0,0);border:1px solid #5e6d6f;box-shadow:none;max-width:100%}html.theme--documenter-dark .docstring>header{cursor:pointer;display:flex;flex-grow:1;align-items:stretch;padding:0.5rem .75rem;background-color:#282f2f;box-shadow:0 0.125em 0.25em rgba(10,10,10,0.1);box-shadow:none;border-bottom:1px solid #5e6d6f}html.theme--documenter-dark .docstring>header code{background-color:transparent}html.theme--documenter-dark .docstring>header .docstring-article-toggle-button{min-width:1.1rem;padding:0.2rem 0.2rem 0.2rem 0}html.theme--documenter-dark .docstring>header .docstring-binding{margin-right:0.3em}html.theme--documenter-dark .docstring>header .docstring-category{margin-left:0.3em}html.theme--documenter-dark .docstring>section{position:relative;padding:.75rem .75rem;border-bottom:1px solid #5e6d6f}html.theme--documenter-dark .docstring>section:last-child{border-bottom:none}html.theme--documenter-dark .docstring>section>a.docs-sourcelink{transition:opacity 0.3s;opacity:0;position:absolute;right:.375rem;bottom:.375rem}html.theme--documenter-dark .docstring>section>a.docs-sourcelink:focus{opacity:1 !important}html.theme--documenter-dark .docstring:hover>section>a.docs-sourcelink{opacity:0.2}html.theme--documenter-dark .docstring:focus-within>section>a.docs-sourcelink{opacity:0.2}html.theme--documenter-dark .docstring>section:hover a.docs-sourcelink{opacity:1}html.theme--documenter-dark .documenter-example-output{background-color:#1f2424}html.theme--documenter-dark .outdated-warning-overlay{position:fixed;top:0;left:0;right:0;box-shadow:0 0 10px rgba(0,0,0,0.3);z-index:999;background-color:#282f2f;color:#fff;border-bottom:3px solid #9e1b0d;padding:10px 35px;text-align:center;font-size:15px}html.theme--documenter-dark .outdated-warning-overlay .outdated-warning-closer{position:absolute;top:calc(50% - 10px);right:18px;cursor:pointer;width:12px}html.theme--documenter-dark .outdated-warning-overlay a{color:#1abc9c}html.theme--documenter-dark .outdated-warning-overlay a:hover{color:#1dd2af}html.theme--documenter-dark .content pre{border:1px solid #5e6d6f}html.theme--documenter-dark .content code{font-weight:inherit}html.theme--documenter-dark .content a code{color:#1abc9c}html.theme--documenter-dark .content h1 code,html.theme--documenter-dark .content h2 code,html.theme--documenter-dark .content h3 code,html.theme--documenter-dark .content h4 code,html.theme--documenter-dark .content h5 code,html.theme--documenter-dark .content h6 code{color:#f2f2f2}html.theme--documenter-dark .content table{display:block;width:initial;max-width:100%;overflow-x:auto}html.theme--documenter-dark .content blockquote>ul:first-child,html.theme--documenter-dark .content blockquote>ol:first-child,html.theme--documenter-dark .content .admonition-body>ul:first-child,html.theme--documenter-dark .content .admonition-body>ol:first-child{margin-top:0}html.theme--documenter-dark pre,html.theme--documenter-dark code{font-variant-ligatures:no-contextual}html.theme--documenter-dark .breadcrumb a.is-disabled{cursor:default;pointer-events:none}html.theme--documenter-dark .breadcrumb a.is-disabled,html.theme--documenter-dark .breadcrumb a.is-disabled:hover{color:#f2f2f2}html.theme--documenter-dark .hljs{background:initial !important}html.theme--documenter-dark .katex .katex-mathml{top:0;right:0}html.theme--documenter-dark .katex-display,html.theme--documenter-dark mjx-container,html.theme--documenter-dark .MathJax_Display{margin:0.5em 0 !important}html.theme--documenter-dark html{-moz-osx-font-smoothing:auto;-webkit-font-smoothing:auto}html.theme--documenter-dark li.no-marker{list-style:none}html.theme--documenter-dark #documenter .docs-main>article{overflow-wrap:break-word}html.theme--documenter-dark #documenter .docs-main>article .math-container{overflow-x:auto;overflow-y:hidden}@media screen and (min-width: 1056px){html.theme--documenter-dark #documenter .docs-main{max-width:52rem;margin-left:20rem;padding-right:1rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-main{width:100%}html.theme--documenter-dark #documenter .docs-main>article{max-width:52rem;margin-left:auto;margin-right:auto;margin-bottom:1rem;padding:0 1rem}html.theme--documenter-dark #documenter .docs-main>header,html.theme--documenter-dark #documenter .docs-main>nav{max-width:100%;width:100%;margin:0}}html.theme--documenter-dark #documenter .docs-main header.docs-navbar{background-color:#1f2424;border-bottom:1px solid #5e6d6f;z-index:2;min-height:4rem;margin-bottom:1rem;display:flex}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .breadcrumb{flex-grow:1}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-sidebar-button{display:block;font-size:1.5rem;padding-bottom:0.1rem;margin-right:1rem}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right{display:flex;white-space:nowrap;gap:1rem;align-items:center}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right .docs-icon,html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right .docs-label{display:inline-block}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right .docs-label{padding:0;margin-left:0.3em}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right .docs-navbar-link{margin-left:0.4rem;margin-right:0.4rem}}html.theme--documenter-dark #documenter .docs-main header.docs-navbar>*{margin:auto 0}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-main header.docs-navbar{position:sticky;top:0;padding:0 1rem;transition-property:top, box-shadow;-webkit-transition-property:top, box-shadow;transition-duration:0.3s;-webkit-transition-duration:0.3s}html.theme--documenter-dark #documenter .docs-main header.docs-navbar.headroom--not-top{box-shadow:.2rem 0rem .4rem #171717;transition-duration:0.7s;-webkit-transition-duration:0.7s}html.theme--documenter-dark #documenter .docs-main header.docs-navbar.headroom--unpinned.headroom--not-top.headroom--not-bottom{top:-4.5rem;transition-duration:0.7s;-webkit-transition-duration:0.7s}}html.theme--documenter-dark #documenter .docs-main section.footnotes{border-top:1px solid #5e6d6f}html.theme--documenter-dark #documenter .docs-main section.footnotes li .tag:first-child,html.theme--documenter-dark #documenter .docs-main section.footnotes li .docstring>section>a.docs-sourcelink:first-child,html.theme--documenter-dark #documenter .docs-main section.footnotes li .content kbd:first-child,html.theme--documenter-dark .content #documenter .docs-main section.footnotes li kbd:first-child{margin-right:1em;margin-bottom:0.4em}html.theme--documenter-dark #documenter .docs-main .docs-footer{display:flex;flex-wrap:wrap;margin-left:0;margin-right:0;border-top:1px solid #5e6d6f;padding-top:1rem;padding-bottom:1rem}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-main .docs-footer{padding-left:1rem;padding-right:1rem}}html.theme--documenter-dark #documenter .docs-main .docs-footer .docs-footer-nextpage,html.theme--documenter-dark #documenter .docs-main .docs-footer .docs-footer-prevpage{flex-grow:1}html.theme--documenter-dark #documenter .docs-main .docs-footer .docs-footer-nextpage{text-align:right}html.theme--documenter-dark #documenter .docs-main .docs-footer .flexbox-break{flex-basis:100%;height:0}html.theme--documenter-dark #documenter .docs-main .docs-footer .footer-message{font-size:0.8em;margin:0.5em auto 0 auto;text-align:center}html.theme--documenter-dark #documenter .docs-sidebar{display:flex;flex-direction:column;color:#fff;background-color:#282f2f;border-right:1px solid #5e6d6f;padding:0;flex:0 0 18rem;z-index:5;font-size:1rem;position:fixed;left:-18rem;width:18rem;height:100%;transition:left 0.3s}html.theme--documenter-dark #documenter .docs-sidebar.visible{left:0;box-shadow:.4rem 0rem .8rem #171717}@media screen and (min-width: 1056px){html.theme--documenter-dark #documenter .docs-sidebar.visible{box-shadow:none}}@media screen and (min-width: 1056px){html.theme--documenter-dark #documenter .docs-sidebar{left:0;top:0}}html.theme--documenter-dark #documenter .docs-sidebar .docs-logo{margin-top:1rem;padding:0 1rem}html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img{max-height:6rem;margin:auto}html.theme--documenter-dark #documenter .docs-sidebar .docs-package-name{flex-shrink:0;font-size:1.5rem;font-weight:700;text-align:center;white-space:nowrap;overflow:hidden;padding:0.5rem 0}html.theme--documenter-dark #documenter .docs-sidebar .docs-package-name .docs-autofit{max-width:16.2rem}html.theme--documenter-dark #documenter .docs-sidebar .docs-package-name a,html.theme--documenter-dark #documenter .docs-sidebar .docs-package-name a:hover{color:#fff}html.theme--documenter-dark #documenter .docs-sidebar .docs-version-selector{border-top:1px solid #5e6d6f;display:none;padding:0.5rem}html.theme--documenter-dark #documenter .docs-sidebar .docs-version-selector.visible{display:flex}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu{flex-grow:1;user-select:none;border-top:1px solid #5e6d6f;padding-bottom:1.5rem}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu>li>.tocitem{font-weight:bold}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu>li li{font-size:.95rem;margin-left:1em;border-left:1px solid #5e6d6f}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu input.collapse-toggle{display:none}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.collapsed{display:none}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu input:checked~ul.collapsed{display:block}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem{display:flex}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem .docs-label{flex-grow:2}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem .docs-chevron{display:inline-block;font-style:normal;font-variant:normal;text-rendering:auto;line-height:1;font-size:.75rem;margin-left:1rem;margin-top:auto;margin-bottom:auto}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem .docs-chevron::before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f054"}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu input:checked~label.tocitem .docs-chevron::before{content:"\f078"}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu .tocitem{display:block;padding:0.5rem 0.5rem}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu .tocitem,html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu .tocitem:hover{color:#fff;background:#282f2f}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu a.tocitem:hover,html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem:hover{color:#fff;background-color:#32393a}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu li.is-active{border-top:1px solid #5e6d6f;border-bottom:1px solid #5e6d6f;background-color:#1f2424}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu li.is-active .tocitem,html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu li.is-active .tocitem:hover{background-color:#1f2424;color:#fff}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu li.is-active ul.internal .tocitem:hover{background-color:#32393a;color:#fff}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu>li.is-active:first-child{border-top:none}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.internal{margin:0 0.5rem 0.5rem;border-top:1px solid #5e6d6f}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.internal li{font-size:.85rem;border-left:none;margin-left:0;margin-top:0.5rem}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.internal .tocitem{width:100%;padding:0}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.internal .tocitem::before{content:"⚬";margin-right:0.4em}html.theme--documenter-dark #documenter .docs-sidebar form.docs-search{margin:auto;margin-top:0.5rem;margin-bottom:0.5rem}html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{width:14.4rem}html.theme--documenter-dark #documenter .docs-sidebar #documenter-search-query{color:#868c98;width:14.4rem;box-shadow:inset 0 1px 2px rgba(10,10,10,0.1)}@media screen and (min-width: 1056px){html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu{overflow-y:auto;-webkit-overflow-scroll:touch}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar{width:.3rem;background:none}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar-thumb{border-radius:5px 0px 0px 5px;background:#3b4445}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar-thumb:hover{background:#4e5a5c}}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-sidebar{overflow-y:auto;-webkit-overflow-scroll:touch}html.theme--documenter-dark #documenter .docs-sidebar::-webkit-scrollbar{width:.3rem;background:none}html.theme--documenter-dark #documenter .docs-sidebar::-webkit-scrollbar-thumb{border-radius:5px 0px 0px 5px;background:#3b4445}html.theme--documenter-dark #documenter .docs-sidebar::-webkit-scrollbar-thumb:hover{background:#4e5a5c}}html.theme--documenter-dark kbd.search-modal-key-hints{border-radius:0.25rem;border:1px solid rgba(245,245,245,0.6);box-shadow:0 2px 0 1px rgba(245,245,245,0.6);cursor:default;font-size:0.9rem;line-height:1.5;min-width:0.75rem;text-align:center;padding:0.1rem 0.3rem;position:relative;top:-1px}html.theme--documenter-dark .search-min-width-50{min-width:50%}html.theme--documenter-dark .search-min-height-100{min-height:100%}html.theme--documenter-dark .search-modal-card-body{max-height:calc(100vh - 15rem)}html.theme--documenter-dark .search-result-link{border-radius:0.7em;transition:all 300ms}html.theme--documenter-dark .search-result-link:hover,html.theme--documenter-dark .search-result-link:focus{background-color:rgba(0,128,128,0.1)}html.theme--documenter-dark .search-result-link .property-search-result-badge,html.theme--documenter-dark .search-result-link .search-filter{transition:all 300ms}html.theme--documenter-dark .property-search-result-badge,html.theme--documenter-dark .search-filter{padding:0.15em 0.5em;font-size:0.8em;font-style:italic;text-transform:none !important;line-height:1.5;color:#f5f5f5;background-color:rgba(51,65,85,0.501961);border-radius:0.6rem}html.theme--documenter-dark .search-result-link:hover .property-search-result-badge,html.theme--documenter-dark .search-result-link:hover .search-filter,html.theme--documenter-dark .search-result-link:focus .property-search-result-badge,html.theme--documenter-dark .search-result-link:focus .search-filter{color:#333;background-color:#f1f5f9}html.theme--documenter-dark .search-filter{color:#333;background-color:#f5f5f5;transition:all 300ms}html.theme--documenter-dark .search-filter:hover,html.theme--documenter-dark .search-filter:focus{color:#333}html.theme--documenter-dark .search-filter-selected{color:#f5f5f5;background-color:rgba(139,0,139,0.5)}html.theme--documenter-dark .search-filter-selected:hover,html.theme--documenter-dark .search-filter-selected:focus{color:#f5f5f5}html.theme--documenter-dark .search-result-highlight{background-color:#ffdd57;color:black}html.theme--documenter-dark .search-divider{border-bottom:1px solid #5e6d6f}html.theme--documenter-dark .search-result-title{width:85%;color:#f5f5f5}html.theme--documenter-dark .search-result-code-title{font-size:0.875rem;font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace}html.theme--documenter-dark #search-modal .modal-card-body::-webkit-scrollbar,html.theme--documenter-dark #search-modal .filter-tabs::-webkit-scrollbar{height:10px;width:10px;background-color:transparent}html.theme--documenter-dark #search-modal .modal-card-body::-webkit-scrollbar-thumb,html.theme--documenter-dark #search-modal .filter-tabs::-webkit-scrollbar-thumb{background-color:gray;border-radius:1rem}html.theme--documenter-dark #search-modal .modal-card-body::-webkit-scrollbar-track,html.theme--documenter-dark #search-modal .filter-tabs::-webkit-scrollbar-track{-webkit-box-shadow:inset 0 0 6px rgba(0,0,0,0.6);background-color:transparent}html.theme--documenter-dark .w-100{width:100%}html.theme--documenter-dark .gap-2{gap:0.5rem}html.theme--documenter-dark .gap-4{gap:1rem}html.theme--documenter-dark .gap-8{gap:2rem}html.theme--documenter-dark{background-color:#1f2424;font-size:16px;min-width:300px;overflow-x:auto;overflow-y:scroll;text-rendering:optimizeLegibility;text-size-adjust:100%}html.theme--documenter-dark .ansi span.sgr1{font-weight:bolder}html.theme--documenter-dark .ansi span.sgr2{font-weight:lighter}html.theme--documenter-dark .ansi span.sgr3{font-style:italic}html.theme--documenter-dark .ansi span.sgr4{text-decoration:underline}html.theme--documenter-dark .ansi span.sgr7{color:#1f2424;background-color:#fff}html.theme--documenter-dark .ansi span.sgr8{color:transparent}html.theme--documenter-dark .ansi span.sgr8 span{color:transparent}html.theme--documenter-dark .ansi span.sgr9{text-decoration:line-through}html.theme--documenter-dark .ansi span.sgr30{color:#242424}html.theme--documenter-dark .ansi span.sgr31{color:#f6705f}html.theme--documenter-dark .ansi span.sgr32{color:#4fb43a}html.theme--documenter-dark .ansi span.sgr33{color:#f4c72f}html.theme--documenter-dark .ansi span.sgr34{color:#7587f0}html.theme--documenter-dark .ansi span.sgr35{color:#bc89d3}html.theme--documenter-dark .ansi span.sgr36{color:#49b6ca}html.theme--documenter-dark .ansi span.sgr37{color:#b3bdbe}html.theme--documenter-dark .ansi span.sgr40{background-color:#242424}html.theme--documenter-dark .ansi span.sgr41{background-color:#f6705f}html.theme--documenter-dark .ansi span.sgr42{background-color:#4fb43a}html.theme--documenter-dark .ansi span.sgr43{background-color:#f4c72f}html.theme--documenter-dark .ansi span.sgr44{background-color:#7587f0}html.theme--documenter-dark .ansi span.sgr45{background-color:#bc89d3}html.theme--documenter-dark .ansi span.sgr46{background-color:#49b6ca}html.theme--documenter-dark .ansi span.sgr47{background-color:#b3bdbe}html.theme--documenter-dark .ansi span.sgr90{color:#92a0a2}html.theme--documenter-dark .ansi span.sgr91{color:#ff8674}html.theme--documenter-dark .ansi span.sgr92{color:#79d462}html.theme--documenter-dark .ansi span.sgr93{color:#ffe76b}html.theme--documenter-dark .ansi span.sgr94{color:#8a98ff}html.theme--documenter-dark .ansi span.sgr95{color:#d2a4e6}html.theme--documenter-dark .ansi span.sgr96{color:#6bc8db}html.theme--documenter-dark .ansi span.sgr97{color:#ecf0f1}html.theme--documenter-dark .ansi span.sgr100{background-color:#92a0a2}html.theme--documenter-dark .ansi span.sgr101{background-color:#ff8674}html.theme--documenter-dark .ansi span.sgr102{background-color:#79d462}html.theme--documenter-dark .ansi span.sgr103{background-color:#ffe76b}html.theme--documenter-dark .ansi span.sgr104{background-color:#8a98ff}html.theme--documenter-dark .ansi span.sgr105{background-color:#d2a4e6}html.theme--documenter-dark .ansi span.sgr106{background-color:#6bc8db}html.theme--documenter-dark .ansi span.sgr107{background-color:#ecf0f1}html.theme--documenter-dark code.language-julia-repl>span.hljs-meta{color:#4fb43a;font-weight:bolder}html.theme--documenter-dark .hljs{background:#2b2b2b;color:#f8f8f2}html.theme--documenter-dark .hljs-comment,html.theme--documenter-dark .hljs-quote{color:#d4d0ab}html.theme--documenter-dark .hljs-variable,html.theme--documenter-dark .hljs-template-variable,html.theme--documenter-dark .hljs-tag,html.theme--documenter-dark .hljs-name,html.theme--documenter-dark .hljs-selector-id,html.theme--documenter-dark .hljs-selector-class,html.theme--documenter-dark .hljs-regexp,html.theme--documenter-dark .hljs-deletion{color:#ffa07a}html.theme--documenter-dark .hljs-number,html.theme--documenter-dark .hljs-built_in,html.theme--documenter-dark .hljs-literal,html.theme--documenter-dark .hljs-type,html.theme--documenter-dark .hljs-params,html.theme--documenter-dark .hljs-meta,html.theme--documenter-dark .hljs-link{color:#f5ab35}html.theme--documenter-dark .hljs-attribute{color:#ffd700}html.theme--documenter-dark .hljs-string,html.theme--documenter-dark .hljs-symbol,html.theme--documenter-dark .hljs-bullet,html.theme--documenter-dark .hljs-addition{color:#abe338}html.theme--documenter-dark .hljs-title,html.theme--documenter-dark .hljs-section{color:#00e0e0}html.theme--documenter-dark .hljs-keyword,html.theme--documenter-dark .hljs-selector-tag{color:#dcc6e0}html.theme--documenter-dark .hljs-emphasis{font-style:italic}html.theme--documenter-dark .hljs-strong{font-weight:bold}@media screen and (-ms-high-contrast: active){html.theme--documenter-dark .hljs-addition,html.theme--documenter-dark .hljs-attribute,html.theme--documenter-dark .hljs-built_in,html.theme--documenter-dark .hljs-bullet,html.theme--documenter-dark .hljs-comment,html.theme--documenter-dark .hljs-link,html.theme--documenter-dark .hljs-literal,html.theme--documenter-dark .hljs-meta,html.theme--documenter-dark .hljs-number,html.theme--documenter-dark .hljs-params,html.theme--documenter-dark .hljs-string,html.theme--documenter-dark .hljs-symbol,html.theme--documenter-dark .hljs-type,html.theme--documenter-dark .hljs-quote{color:highlight}html.theme--documenter-dark .hljs-keyword,html.theme--documenter-dark .hljs-selector-tag{font-weight:bold}}html.theme--documenter-dark .hljs-subst{color:#f8f8f2}
diff --git a/previews/PR176/assets/themes/documenter-light.css b/previews/PR176/assets/themes/documenter-light.css
new file mode 100644
index 000000000..60a317a4c
--- /dev/null
+++ b/previews/PR176/assets/themes/documenter-light.css
@@ -0,0 +1,9 @@
+.pagination-previous,.pagination-next,.pagination-link,.pagination-ellipsis,.file-cta,.file-name,.select select,.textarea,.input,#documenter .docs-sidebar form.docs-search>input,.button{-moz-appearance:none;-webkit-appearance:none;align-items:center;border:1px solid transparent;border-radius:4px;box-shadow:none;display:inline-flex;font-size:1rem;height:2.5em;justify-content:flex-start;line-height:1.5;padding-bottom:calc(0.5em - 1px);padding-left:calc(0.75em - 1px);padding-right:calc(0.75em - 1px);padding-top:calc(0.5em - 1px);position:relative;vertical-align:top}.pagination-previous:focus,.pagination-next:focus,.pagination-link:focus,.pagination-ellipsis:focus,.file-cta:focus,.file-name:focus,.select select:focus,.textarea:focus,.input:focus,#documenter .docs-sidebar form.docs-search>input:focus,.button:focus,.is-focused.pagination-previous,.is-focused.pagination-next,.is-focused.pagination-link,.is-focused.pagination-ellipsis,.is-focused.file-cta,.is-focused.file-name,.select select.is-focused,.is-focused.textarea,.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-focused.button,.pagination-previous:active,.pagination-next:active,.pagination-link:active,.pagination-ellipsis:active,.file-cta:active,.file-name:active,.select select:active,.textarea:active,.input:active,#documenter .docs-sidebar form.docs-search>input:active,.button:active,.is-active.pagination-previous,.is-active.pagination-next,.is-active.pagination-link,.is-active.pagination-ellipsis,.is-active.file-cta,.is-active.file-name,.select select.is-active,.is-active.textarea,.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active,.is-active.button{outline:none}.pagination-previous[disabled],.pagination-next[disabled],.pagination-link[disabled],.pagination-ellipsis[disabled],.file-cta[disabled],.file-name[disabled],.select select[disabled],.textarea[disabled],.input[disabled],#documenter .docs-sidebar form.docs-search>input[disabled],.button[disabled],fieldset[disabled] .pagination-previous,fieldset[disabled] .pagination-next,fieldset[disabled] .pagination-link,fieldset[disabled] .pagination-ellipsis,fieldset[disabled] .file-cta,fieldset[disabled] .file-name,fieldset[disabled] .select select,.select fieldset[disabled] select,fieldset[disabled] .textarea,fieldset[disabled] .input,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input,fieldset[disabled] .button{cursor:not-allowed}.tabs,.pagination-previous,.pagination-next,.pagination-link,.pagination-ellipsis,.breadcrumb,.file,.button,.is-unselectable{-webkit-touch-callout:none;-webkit-user-select:none;-moz-user-select:none;-ms-user-select:none;user-select:none}.navbar-link:not(.is-arrowless)::after,.select:not(.is-multiple):not(.is-loading)::after{border:3px solid rgba(0,0,0,0);border-radius:2px;border-right:0;border-top:0;content:" ";display:block;height:0.625em;margin-top:-0.4375em;pointer-events:none;position:absolute;top:50%;transform:rotate(-45deg);transform-origin:center;width:0.625em}.admonition:not(:last-child),.tabs:not(:last-child),.pagination:not(:last-child),.message:not(:last-child),.level:not(:last-child),.breadcrumb:not(:last-child),.block:not(:last-child),.title:not(:last-child),.subtitle:not(:last-child),.table-container:not(:last-child),.table:not(:last-child),.progress:not(:last-child),.notification:not(:last-child),.content:not(:last-child),.box:not(:last-child){margin-bottom:1.5rem}.modal-close,.delete{-webkit-touch-callout:none;-webkit-user-select:none;-moz-user-select:none;-ms-user-select:none;user-select:none;-moz-appearance:none;-webkit-appearance:none;background-color:rgba(10,10,10,0.2);border:none;border-radius:9999px;cursor:pointer;pointer-events:auto;display:inline-block;flex-grow:0;flex-shrink:0;font-size:0;height:20px;max-height:20px;max-width:20px;min-height:20px;min-width:20px;outline:none;position:relative;vertical-align:top;width:20px}.modal-close::before,.delete::before,.modal-close::after,.delete::after{background-color:#fff;content:"";display:block;left:50%;position:absolute;top:50%;transform:translateX(-50%) translateY(-50%) rotate(45deg);transform-origin:center center}.modal-close::before,.delete::before{height:2px;width:50%}.modal-close::after,.delete::after{height:50%;width:2px}.modal-close:hover,.delete:hover,.modal-close:focus,.delete:focus{background-color:rgba(10,10,10,0.3)}.modal-close:active,.delete:active{background-color:rgba(10,10,10,0.4)}.is-small.modal-close,#documenter .docs-sidebar form.docs-search>input.modal-close,.is-small.delete,#documenter .docs-sidebar form.docs-search>input.delete{height:16px;max-height:16px;max-width:16px;min-height:16px;min-width:16px;width:16px}.is-medium.modal-close,.is-medium.delete{height:24px;max-height:24px;max-width:24px;min-height:24px;min-width:24px;width:24px}.is-large.modal-close,.is-large.delete{height:32px;max-height:32px;max-width:32px;min-height:32px;min-width:32px;width:32px}.control.is-loading::after,.select.is-loading::after,.loader,.button.is-loading::after{animation:spinAround 500ms infinite linear;border:2px solid #dbdbdb;border-radius:9999px;border-right-color:transparent;border-top-color:transparent;content:"";display:block;height:1em;position:relative;width:1em}.hero-video,.modal-background,.modal,.image.is-square img,#documenter .docs-sidebar .docs-logo>img.is-square img,.image.is-square .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-square .has-ratio,.image.is-1by1 img,#documenter .docs-sidebar .docs-logo>img.is-1by1 img,.image.is-1by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by1 .has-ratio,.image.is-5by4 img,#documenter .docs-sidebar .docs-logo>img.is-5by4 img,.image.is-5by4 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-5by4 .has-ratio,.image.is-4by3 img,#documenter .docs-sidebar .docs-logo>img.is-4by3 img,.image.is-4by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-4by3 .has-ratio,.image.is-3by2 img,#documenter .docs-sidebar .docs-logo>img.is-3by2 img,.image.is-3by2 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by2 .has-ratio,.image.is-5by3 img,#documenter .docs-sidebar .docs-logo>img.is-5by3 img,.image.is-5by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-5by3 .has-ratio,.image.is-16by9 img,#documenter .docs-sidebar .docs-logo>img.is-16by9 img,.image.is-16by9 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-16by9 .has-ratio,.image.is-2by1 img,#documenter .docs-sidebar .docs-logo>img.is-2by1 img,.image.is-2by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-2by1 .has-ratio,.image.is-3by1 img,#documenter .docs-sidebar .docs-logo>img.is-3by1 img,.image.is-3by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by1 .has-ratio,.image.is-4by5 img,#documenter .docs-sidebar .docs-logo>img.is-4by5 img,.image.is-4by5 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-4by5 .has-ratio,.image.is-3by4 img,#documenter .docs-sidebar .docs-logo>img.is-3by4 img,.image.is-3by4 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by4 .has-ratio,.image.is-2by3 img,#documenter .docs-sidebar .docs-logo>img.is-2by3 img,.image.is-2by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-2by3 .has-ratio,.image.is-3by5 img,#documenter .docs-sidebar .docs-logo>img.is-3by5 img,.image.is-3by5 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by5 .has-ratio,.image.is-9by16 img,#documenter .docs-sidebar .docs-logo>img.is-9by16 img,.image.is-9by16 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-9by16 .has-ratio,.image.is-1by2 img,#documenter .docs-sidebar .docs-logo>img.is-1by2 img,.image.is-1by2 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by2 .has-ratio,.image.is-1by3 img,#documenter .docs-sidebar .docs-logo>img.is-1by3 img,.image.is-1by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by3 .has-ratio,.is-overlay{bottom:0;left:0;position:absolute;right:0;top:0}.navbar-burger{-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;color:currentColor;font-family:inherit;font-size:1em;margin:0;padding:0}.has-text-white{color:#fff !important}a.has-text-white:hover,a.has-text-white:focus{color:#e6e6e6 !important}.has-background-white{background-color:#fff !important}.has-text-black{color:#0a0a0a !important}a.has-text-black:hover,a.has-text-black:focus{color:#000 !important}.has-background-black{background-color:#0a0a0a !important}.has-text-light{color:#f5f5f5 !important}a.has-text-light:hover,a.has-text-light:focus{color:#dbdbdb !important}.has-background-light{background-color:#f5f5f5 !important}.has-text-dark{color:#363636 !important}a.has-text-dark:hover,a.has-text-dark:focus{color:#1c1c1c !important}.has-background-dark{background-color:#363636 !important}.has-text-primary{color:#4eb5de !important}a.has-text-primary:hover,a.has-text-primary:focus{color:#27a1d2 !important}.has-background-primary{background-color:#4eb5de !important}.has-text-primary-light{color:#eef8fc !important}a.has-text-primary-light:hover,a.has-text-primary-light:focus{color:#c3e6f4 !important}.has-background-primary-light{background-color:#eef8fc !important}.has-text-primary-dark{color:#1a6d8e !important}a.has-text-primary-dark:hover,a.has-text-primary-dark:focus{color:#228eb9 !important}.has-background-primary-dark{background-color:#1a6d8e !important}.has-text-link{color:#2e63b8 !important}a.has-text-link:hover,a.has-text-link:focus{color:#244d8f !important}.has-background-link{background-color:#2e63b8 !important}.has-text-link-light{color:#eff3fb !important}a.has-text-link-light:hover,a.has-text-link-light:focus{color:#c6d6f1 !important}.has-background-link-light{background-color:#eff3fb !important}.has-text-link-dark{color:#3169c4 !important}a.has-text-link-dark:hover,a.has-text-link-dark:focus{color:#5485d4 !important}.has-background-link-dark{background-color:#3169c4 !important}.has-text-info{color:#209cee !important}a.has-text-info:hover,a.has-text-info:focus{color:#1081cb !important}.has-background-info{background-color:#209cee !important}.has-text-info-light{color:#ecf7fe !important}a.has-text-info-light:hover,a.has-text-info-light:focus{color:#bde2fa !important}.has-background-info-light{background-color:#ecf7fe !important}.has-text-info-dark{color:#0e72b4 !important}a.has-text-info-dark:hover,a.has-text-info-dark:focus{color:#1190e3 !important}.has-background-info-dark{background-color:#0e72b4 !important}.has-text-success{color:#22c35b !important}a.has-text-success:hover,a.has-text-success:focus{color:#1a9847 !important}.has-background-success{background-color:#22c35b !important}.has-text-success-light{color:#eefcf3 !important}a.has-text-success-light:hover,a.has-text-success-light:focus{color:#c2f4d4 !important}.has-background-success-light{background-color:#eefcf3 !important}.has-text-success-dark{color:#198f43 !important}a.has-text-success-dark:hover,a.has-text-success-dark:focus{color:#21bb57 !important}.has-background-success-dark{background-color:#198f43 !important}.has-text-warning{color:#ffdd57 !important}a.has-text-warning:hover,a.has-text-warning:focus{color:#ffd324 !important}.has-background-warning{background-color:#ffdd57 !important}.has-text-warning-light{color:#fffbeb !important}a.has-text-warning-light:hover,a.has-text-warning-light:focus{color:#fff1b8 !important}.has-background-warning-light{background-color:#fffbeb !important}.has-text-warning-dark{color:#947600 !important}a.has-text-warning-dark:hover,a.has-text-warning-dark:focus{color:#c79f00 !important}.has-background-warning-dark{background-color:#947600 !important}.has-text-danger{color:#da0b00 !important}a.has-text-danger:hover,a.has-text-danger:focus{color:#a70800 !important}.has-background-danger{background-color:#da0b00 !important}.has-text-danger-light{color:#ffeceb !important}a.has-text-danger-light:hover,a.has-text-danger-light:focus{color:#ffbbb8 !important}.has-background-danger-light{background-color:#ffeceb !important}.has-text-danger-dark{color:#f50c00 !important}a.has-text-danger-dark:hover,a.has-text-danger-dark:focus{color:#ff3429 !important}.has-background-danger-dark{background-color:#f50c00 !important}.has-text-black-bis{color:#121212 !important}.has-background-black-bis{background-color:#121212 !important}.has-text-black-ter{color:#242424 !important}.has-background-black-ter{background-color:#242424 !important}.has-text-grey-darker{color:#363636 !important}.has-background-grey-darker{background-color:#363636 !important}.has-text-grey-dark{color:#4a4a4a !important}.has-background-grey-dark{background-color:#4a4a4a !important}.has-text-grey{color:#6b6b6b !important}.has-background-grey{background-color:#6b6b6b !important}.has-text-grey-light{color:#b5b5b5 !important}.has-background-grey-light{background-color:#b5b5b5 !important}.has-text-grey-lighter{color:#dbdbdb !important}.has-background-grey-lighter{background-color:#dbdbdb !important}.has-text-white-ter{color:#f5f5f5 !important}.has-background-white-ter{background-color:#f5f5f5 !important}.has-text-white-bis{color:#fafafa !important}.has-background-white-bis{background-color:#fafafa !important}.is-flex-direction-row{flex-direction:row !important}.is-flex-direction-row-reverse{flex-direction:row-reverse !important}.is-flex-direction-column{flex-direction:column !important}.is-flex-direction-column-reverse{flex-direction:column-reverse !important}.is-flex-wrap-nowrap{flex-wrap:nowrap !important}.is-flex-wrap-wrap{flex-wrap:wrap !important}.is-flex-wrap-wrap-reverse{flex-wrap:wrap-reverse !important}.is-justify-content-flex-start{justify-content:flex-start !important}.is-justify-content-flex-end{justify-content:flex-end !important}.is-justify-content-center{justify-content:center !important}.is-justify-content-space-between{justify-content:space-between !important}.is-justify-content-space-around{justify-content:space-around !important}.is-justify-content-space-evenly{justify-content:space-evenly !important}.is-justify-content-start{justify-content:start !important}.is-justify-content-end{justify-content:end !important}.is-justify-content-left{justify-content:left !important}.is-justify-content-right{justify-content:right !important}.is-align-content-flex-start{align-content:flex-start !important}.is-align-content-flex-end{align-content:flex-end !important}.is-align-content-center{align-content:center !important}.is-align-content-space-between{align-content:space-between !important}.is-align-content-space-around{align-content:space-around !important}.is-align-content-space-evenly{align-content:space-evenly !important}.is-align-content-stretch{align-content:stretch !important}.is-align-content-start{align-content:start !important}.is-align-content-end{align-content:end !important}.is-align-content-baseline{align-content:baseline !important}.is-align-items-stretch{align-items:stretch !important}.is-align-items-flex-start{align-items:flex-start !important}.is-align-items-flex-end{align-items:flex-end !important}.is-align-items-center{align-items:center !important}.is-align-items-baseline{align-items:baseline !important}.is-align-items-start{align-items:start !important}.is-align-items-end{align-items:end !important}.is-align-items-self-start{align-items:self-start !important}.is-align-items-self-end{align-items:self-end !important}.is-align-self-auto{align-self:auto !important}.is-align-self-flex-start{align-self:flex-start !important}.is-align-self-flex-end{align-self:flex-end !important}.is-align-self-center{align-self:center !important}.is-align-self-baseline{align-self:baseline !important}.is-align-self-stretch{align-self:stretch !important}.is-flex-grow-0{flex-grow:0 !important}.is-flex-grow-1{flex-grow:1 !important}.is-flex-grow-2{flex-grow:2 !important}.is-flex-grow-3{flex-grow:3 !important}.is-flex-grow-4{flex-grow:4 !important}.is-flex-grow-5{flex-grow:5 !important}.is-flex-shrink-0{flex-shrink:0 !important}.is-flex-shrink-1{flex-shrink:1 !important}.is-flex-shrink-2{flex-shrink:2 !important}.is-flex-shrink-3{flex-shrink:3 !important}.is-flex-shrink-4{flex-shrink:4 !important}.is-flex-shrink-5{flex-shrink:5 !important}.is-clearfix::after{clear:both;content:" ";display:table}.is-pulled-left{float:left !important}.is-pulled-right{float:right !important}.is-radiusless{border-radius:0 !important}.is-shadowless{box-shadow:none !important}.is-clickable{cursor:pointer !important;pointer-events:all !important}.is-clipped{overflow:hidden !important}.is-relative{position:relative !important}.is-marginless{margin:0 !important}.is-paddingless{padding:0 !important}.m-0{margin:0 !important}.mt-0{margin-top:0 !important}.mr-0{margin-right:0 !important}.mb-0{margin-bottom:0 !important}.ml-0{margin-left:0 !important}.mx-0{margin-left:0 !important;margin-right:0 !important}.my-0{margin-top:0 !important;margin-bottom:0 !important}.m-1{margin:.25rem !important}.mt-1{margin-top:.25rem !important}.mr-1{margin-right:.25rem !important}.mb-1{margin-bottom:.25rem !important}.ml-1{margin-left:.25rem !important}.mx-1{margin-left:.25rem !important;margin-right:.25rem !important}.my-1{margin-top:.25rem !important;margin-bottom:.25rem !important}.m-2{margin:.5rem !important}.mt-2{margin-top:.5rem !important}.mr-2{margin-right:.5rem !important}.mb-2{margin-bottom:.5rem !important}.ml-2{margin-left:.5rem !important}.mx-2{margin-left:.5rem !important;margin-right:.5rem !important}.my-2{margin-top:.5rem !important;margin-bottom:.5rem !important}.m-3{margin:.75rem !important}.mt-3{margin-top:.75rem !important}.mr-3{margin-right:.75rem !important}.mb-3{margin-bottom:.75rem !important}.ml-3{margin-left:.75rem !important}.mx-3{margin-left:.75rem !important;margin-right:.75rem !important}.my-3{margin-top:.75rem !important;margin-bottom:.75rem !important}.m-4{margin:1rem !important}.mt-4{margin-top:1rem !important}.mr-4{margin-right:1rem !important}.mb-4{margin-bottom:1rem !important}.ml-4{margin-left:1rem !important}.mx-4{margin-left:1rem !important;margin-right:1rem !important}.my-4{margin-top:1rem !important;margin-bottom:1rem !important}.m-5{margin:1.5rem !important}.mt-5{margin-top:1.5rem !important}.mr-5{margin-right:1.5rem !important}.mb-5{margin-bottom:1.5rem !important}.ml-5{margin-left:1.5rem !important}.mx-5{margin-left:1.5rem !important;margin-right:1.5rem !important}.my-5{margin-top:1.5rem !important;margin-bottom:1.5rem !important}.m-6{margin:3rem !important}.mt-6{margin-top:3rem !important}.mr-6{margin-right:3rem !important}.mb-6{margin-bottom:3rem !important}.ml-6{margin-left:3rem !important}.mx-6{margin-left:3rem !important;margin-right:3rem !important}.my-6{margin-top:3rem !important;margin-bottom:3rem !important}.m-auto{margin:auto !important}.mt-auto{margin-top:auto !important}.mr-auto{margin-right:auto !important}.mb-auto{margin-bottom:auto !important}.ml-auto{margin-left:auto !important}.mx-auto{margin-left:auto !important;margin-right:auto !important}.my-auto{margin-top:auto !important;margin-bottom:auto !important}.p-0{padding:0 !important}.pt-0{padding-top:0 !important}.pr-0{padding-right:0 !important}.pb-0{padding-bottom:0 !important}.pl-0{padding-left:0 !important}.px-0{padding-left:0 !important;padding-right:0 !important}.py-0{padding-top:0 !important;padding-bottom:0 !important}.p-1{padding:.25rem !important}.pt-1{padding-top:.25rem !important}.pr-1{padding-right:.25rem !important}.pb-1{padding-bottom:.25rem !important}.pl-1{padding-left:.25rem !important}.px-1{padding-left:.25rem !important;padding-right:.25rem !important}.py-1{padding-top:.25rem !important;padding-bottom:.25rem !important}.p-2{padding:.5rem !important}.pt-2{padding-top:.5rem !important}.pr-2{padding-right:.5rem !important}.pb-2{padding-bottom:.5rem !important}.pl-2{padding-left:.5rem !important}.px-2{padding-left:.5rem !important;padding-right:.5rem !important}.py-2{padding-top:.5rem !important;padding-bottom:.5rem !important}.p-3{padding:.75rem !important}.pt-3{padding-top:.75rem !important}.pr-3{padding-right:.75rem !important}.pb-3{padding-bottom:.75rem !important}.pl-3{padding-left:.75rem !important}.px-3{padding-left:.75rem !important;padding-right:.75rem !important}.py-3{padding-top:.75rem !important;padding-bottom:.75rem !important}.p-4{padding:1rem !important}.pt-4{padding-top:1rem !important}.pr-4{padding-right:1rem !important}.pb-4{padding-bottom:1rem !important}.pl-4{padding-left:1rem !important}.px-4{padding-left:1rem !important;padding-right:1rem !important}.py-4{padding-top:1rem !important;padding-bottom:1rem !important}.p-5{padding:1.5rem !important}.pt-5{padding-top:1.5rem !important}.pr-5{padding-right:1.5rem !important}.pb-5{padding-bottom:1.5rem !important}.pl-5{padding-left:1.5rem !important}.px-5{padding-left:1.5rem !important;padding-right:1.5rem !important}.py-5{padding-top:1.5rem !important;padding-bottom:1.5rem !important}.p-6{padding:3rem !important}.pt-6{padding-top:3rem !important}.pr-6{padding-right:3rem !important}.pb-6{padding-bottom:3rem !important}.pl-6{padding-left:3rem !important}.px-6{padding-left:3rem !important;padding-right:3rem !important}.py-6{padding-top:3rem !important;padding-bottom:3rem !important}.p-auto{padding:auto !important}.pt-auto{padding-top:auto !important}.pr-auto{padding-right:auto !important}.pb-auto{padding-bottom:auto !important}.pl-auto{padding-left:auto !important}.px-auto{padding-left:auto !important;padding-right:auto !important}.py-auto{padding-top:auto !important;padding-bottom:auto !important}.is-size-1{font-size:3rem !important}.is-size-2{font-size:2.5rem !important}.is-size-3{font-size:2rem !important}.is-size-4{font-size:1.5rem !important}.is-size-5{font-size:1.25rem !important}.is-size-6{font-size:1rem !important}.is-size-7,.docstring>section>a.docs-sourcelink{font-size:.75rem !important}@media screen and (max-width: 768px){.is-size-1-mobile{font-size:3rem !important}.is-size-2-mobile{font-size:2.5rem !important}.is-size-3-mobile{font-size:2rem !important}.is-size-4-mobile{font-size:1.5rem !important}.is-size-5-mobile{font-size:1.25rem !important}.is-size-6-mobile{font-size:1rem !important}.is-size-7-mobile{font-size:.75rem !important}}@media screen and (min-width: 769px),print{.is-size-1-tablet{font-size:3rem !important}.is-size-2-tablet{font-size:2.5rem !important}.is-size-3-tablet{font-size:2rem !important}.is-size-4-tablet{font-size:1.5rem !important}.is-size-5-tablet{font-size:1.25rem !important}.is-size-6-tablet{font-size:1rem !important}.is-size-7-tablet{font-size:.75rem !important}}@media screen and (max-width: 1055px){.is-size-1-touch{font-size:3rem !important}.is-size-2-touch{font-size:2.5rem !important}.is-size-3-touch{font-size:2rem !important}.is-size-4-touch{font-size:1.5rem !important}.is-size-5-touch{font-size:1.25rem !important}.is-size-6-touch{font-size:1rem !important}.is-size-7-touch{font-size:.75rem !important}}@media screen and (min-width: 1056px){.is-size-1-desktop{font-size:3rem !important}.is-size-2-desktop{font-size:2.5rem !important}.is-size-3-desktop{font-size:2rem !important}.is-size-4-desktop{font-size:1.5rem !important}.is-size-5-desktop{font-size:1.25rem !important}.is-size-6-desktop{font-size:1rem !important}.is-size-7-desktop{font-size:.75rem !important}}@media screen and (min-width: 1216px){.is-size-1-widescreen{font-size:3rem !important}.is-size-2-widescreen{font-size:2.5rem !important}.is-size-3-widescreen{font-size:2rem !important}.is-size-4-widescreen{font-size:1.5rem !important}.is-size-5-widescreen{font-size:1.25rem !important}.is-size-6-widescreen{font-size:1rem !important}.is-size-7-widescreen{font-size:.75rem !important}}@media screen and (min-width: 1408px){.is-size-1-fullhd{font-size:3rem !important}.is-size-2-fullhd{font-size:2.5rem !important}.is-size-3-fullhd{font-size:2rem !important}.is-size-4-fullhd{font-size:1.5rem !important}.is-size-5-fullhd{font-size:1.25rem !important}.is-size-6-fullhd{font-size:1rem !important}.is-size-7-fullhd{font-size:.75rem !important}}.has-text-centered{text-align:center !important}.has-text-justified{text-align:justify !important}.has-text-left{text-align:left !important}.has-text-right{text-align:right !important}@media screen and (max-width: 768px){.has-text-centered-mobile{text-align:center !important}}@media screen and (min-width: 769px),print{.has-text-centered-tablet{text-align:center !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-centered-tablet-only{text-align:center !important}}@media screen and (max-width: 1055px){.has-text-centered-touch{text-align:center !important}}@media screen and (min-width: 1056px){.has-text-centered-desktop{text-align:center !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-centered-desktop-only{text-align:center !important}}@media screen and (min-width: 1216px){.has-text-centered-widescreen{text-align:center !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-centered-widescreen-only{text-align:center !important}}@media screen and (min-width: 1408px){.has-text-centered-fullhd{text-align:center !important}}@media screen and (max-width: 768px){.has-text-justified-mobile{text-align:justify !important}}@media screen and (min-width: 769px),print{.has-text-justified-tablet{text-align:justify !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-justified-tablet-only{text-align:justify !important}}@media screen and (max-width: 1055px){.has-text-justified-touch{text-align:justify !important}}@media screen and (min-width: 1056px){.has-text-justified-desktop{text-align:justify !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-justified-desktop-only{text-align:justify !important}}@media screen and (min-width: 1216px){.has-text-justified-widescreen{text-align:justify !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-justified-widescreen-only{text-align:justify !important}}@media screen and (min-width: 1408px){.has-text-justified-fullhd{text-align:justify !important}}@media screen and (max-width: 768px){.has-text-left-mobile{text-align:left !important}}@media screen and (min-width: 769px),print{.has-text-left-tablet{text-align:left !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-left-tablet-only{text-align:left !important}}@media screen and (max-width: 1055px){.has-text-left-touch{text-align:left !important}}@media screen and (min-width: 1056px){.has-text-left-desktop{text-align:left !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-left-desktop-only{text-align:left !important}}@media screen and (min-width: 1216px){.has-text-left-widescreen{text-align:left !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-left-widescreen-only{text-align:left !important}}@media screen and (min-width: 1408px){.has-text-left-fullhd{text-align:left !important}}@media screen and (max-width: 768px){.has-text-right-mobile{text-align:right !important}}@media screen and (min-width: 769px),print{.has-text-right-tablet{text-align:right !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-right-tablet-only{text-align:right !important}}@media screen and (max-width: 1055px){.has-text-right-touch{text-align:right !important}}@media screen and (min-width: 1056px){.has-text-right-desktop{text-align:right !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-right-desktop-only{text-align:right !important}}@media screen and (min-width: 1216px){.has-text-right-widescreen{text-align:right !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-right-widescreen-only{text-align:right !important}}@media screen and (min-width: 1408px){.has-text-right-fullhd{text-align:right !important}}.is-capitalized{text-transform:capitalize !important}.is-lowercase{text-transform:lowercase !important}.is-uppercase{text-transform:uppercase !important}.is-italic{font-style:italic !important}.is-underlined{text-decoration:underline !important}.has-text-weight-light{font-weight:300 !important}.has-text-weight-normal{font-weight:400 !important}.has-text-weight-medium{font-weight:500 !important}.has-text-weight-semibold{font-weight:600 !important}.has-text-weight-bold{font-weight:700 !important}.is-family-primary{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-secondary{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-sans-serif{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-monospace{font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace !important}.is-family-code{font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace !important}.is-block{display:block !important}@media screen and (max-width: 768px){.is-block-mobile{display:block !important}}@media screen and (min-width: 769px),print{.is-block-tablet{display:block !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-block-tablet-only{display:block !important}}@media screen and (max-width: 1055px){.is-block-touch{display:block !important}}@media screen and (min-width: 1056px){.is-block-desktop{display:block !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-block-desktop-only{display:block !important}}@media screen and (min-width: 1216px){.is-block-widescreen{display:block !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-block-widescreen-only{display:block !important}}@media screen and (min-width: 1408px){.is-block-fullhd{display:block !important}}.is-flex{display:flex !important}@media screen and (max-width: 768px){.is-flex-mobile{display:flex !important}}@media screen and (min-width: 769px),print{.is-flex-tablet{display:flex !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-flex-tablet-only{display:flex !important}}@media screen and (max-width: 1055px){.is-flex-touch{display:flex !important}}@media screen and (min-width: 1056px){.is-flex-desktop{display:flex !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-flex-desktop-only{display:flex !important}}@media screen and (min-width: 1216px){.is-flex-widescreen{display:flex !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-flex-widescreen-only{display:flex !important}}@media screen and (min-width: 1408px){.is-flex-fullhd{display:flex !important}}.is-inline{display:inline !important}@media screen and (max-width: 768px){.is-inline-mobile{display:inline !important}}@media screen and (min-width: 769px),print{.is-inline-tablet{display:inline !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-tablet-only{display:inline !important}}@media screen and (max-width: 1055px){.is-inline-touch{display:inline !important}}@media screen and (min-width: 1056px){.is-inline-desktop{display:inline !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-desktop-only{display:inline !important}}@media screen and (min-width: 1216px){.is-inline-widescreen{display:inline !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-widescreen-only{display:inline !important}}@media screen and (min-width: 1408px){.is-inline-fullhd{display:inline !important}}.is-inline-block{display:inline-block !important}@media screen and (max-width: 768px){.is-inline-block-mobile{display:inline-block !important}}@media screen and (min-width: 769px),print{.is-inline-block-tablet{display:inline-block !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-block-tablet-only{display:inline-block !important}}@media screen and (max-width: 1055px){.is-inline-block-touch{display:inline-block !important}}@media screen and (min-width: 1056px){.is-inline-block-desktop{display:inline-block !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-block-desktop-only{display:inline-block !important}}@media screen and (min-width: 1216px){.is-inline-block-widescreen{display:inline-block !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-block-widescreen-only{display:inline-block !important}}@media screen and (min-width: 1408px){.is-inline-block-fullhd{display:inline-block !important}}.is-inline-flex{display:inline-flex !important}@media screen and (max-width: 768px){.is-inline-flex-mobile{display:inline-flex !important}}@media screen and (min-width: 769px),print{.is-inline-flex-tablet{display:inline-flex !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-flex-tablet-only{display:inline-flex !important}}@media screen and (max-width: 1055px){.is-inline-flex-touch{display:inline-flex !important}}@media screen and (min-width: 1056px){.is-inline-flex-desktop{display:inline-flex !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-flex-desktop-only{display:inline-flex !important}}@media screen and (min-width: 1216px){.is-inline-flex-widescreen{display:inline-flex !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-flex-widescreen-only{display:inline-flex !important}}@media screen and (min-width: 1408px){.is-inline-flex-fullhd{display:inline-flex !important}}.is-hidden{display:none !important}.is-sr-only{border:none !important;clip:rect(0, 0, 0, 0) !important;height:0.01em !important;overflow:hidden !important;padding:0 !important;position:absolute !important;white-space:nowrap !important;width:0.01em !important}@media screen and (max-width: 768px){.is-hidden-mobile{display:none !important}}@media screen and (min-width: 769px),print{.is-hidden-tablet{display:none !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-hidden-tablet-only{display:none !important}}@media screen and (max-width: 1055px){.is-hidden-touch{display:none !important}}@media screen and (min-width: 1056px){.is-hidden-desktop{display:none !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-hidden-desktop-only{display:none !important}}@media screen and (min-width: 1216px){.is-hidden-widescreen{display:none !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-hidden-widescreen-only{display:none !important}}@media screen and (min-width: 1408px){.is-hidden-fullhd{display:none !important}}.is-invisible{visibility:hidden !important}@media screen and (max-width: 768px){.is-invisible-mobile{visibility:hidden !important}}@media screen and (min-width: 769px),print{.is-invisible-tablet{visibility:hidden !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-invisible-tablet-only{visibility:hidden !important}}@media screen and (max-width: 1055px){.is-invisible-touch{visibility:hidden !important}}@media screen and (min-width: 1056px){.is-invisible-desktop{visibility:hidden !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-invisible-desktop-only{visibility:hidden !important}}@media screen and (min-width: 1216px){.is-invisible-widescreen{visibility:hidden !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-invisible-widescreen-only{visibility:hidden !important}}@media screen and (min-width: 1408px){.is-invisible-fullhd{visibility:hidden !important}}/*! minireset.css v0.0.6 | MIT License | github.com/jgthms/minireset.css */html,body,p,ol,ul,li,dl,dt,dd,blockquote,figure,fieldset,legend,textarea,pre,iframe,hr,h1,h2,h3,h4,h5,h6{margin:0;padding:0}h1,h2,h3,h4,h5,h6{font-size:100%;font-weight:normal}ul{list-style:none}button,input,select,textarea{margin:0}html{box-sizing:border-box}*,*::before,*::after{box-sizing:inherit}img,video{height:auto;max-width:100%}iframe{border:0}table{border-collapse:collapse;border-spacing:0}td,th{padding:0}td:not([align]),th:not([align]){text-align:inherit}html{background-color:#fff;font-size:16px;-moz-osx-font-smoothing:grayscale;-webkit-font-smoothing:antialiased;min-width:300px;overflow-x:auto;overflow-y:scroll;text-rendering:optimizeLegibility;text-size-adjust:100%}article,aside,figure,footer,header,hgroup,section{display:block}body,button,input,optgroup,select,textarea{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif}code,pre{-moz-osx-font-smoothing:auto;-webkit-font-smoothing:auto;font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace}body{color:#222;font-size:1em;font-weight:400;line-height:1.5}a{color:#2e63b8;cursor:pointer;text-decoration:none}a strong{color:currentColor}a:hover{color:#363636}code{background-color:rgba(0,0,0,0.05);color:#000;font-size:.875em;font-weight:normal;padding:.1em}hr{background-color:#f5f5f5;border:none;display:block;height:2px;margin:1.5rem 0}img{height:auto;max-width:100%}input[type="checkbox"],input[type="radio"]{vertical-align:baseline}small{font-size:.875em}span{font-style:inherit;font-weight:inherit}strong{color:#222;font-weight:700}fieldset{border:none}pre{-webkit-overflow-scrolling:touch;background-color:#f5f5f5;color:#222;font-size:.875em;overflow-x:auto;padding:1.25rem 1.5rem;white-space:pre;word-wrap:normal}pre code{background-color:transparent;color:currentColor;font-size:1em;padding:0}table td,table th{vertical-align:top}table td:not([align]),table th:not([align]){text-align:inherit}table th{color:#222}@keyframes spinAround{from{transform:rotate(0deg)}to{transform:rotate(359deg)}}.box{background-color:#fff;border-radius:6px;box-shadow:#bbb;color:#222;display:block;padding:1.25rem}a.box:hover,a.box:focus{box-shadow:0 0.5em 1em -0.125em rgba(10,10,10,0.1),0 0 0 1px #2e63b8}a.box:active{box-shadow:inset 0 1px 2px rgba(10,10,10,0.2),0 0 0 1px #2e63b8}.button{background-color:#fff;border-color:#dbdbdb;border-width:1px;color:#222;cursor:pointer;justify-content:center;padding-bottom:calc(0.5em - 1px);padding-left:1em;padding-right:1em;padding-top:calc(0.5em - 1px);text-align:center;white-space:nowrap}.button strong{color:inherit}.button .icon,.button .icon.is-small,.button #documenter .docs-sidebar form.docs-search>input.icon,#documenter .docs-sidebar .button form.docs-search>input.icon,.button .icon.is-medium,.button .icon.is-large{height:1.5em;width:1.5em}.button .icon:first-child:not(:last-child){margin-left:calc(-0.5em - 1px);margin-right:.25em}.button .icon:last-child:not(:first-child){margin-left:.25em;margin-right:calc(-0.5em - 1px)}.button .icon:first-child:last-child{margin-left:calc(-0.5em - 1px);margin-right:calc(-0.5em - 1px)}.button:hover,.button.is-hovered{border-color:#b5b5b5;color:#363636}.button:focus,.button.is-focused{border-color:#3c5dcd;color:#363636}.button:focus:not(:active),.button.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.button:active,.button.is-active{border-color:#4a4a4a;color:#363636}.button.is-text{background-color:transparent;border-color:transparent;color:#222;text-decoration:underline}.button.is-text:hover,.button.is-text.is-hovered,.button.is-text:focus,.button.is-text.is-focused{background-color:#f5f5f5;color:#222}.button.is-text:active,.button.is-text.is-active{background-color:#e8e8e8;color:#222}.button.is-text[disabled],fieldset[disabled] .button.is-text{background-color:transparent;border-color:transparent;box-shadow:none}.button.is-ghost{background:none;border-color:rgba(0,0,0,0);color:#2e63b8;text-decoration:none}.button.is-ghost:hover,.button.is-ghost.is-hovered{color:#2e63b8;text-decoration:underline}.button.is-white{background-color:#fff;border-color:transparent;color:#0a0a0a}.button.is-white:hover,.button.is-white.is-hovered{background-color:#f9f9f9;border-color:transparent;color:#0a0a0a}.button.is-white:focus,.button.is-white.is-focused{border-color:transparent;color:#0a0a0a}.button.is-white:focus:not(:active),.button.is-white.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}.button.is-white:active,.button.is-white.is-active{background-color:#f2f2f2;border-color:transparent;color:#0a0a0a}.button.is-white[disabled],fieldset[disabled] .button.is-white{background-color:#fff;border-color:#fff;box-shadow:none}.button.is-white.is-inverted{background-color:#0a0a0a;color:#fff}.button.is-white.is-inverted:hover,.button.is-white.is-inverted.is-hovered{background-color:#000}.button.is-white.is-inverted[disabled],fieldset[disabled] .button.is-white.is-inverted{background-color:#0a0a0a;border-color:transparent;box-shadow:none;color:#fff}.button.is-white.is-loading::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}.button.is-white.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-white.is-outlined:hover,.button.is-white.is-outlined.is-hovered,.button.is-white.is-outlined:focus,.button.is-white.is-outlined.is-focused{background-color:#fff;border-color:#fff;color:#0a0a0a}.button.is-white.is-outlined.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-white.is-outlined.is-loading:hover::after,.button.is-white.is-outlined.is-loading.is-hovered::after,.button.is-white.is-outlined.is-loading:focus::after,.button.is-white.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}.button.is-white.is-outlined[disabled],fieldset[disabled] .button.is-white.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-white.is-inverted.is-outlined{background-color:transparent;border-color:#0a0a0a;color:#0a0a0a}.button.is-white.is-inverted.is-outlined:hover,.button.is-white.is-inverted.is-outlined.is-hovered,.button.is-white.is-inverted.is-outlined:focus,.button.is-white.is-inverted.is-outlined.is-focused{background-color:#0a0a0a;color:#fff}.button.is-white.is-inverted.is-outlined.is-loading:hover::after,.button.is-white.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-white.is-inverted.is-outlined.is-loading:focus::after,.button.is-white.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-white.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-white.is-inverted.is-outlined{background-color:transparent;border-color:#0a0a0a;box-shadow:none;color:#0a0a0a}.button.is-black{background-color:#0a0a0a;border-color:transparent;color:#fff}.button.is-black:hover,.button.is-black.is-hovered{background-color:#040404;border-color:transparent;color:#fff}.button.is-black:focus,.button.is-black.is-focused{border-color:transparent;color:#fff}.button.is-black:focus:not(:active),.button.is-black.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}.button.is-black:active,.button.is-black.is-active{background-color:#000;border-color:transparent;color:#fff}.button.is-black[disabled],fieldset[disabled] .button.is-black{background-color:#0a0a0a;border-color:#0a0a0a;box-shadow:none}.button.is-black.is-inverted{background-color:#fff;color:#0a0a0a}.button.is-black.is-inverted:hover,.button.is-black.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-black.is-inverted[disabled],fieldset[disabled] .button.is-black.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#0a0a0a}.button.is-black.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-black.is-outlined{background-color:transparent;border-color:#0a0a0a;color:#0a0a0a}.button.is-black.is-outlined:hover,.button.is-black.is-outlined.is-hovered,.button.is-black.is-outlined:focus,.button.is-black.is-outlined.is-focused{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}.button.is-black.is-outlined.is-loading::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}.button.is-black.is-outlined.is-loading:hover::after,.button.is-black.is-outlined.is-loading.is-hovered::after,.button.is-black.is-outlined.is-loading:focus::after,.button.is-black.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-black.is-outlined[disabled],fieldset[disabled] .button.is-black.is-outlined{background-color:transparent;border-color:#0a0a0a;box-shadow:none;color:#0a0a0a}.button.is-black.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-black.is-inverted.is-outlined:hover,.button.is-black.is-inverted.is-outlined.is-hovered,.button.is-black.is-inverted.is-outlined:focus,.button.is-black.is-inverted.is-outlined.is-focused{background-color:#fff;color:#0a0a0a}.button.is-black.is-inverted.is-outlined.is-loading:hover::after,.button.is-black.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-black.is-inverted.is-outlined.is-loading:focus::after,.button.is-black.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}.button.is-black.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-black.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-light{background-color:#f5f5f5;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-light:hover,.button.is-light.is-hovered{background-color:#eee;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-light:focus,.button.is-light.is-focused{border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-light:focus:not(:active),.button.is-light.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(245,245,245,0.25)}.button.is-light:active,.button.is-light.is-active{background-color:#e8e8e8;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-light[disabled],fieldset[disabled] .button.is-light{background-color:#f5f5f5;border-color:#f5f5f5;box-shadow:none}.button.is-light.is-inverted{background-color:rgba(0,0,0,0.7);color:#f5f5f5}.button.is-light.is-inverted:hover,.button.is-light.is-inverted.is-hovered{background-color:rgba(0,0,0,0.7)}.button.is-light.is-inverted[disabled],fieldset[disabled] .button.is-light.is-inverted{background-color:rgba(0,0,0,0.7);border-color:transparent;box-shadow:none;color:#f5f5f5}.button.is-light.is-loading::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}.button.is-light.is-outlined{background-color:transparent;border-color:#f5f5f5;color:#f5f5f5}.button.is-light.is-outlined:hover,.button.is-light.is-outlined.is-hovered,.button.is-light.is-outlined:focus,.button.is-light.is-outlined.is-focused{background-color:#f5f5f5;border-color:#f5f5f5;color:rgba(0,0,0,0.7)}.button.is-light.is-outlined.is-loading::after{border-color:transparent transparent #f5f5f5 #f5f5f5 !important}.button.is-light.is-outlined.is-loading:hover::after,.button.is-light.is-outlined.is-loading.is-hovered::after,.button.is-light.is-outlined.is-loading:focus::after,.button.is-light.is-outlined.is-loading.is-focused::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}.button.is-light.is-outlined[disabled],fieldset[disabled] .button.is-light.is-outlined{background-color:transparent;border-color:#f5f5f5;box-shadow:none;color:#f5f5f5}.button.is-light.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);color:rgba(0,0,0,0.7)}.button.is-light.is-inverted.is-outlined:hover,.button.is-light.is-inverted.is-outlined.is-hovered,.button.is-light.is-inverted.is-outlined:focus,.button.is-light.is-inverted.is-outlined.is-focused{background-color:rgba(0,0,0,0.7);color:#f5f5f5}.button.is-light.is-inverted.is-outlined.is-loading:hover::after,.button.is-light.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-light.is-inverted.is-outlined.is-loading:focus::after,.button.is-light.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #f5f5f5 #f5f5f5 !important}.button.is-light.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-light.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);box-shadow:none;color:rgba(0,0,0,0.7)}.button.is-dark,.content kbd.button{background-color:#363636;border-color:transparent;color:#fff}.button.is-dark:hover,.content kbd.button:hover,.button.is-dark.is-hovered,.content kbd.button.is-hovered{background-color:#2f2f2f;border-color:transparent;color:#fff}.button.is-dark:focus,.content kbd.button:focus,.button.is-dark.is-focused,.content kbd.button.is-focused{border-color:transparent;color:#fff}.button.is-dark:focus:not(:active),.content kbd.button:focus:not(:active),.button.is-dark.is-focused:not(:active),.content kbd.button.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(54,54,54,0.25)}.button.is-dark:active,.content kbd.button:active,.button.is-dark.is-active,.content kbd.button.is-active{background-color:#292929;border-color:transparent;color:#fff}.button.is-dark[disabled],.content kbd.button[disabled],fieldset[disabled] .button.is-dark,fieldset[disabled] .content kbd.button,.content fieldset[disabled] kbd.button{background-color:#363636;border-color:#363636;box-shadow:none}.button.is-dark.is-inverted,.content kbd.button.is-inverted{background-color:#fff;color:#363636}.button.is-dark.is-inverted:hover,.content kbd.button.is-inverted:hover,.button.is-dark.is-inverted.is-hovered,.content kbd.button.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-dark.is-inverted[disabled],.content kbd.button.is-inverted[disabled],fieldset[disabled] .button.is-dark.is-inverted,fieldset[disabled] .content kbd.button.is-inverted,.content fieldset[disabled] kbd.button.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#363636}.button.is-dark.is-loading::after,.content kbd.button.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-dark.is-outlined,.content kbd.button.is-outlined{background-color:transparent;border-color:#363636;color:#363636}.button.is-dark.is-outlined:hover,.content kbd.button.is-outlined:hover,.button.is-dark.is-outlined.is-hovered,.content kbd.button.is-outlined.is-hovered,.button.is-dark.is-outlined:focus,.content kbd.button.is-outlined:focus,.button.is-dark.is-outlined.is-focused,.content kbd.button.is-outlined.is-focused{background-color:#363636;border-color:#363636;color:#fff}.button.is-dark.is-outlined.is-loading::after,.content kbd.button.is-outlined.is-loading::after{border-color:transparent transparent #363636 #363636 !important}.button.is-dark.is-outlined.is-loading:hover::after,.content kbd.button.is-outlined.is-loading:hover::after,.button.is-dark.is-outlined.is-loading.is-hovered::after,.content kbd.button.is-outlined.is-loading.is-hovered::after,.button.is-dark.is-outlined.is-loading:focus::after,.content kbd.button.is-outlined.is-loading:focus::after,.button.is-dark.is-outlined.is-loading.is-focused::after,.content kbd.button.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-dark.is-outlined[disabled],.content kbd.button.is-outlined[disabled],fieldset[disabled] .button.is-dark.is-outlined,fieldset[disabled] .content kbd.button.is-outlined,.content fieldset[disabled] kbd.button.is-outlined{background-color:transparent;border-color:#363636;box-shadow:none;color:#363636}.button.is-dark.is-inverted.is-outlined,.content kbd.button.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-dark.is-inverted.is-outlined:hover,.content kbd.button.is-inverted.is-outlined:hover,.button.is-dark.is-inverted.is-outlined.is-hovered,.content kbd.button.is-inverted.is-outlined.is-hovered,.button.is-dark.is-inverted.is-outlined:focus,.content kbd.button.is-inverted.is-outlined:focus,.button.is-dark.is-inverted.is-outlined.is-focused,.content kbd.button.is-inverted.is-outlined.is-focused{background-color:#fff;color:#363636}.button.is-dark.is-inverted.is-outlined.is-loading:hover::after,.content kbd.button.is-inverted.is-outlined.is-loading:hover::after,.button.is-dark.is-inverted.is-outlined.is-loading.is-hovered::after,.content kbd.button.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-dark.is-inverted.is-outlined.is-loading:focus::after,.content kbd.button.is-inverted.is-outlined.is-loading:focus::after,.button.is-dark.is-inverted.is-outlined.is-loading.is-focused::after,.content kbd.button.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #363636 #363636 !important}.button.is-dark.is-inverted.is-outlined[disabled],.content kbd.button.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-dark.is-inverted.is-outlined,fieldset[disabled] .content kbd.button.is-inverted.is-outlined,.content fieldset[disabled] kbd.button.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-primary,.docstring>section>a.button.docs-sourcelink{background-color:#4eb5de;border-color:transparent;color:#fff}.button.is-primary:hover,.docstring>section>a.button.docs-sourcelink:hover,.button.is-primary.is-hovered,.docstring>section>a.button.is-hovered.docs-sourcelink{background-color:#43b1dc;border-color:transparent;color:#fff}.button.is-primary:focus,.docstring>section>a.button.docs-sourcelink:focus,.button.is-primary.is-focused,.docstring>section>a.button.is-focused.docs-sourcelink{border-color:transparent;color:#fff}.button.is-primary:focus:not(:active),.docstring>section>a.button.docs-sourcelink:focus:not(:active),.button.is-primary.is-focused:not(:active),.docstring>section>a.button.is-focused.docs-sourcelink:not(:active){box-shadow:0 0 0 0.125em rgba(78,181,222,0.25)}.button.is-primary:active,.docstring>section>a.button.docs-sourcelink:active,.button.is-primary.is-active,.docstring>section>a.button.is-active.docs-sourcelink{background-color:#39acda;border-color:transparent;color:#fff}.button.is-primary[disabled],.docstring>section>a.button.docs-sourcelink[disabled],fieldset[disabled] .button.is-primary,fieldset[disabled] .docstring>section>a.button.docs-sourcelink{background-color:#4eb5de;border-color:#4eb5de;box-shadow:none}.button.is-primary.is-inverted,.docstring>section>a.button.is-inverted.docs-sourcelink{background-color:#fff;color:#4eb5de}.button.is-primary.is-inverted:hover,.docstring>section>a.button.is-inverted.docs-sourcelink:hover,.button.is-primary.is-inverted.is-hovered,.docstring>section>a.button.is-inverted.is-hovered.docs-sourcelink{background-color:#f2f2f2}.button.is-primary.is-inverted[disabled],.docstring>section>a.button.is-inverted.docs-sourcelink[disabled],fieldset[disabled] .button.is-primary.is-inverted,fieldset[disabled] .docstring>section>a.button.is-inverted.docs-sourcelink{background-color:#fff;border-color:transparent;box-shadow:none;color:#4eb5de}.button.is-primary.is-loading::after,.docstring>section>a.button.is-loading.docs-sourcelink::after{border-color:transparent transparent #fff #fff !important}.button.is-primary.is-outlined,.docstring>section>a.button.is-outlined.docs-sourcelink{background-color:transparent;border-color:#4eb5de;color:#4eb5de}.button.is-primary.is-outlined:hover,.docstring>section>a.button.is-outlined.docs-sourcelink:hover,.button.is-primary.is-outlined.is-hovered,.docstring>section>a.button.is-outlined.is-hovered.docs-sourcelink,.button.is-primary.is-outlined:focus,.docstring>section>a.button.is-outlined.docs-sourcelink:focus,.button.is-primary.is-outlined.is-focused,.docstring>section>a.button.is-outlined.is-focused.docs-sourcelink{background-color:#4eb5de;border-color:#4eb5de;color:#fff}.button.is-primary.is-outlined.is-loading::after,.docstring>section>a.button.is-outlined.is-loading.docs-sourcelink::after{border-color:transparent transparent #4eb5de #4eb5de !important}.button.is-primary.is-outlined.is-loading:hover::after,.docstring>section>a.button.is-outlined.is-loading.docs-sourcelink:hover::after,.button.is-primary.is-outlined.is-loading.is-hovered::after,.docstring>section>a.button.is-outlined.is-loading.is-hovered.docs-sourcelink::after,.button.is-primary.is-outlined.is-loading:focus::after,.docstring>section>a.button.is-outlined.is-loading.docs-sourcelink:focus::after,.button.is-primary.is-outlined.is-loading.is-focused::after,.docstring>section>a.button.is-outlined.is-loading.is-focused.docs-sourcelink::after{border-color:transparent transparent #fff #fff !important}.button.is-primary.is-outlined[disabled],.docstring>section>a.button.is-outlined.docs-sourcelink[disabled],fieldset[disabled] .button.is-primary.is-outlined,fieldset[disabled] .docstring>section>a.button.is-outlined.docs-sourcelink{background-color:transparent;border-color:#4eb5de;box-shadow:none;color:#4eb5de}.button.is-primary.is-inverted.is-outlined,.docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink{background-color:transparent;border-color:#fff;color:#fff}.button.is-primary.is-inverted.is-outlined:hover,.docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink:hover,.button.is-primary.is-inverted.is-outlined.is-hovered,.docstring>section>a.button.is-inverted.is-outlined.is-hovered.docs-sourcelink,.button.is-primary.is-inverted.is-outlined:focus,.docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink:focus,.button.is-primary.is-inverted.is-outlined.is-focused,.docstring>section>a.button.is-inverted.is-outlined.is-focused.docs-sourcelink{background-color:#fff;color:#4eb5de}.button.is-primary.is-inverted.is-outlined.is-loading:hover::after,.docstring>section>a.button.is-inverted.is-outlined.is-loading.docs-sourcelink:hover::after,.button.is-primary.is-inverted.is-outlined.is-loading.is-hovered::after,.docstring>section>a.button.is-inverted.is-outlined.is-loading.is-hovered.docs-sourcelink::after,.button.is-primary.is-inverted.is-outlined.is-loading:focus::after,.docstring>section>a.button.is-inverted.is-outlined.is-loading.docs-sourcelink:focus::after,.button.is-primary.is-inverted.is-outlined.is-loading.is-focused::after,.docstring>section>a.button.is-inverted.is-outlined.is-loading.is-focused.docs-sourcelink::after{border-color:transparent transparent #4eb5de #4eb5de !important}.button.is-primary.is-inverted.is-outlined[disabled],.docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink[disabled],fieldset[disabled] .button.is-primary.is-inverted.is-outlined,fieldset[disabled] .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-primary.is-light,.docstring>section>a.button.is-light.docs-sourcelink{background-color:#eef8fc;color:#1a6d8e}.button.is-primary.is-light:hover,.docstring>section>a.button.is-light.docs-sourcelink:hover,.button.is-primary.is-light.is-hovered,.docstring>section>a.button.is-light.is-hovered.docs-sourcelink{background-color:#e3f3fa;border-color:transparent;color:#1a6d8e}.button.is-primary.is-light:active,.docstring>section>a.button.is-light.docs-sourcelink:active,.button.is-primary.is-light.is-active,.docstring>section>a.button.is-light.is-active.docs-sourcelink{background-color:#d8eff8;border-color:transparent;color:#1a6d8e}.button.is-link{background-color:#2e63b8;border-color:transparent;color:#fff}.button.is-link:hover,.button.is-link.is-hovered{background-color:#2b5eae;border-color:transparent;color:#fff}.button.is-link:focus,.button.is-link.is-focused{border-color:transparent;color:#fff}.button.is-link:focus:not(:active),.button.is-link.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.button.is-link:active,.button.is-link.is-active{background-color:#2958a4;border-color:transparent;color:#fff}.button.is-link[disabled],fieldset[disabled] .button.is-link{background-color:#2e63b8;border-color:#2e63b8;box-shadow:none}.button.is-link.is-inverted{background-color:#fff;color:#2e63b8}.button.is-link.is-inverted:hover,.button.is-link.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-link.is-inverted[disabled],fieldset[disabled] .button.is-link.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#2e63b8}.button.is-link.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-link.is-outlined{background-color:transparent;border-color:#2e63b8;color:#2e63b8}.button.is-link.is-outlined:hover,.button.is-link.is-outlined.is-hovered,.button.is-link.is-outlined:focus,.button.is-link.is-outlined.is-focused{background-color:#2e63b8;border-color:#2e63b8;color:#fff}.button.is-link.is-outlined.is-loading::after{border-color:transparent transparent #2e63b8 #2e63b8 !important}.button.is-link.is-outlined.is-loading:hover::after,.button.is-link.is-outlined.is-loading.is-hovered::after,.button.is-link.is-outlined.is-loading:focus::after,.button.is-link.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-link.is-outlined[disabled],fieldset[disabled] .button.is-link.is-outlined{background-color:transparent;border-color:#2e63b8;box-shadow:none;color:#2e63b8}.button.is-link.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-link.is-inverted.is-outlined:hover,.button.is-link.is-inverted.is-outlined.is-hovered,.button.is-link.is-inverted.is-outlined:focus,.button.is-link.is-inverted.is-outlined.is-focused{background-color:#fff;color:#2e63b8}.button.is-link.is-inverted.is-outlined.is-loading:hover::after,.button.is-link.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-link.is-inverted.is-outlined.is-loading:focus::after,.button.is-link.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #2e63b8 #2e63b8 !important}.button.is-link.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-link.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-link.is-light{background-color:#eff3fb;color:#3169c4}.button.is-link.is-light:hover,.button.is-link.is-light.is-hovered{background-color:#e4ecf8;border-color:transparent;color:#3169c4}.button.is-link.is-light:active,.button.is-link.is-light.is-active{background-color:#dae5f6;border-color:transparent;color:#3169c4}.button.is-info{background-color:#209cee;border-color:transparent;color:#fff}.button.is-info:hover,.button.is-info.is-hovered{background-color:#1497ed;border-color:transparent;color:#fff}.button.is-info:focus,.button.is-info.is-focused{border-color:transparent;color:#fff}.button.is-info:focus:not(:active),.button.is-info.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(32,156,238,0.25)}.button.is-info:active,.button.is-info.is-active{background-color:#1190e3;border-color:transparent;color:#fff}.button.is-info[disabled],fieldset[disabled] .button.is-info{background-color:#209cee;border-color:#209cee;box-shadow:none}.button.is-info.is-inverted{background-color:#fff;color:#209cee}.button.is-info.is-inverted:hover,.button.is-info.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-info.is-inverted[disabled],fieldset[disabled] .button.is-info.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#209cee}.button.is-info.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-info.is-outlined{background-color:transparent;border-color:#209cee;color:#209cee}.button.is-info.is-outlined:hover,.button.is-info.is-outlined.is-hovered,.button.is-info.is-outlined:focus,.button.is-info.is-outlined.is-focused{background-color:#209cee;border-color:#209cee;color:#fff}.button.is-info.is-outlined.is-loading::after{border-color:transparent transparent #209cee #209cee !important}.button.is-info.is-outlined.is-loading:hover::after,.button.is-info.is-outlined.is-loading.is-hovered::after,.button.is-info.is-outlined.is-loading:focus::after,.button.is-info.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-info.is-outlined[disabled],fieldset[disabled] .button.is-info.is-outlined{background-color:transparent;border-color:#209cee;box-shadow:none;color:#209cee}.button.is-info.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-info.is-inverted.is-outlined:hover,.button.is-info.is-inverted.is-outlined.is-hovered,.button.is-info.is-inverted.is-outlined:focus,.button.is-info.is-inverted.is-outlined.is-focused{background-color:#fff;color:#209cee}.button.is-info.is-inverted.is-outlined.is-loading:hover::after,.button.is-info.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-info.is-inverted.is-outlined.is-loading:focus::after,.button.is-info.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #209cee #209cee !important}.button.is-info.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-info.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-info.is-light{background-color:#ecf7fe;color:#0e72b4}.button.is-info.is-light:hover,.button.is-info.is-light.is-hovered{background-color:#e0f1fd;border-color:transparent;color:#0e72b4}.button.is-info.is-light:active,.button.is-info.is-light.is-active{background-color:#d4ecfc;border-color:transparent;color:#0e72b4}.button.is-success{background-color:#22c35b;border-color:transparent;color:#fff}.button.is-success:hover,.button.is-success.is-hovered{background-color:#20b856;border-color:transparent;color:#fff}.button.is-success:focus,.button.is-success.is-focused{border-color:transparent;color:#fff}.button.is-success:focus:not(:active),.button.is-success.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(34,195,91,0.25)}.button.is-success:active,.button.is-success.is-active{background-color:#1ead51;border-color:transparent;color:#fff}.button.is-success[disabled],fieldset[disabled] .button.is-success{background-color:#22c35b;border-color:#22c35b;box-shadow:none}.button.is-success.is-inverted{background-color:#fff;color:#22c35b}.button.is-success.is-inverted:hover,.button.is-success.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-success.is-inverted[disabled],fieldset[disabled] .button.is-success.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#22c35b}.button.is-success.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-success.is-outlined{background-color:transparent;border-color:#22c35b;color:#22c35b}.button.is-success.is-outlined:hover,.button.is-success.is-outlined.is-hovered,.button.is-success.is-outlined:focus,.button.is-success.is-outlined.is-focused{background-color:#22c35b;border-color:#22c35b;color:#fff}.button.is-success.is-outlined.is-loading::after{border-color:transparent transparent #22c35b #22c35b !important}.button.is-success.is-outlined.is-loading:hover::after,.button.is-success.is-outlined.is-loading.is-hovered::after,.button.is-success.is-outlined.is-loading:focus::after,.button.is-success.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-success.is-outlined[disabled],fieldset[disabled] .button.is-success.is-outlined{background-color:transparent;border-color:#22c35b;box-shadow:none;color:#22c35b}.button.is-success.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-success.is-inverted.is-outlined:hover,.button.is-success.is-inverted.is-outlined.is-hovered,.button.is-success.is-inverted.is-outlined:focus,.button.is-success.is-inverted.is-outlined.is-focused{background-color:#fff;color:#22c35b}.button.is-success.is-inverted.is-outlined.is-loading:hover::after,.button.is-success.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-success.is-inverted.is-outlined.is-loading:focus::after,.button.is-success.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #22c35b #22c35b !important}.button.is-success.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-success.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-success.is-light{background-color:#eefcf3;color:#198f43}.button.is-success.is-light:hover,.button.is-success.is-light.is-hovered{background-color:#e3faeb;border-color:transparent;color:#198f43}.button.is-success.is-light:active,.button.is-success.is-light.is-active{background-color:#d8f8e3;border-color:transparent;color:#198f43}.button.is-warning{background-color:#ffdd57;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-warning:hover,.button.is-warning.is-hovered{background-color:#ffda4a;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-warning:focus,.button.is-warning.is-focused{border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-warning:focus:not(:active),.button.is-warning.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(255,221,87,0.25)}.button.is-warning:active,.button.is-warning.is-active{background-color:#ffd83e;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-warning[disabled],fieldset[disabled] .button.is-warning{background-color:#ffdd57;border-color:#ffdd57;box-shadow:none}.button.is-warning.is-inverted{background-color:rgba(0,0,0,0.7);color:#ffdd57}.button.is-warning.is-inverted:hover,.button.is-warning.is-inverted.is-hovered{background-color:rgba(0,0,0,0.7)}.button.is-warning.is-inverted[disabled],fieldset[disabled] .button.is-warning.is-inverted{background-color:rgba(0,0,0,0.7);border-color:transparent;box-shadow:none;color:#ffdd57}.button.is-warning.is-loading::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}.button.is-warning.is-outlined{background-color:transparent;border-color:#ffdd57;color:#ffdd57}.button.is-warning.is-outlined:hover,.button.is-warning.is-outlined.is-hovered,.button.is-warning.is-outlined:focus,.button.is-warning.is-outlined.is-focused{background-color:#ffdd57;border-color:#ffdd57;color:rgba(0,0,0,0.7)}.button.is-warning.is-outlined.is-loading::after{border-color:transparent transparent #ffdd57 #ffdd57 !important}.button.is-warning.is-outlined.is-loading:hover::after,.button.is-warning.is-outlined.is-loading.is-hovered::after,.button.is-warning.is-outlined.is-loading:focus::after,.button.is-warning.is-outlined.is-loading.is-focused::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}.button.is-warning.is-outlined[disabled],fieldset[disabled] .button.is-warning.is-outlined{background-color:transparent;border-color:#ffdd57;box-shadow:none;color:#ffdd57}.button.is-warning.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);color:rgba(0,0,0,0.7)}.button.is-warning.is-inverted.is-outlined:hover,.button.is-warning.is-inverted.is-outlined.is-hovered,.button.is-warning.is-inverted.is-outlined:focus,.button.is-warning.is-inverted.is-outlined.is-focused{background-color:rgba(0,0,0,0.7);color:#ffdd57}.button.is-warning.is-inverted.is-outlined.is-loading:hover::after,.button.is-warning.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-warning.is-inverted.is-outlined.is-loading:focus::after,.button.is-warning.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #ffdd57 #ffdd57 !important}.button.is-warning.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-warning.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);box-shadow:none;color:rgba(0,0,0,0.7)}.button.is-warning.is-light{background-color:#fffbeb;color:#947600}.button.is-warning.is-light:hover,.button.is-warning.is-light.is-hovered{background-color:#fff8de;border-color:transparent;color:#947600}.button.is-warning.is-light:active,.button.is-warning.is-light.is-active{background-color:#fff6d1;border-color:transparent;color:#947600}.button.is-danger{background-color:#da0b00;border-color:transparent;color:#fff}.button.is-danger:hover,.button.is-danger.is-hovered{background-color:#cd0a00;border-color:transparent;color:#fff}.button.is-danger:focus,.button.is-danger.is-focused{border-color:transparent;color:#fff}.button.is-danger:focus:not(:active),.button.is-danger.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(218,11,0,0.25)}.button.is-danger:active,.button.is-danger.is-active{background-color:#c10a00;border-color:transparent;color:#fff}.button.is-danger[disabled],fieldset[disabled] .button.is-danger{background-color:#da0b00;border-color:#da0b00;box-shadow:none}.button.is-danger.is-inverted{background-color:#fff;color:#da0b00}.button.is-danger.is-inverted:hover,.button.is-danger.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-danger.is-inverted[disabled],fieldset[disabled] .button.is-danger.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#da0b00}.button.is-danger.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-danger.is-outlined{background-color:transparent;border-color:#da0b00;color:#da0b00}.button.is-danger.is-outlined:hover,.button.is-danger.is-outlined.is-hovered,.button.is-danger.is-outlined:focus,.button.is-danger.is-outlined.is-focused{background-color:#da0b00;border-color:#da0b00;color:#fff}.button.is-danger.is-outlined.is-loading::after{border-color:transparent transparent #da0b00 #da0b00 !important}.button.is-danger.is-outlined.is-loading:hover::after,.button.is-danger.is-outlined.is-loading.is-hovered::after,.button.is-danger.is-outlined.is-loading:focus::after,.button.is-danger.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-danger.is-outlined[disabled],fieldset[disabled] .button.is-danger.is-outlined{background-color:transparent;border-color:#da0b00;box-shadow:none;color:#da0b00}.button.is-danger.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-danger.is-inverted.is-outlined:hover,.button.is-danger.is-inverted.is-outlined.is-hovered,.button.is-danger.is-inverted.is-outlined:focus,.button.is-danger.is-inverted.is-outlined.is-focused{background-color:#fff;color:#da0b00}.button.is-danger.is-inverted.is-outlined.is-loading:hover::after,.button.is-danger.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-danger.is-inverted.is-outlined.is-loading:focus::after,.button.is-danger.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #da0b00 #da0b00 !important}.button.is-danger.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-danger.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-danger.is-light{background-color:#ffeceb;color:#f50c00}.button.is-danger.is-light:hover,.button.is-danger.is-light.is-hovered{background-color:#ffe0de;border-color:transparent;color:#f50c00}.button.is-danger.is-light:active,.button.is-danger.is-light.is-active{background-color:#ffd3d1;border-color:transparent;color:#f50c00}.button.is-small,#documenter .docs-sidebar form.docs-search>input.button{font-size:.75rem}.button.is-small:not(.is-rounded),#documenter .docs-sidebar form.docs-search>input.button:not(.is-rounded){border-radius:2px}.button.is-normal{font-size:1rem}.button.is-medium{font-size:1.25rem}.button.is-large{font-size:1.5rem}.button[disabled],fieldset[disabled] .button{background-color:#fff;border-color:#dbdbdb;box-shadow:none;opacity:.5}.button.is-fullwidth{display:flex;width:100%}.button.is-loading{color:transparent !important;pointer-events:none}.button.is-loading::after{position:absolute;left:calc(50% - (1em * 0.5));top:calc(50% - (1em * 0.5));position:absolute !important}.button.is-static{background-color:#f5f5f5;border-color:#dbdbdb;color:#6b6b6b;box-shadow:none;pointer-events:none}.button.is-rounded,#documenter .docs-sidebar form.docs-search>input.button{border-radius:9999px;padding-left:calc(1em + 0.25em);padding-right:calc(1em + 0.25em)}.buttons{align-items:center;display:flex;flex-wrap:wrap;justify-content:flex-start}.buttons .button{margin-bottom:0.5rem}.buttons .button:not(:last-child):not(.is-fullwidth){margin-right:.5rem}.buttons:last-child{margin-bottom:-0.5rem}.buttons:not(:last-child){margin-bottom:1rem}.buttons.are-small .button:not(.is-normal):not(.is-medium):not(.is-large){font-size:.75rem}.buttons.are-small .button:not(.is-normal):not(.is-medium):not(.is-large):not(.is-rounded){border-radius:2px}.buttons.are-medium .button:not(.is-small):not(.is-normal):not(.is-large){font-size:1.25rem}.buttons.are-large .button:not(.is-small):not(.is-normal):not(.is-medium){font-size:1.5rem}.buttons.has-addons .button:not(:first-child){border-bottom-left-radius:0;border-top-left-radius:0}.buttons.has-addons .button:not(:last-child){border-bottom-right-radius:0;border-top-right-radius:0;margin-right:-1px}.buttons.has-addons .button:last-child{margin-right:0}.buttons.has-addons .button:hover,.buttons.has-addons .button.is-hovered{z-index:2}.buttons.has-addons .button:focus,.buttons.has-addons .button.is-focused,.buttons.has-addons .button:active,.buttons.has-addons .button.is-active,.buttons.has-addons .button.is-selected{z-index:3}.buttons.has-addons .button:focus:hover,.buttons.has-addons .button.is-focused:hover,.buttons.has-addons .button:active:hover,.buttons.has-addons .button.is-active:hover,.buttons.has-addons .button.is-selected:hover{z-index:4}.buttons.has-addons .button.is-expanded{flex-grow:1;flex-shrink:1}.buttons.is-centered{justify-content:center}.buttons.is-centered:not(.has-addons) .button:not(.is-fullwidth){margin-left:0.25rem;margin-right:0.25rem}.buttons.is-right{justify-content:flex-end}.buttons.is-right:not(.has-addons) .button:not(.is-fullwidth){margin-left:0.25rem;margin-right:0.25rem}@media screen and (max-width: 768px){.button.is-responsive.is-small,#documenter .docs-sidebar form.docs-search>input.is-responsive{font-size:.5625rem}.button.is-responsive,.button.is-responsive.is-normal{font-size:.65625rem}.button.is-responsive.is-medium{font-size:.75rem}.button.is-responsive.is-large{font-size:1rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.button.is-responsive.is-small,#documenter .docs-sidebar form.docs-search>input.is-responsive{font-size:.65625rem}.button.is-responsive,.button.is-responsive.is-normal{font-size:.75rem}.button.is-responsive.is-medium{font-size:1rem}.button.is-responsive.is-large{font-size:1.25rem}}.container{flex-grow:1;margin:0 auto;position:relative;width:auto}.container.is-fluid{max-width:none !important;padding-left:32px;padding-right:32px;width:100%}@media screen and (min-width: 1056px){.container{max-width:992px}}@media screen and (max-width: 1215px){.container.is-widescreen:not(.is-max-desktop){max-width:1152px}}@media screen and (max-width: 1407px){.container.is-fullhd:not(.is-max-desktop):not(.is-max-widescreen){max-width:1344px}}@media screen and (min-width: 1216px){.container:not(.is-max-desktop){max-width:1152px}}@media screen and (min-width: 1408px){.container:not(.is-max-desktop):not(.is-max-widescreen){max-width:1344px}}.content li+li{margin-top:0.25em}.content p:not(:last-child),.content dl:not(:last-child),.content ol:not(:last-child),.content ul:not(:last-child),.content blockquote:not(:last-child),.content pre:not(:last-child),.content table:not(:last-child){margin-bottom:1em}.content h1,.content h2,.content h3,.content h4,.content h5,.content h6{color:#222;font-weight:600;line-height:1.125}.content h1{font-size:2em;margin-bottom:0.5em}.content h1:not(:first-child){margin-top:1em}.content h2{font-size:1.75em;margin-bottom:0.5714em}.content h2:not(:first-child){margin-top:1.1428em}.content h3{font-size:1.5em;margin-bottom:0.6666em}.content h3:not(:first-child){margin-top:1.3333em}.content h4{font-size:1.25em;margin-bottom:0.8em}.content h5{font-size:1.125em;margin-bottom:0.8888em}.content h6{font-size:1em;margin-bottom:1em}.content blockquote{background-color:#f5f5f5;border-left:5px solid #dbdbdb;padding:1.25em 1.5em}.content ol{list-style-position:outside;margin-left:2em;margin-top:1em}.content ol:not([type]){list-style-type:decimal}.content ol.is-lower-alpha:not([type]){list-style-type:lower-alpha}.content ol.is-lower-roman:not([type]){list-style-type:lower-roman}.content ol.is-upper-alpha:not([type]){list-style-type:upper-alpha}.content ol.is-upper-roman:not([type]){list-style-type:upper-roman}.content ul{list-style:disc outside;margin-left:2em;margin-top:1em}.content ul ul{list-style-type:circle;margin-top:0.5em}.content ul ul ul{list-style-type:square}.content dd{margin-left:2em}.content figure{margin-left:2em;margin-right:2em;text-align:center}.content figure:not(:first-child){margin-top:2em}.content figure:not(:last-child){margin-bottom:2em}.content figure img{display:inline-block}.content figure figcaption{font-style:italic}.content pre{-webkit-overflow-scrolling:touch;overflow-x:auto;padding:0;white-space:pre;word-wrap:normal}.content sup,.content sub{font-size:75%}.content table{width:100%}.content table td,.content table th{border:1px solid #dbdbdb;border-width:0 0 1px;padding:0.5em 0.75em;vertical-align:top}.content table th{color:#222}.content table th:not([align]){text-align:inherit}.content table thead td,.content table thead th{border-width:0 0 2px;color:#222}.content table tfoot td,.content table tfoot th{border-width:2px 0 0;color:#222}.content table tbody tr:last-child td,.content table tbody tr:last-child th{border-bottom-width:0}.content .tabs li+li{margin-top:0}.content.is-small,#documenter .docs-sidebar form.docs-search>input.content{font-size:.75rem}.content.is-normal{font-size:1rem}.content.is-medium{font-size:1.25rem}.content.is-large{font-size:1.5rem}.icon{align-items:center;display:inline-flex;justify-content:center;height:1.5rem;width:1.5rem}.icon.is-small,#documenter .docs-sidebar form.docs-search>input.icon{height:1rem;width:1rem}.icon.is-medium{height:2rem;width:2rem}.icon.is-large{height:3rem;width:3rem}.icon-text{align-items:flex-start;color:inherit;display:inline-flex;flex-wrap:wrap;line-height:1.5rem;vertical-align:top}.icon-text .icon{flex-grow:0;flex-shrink:0}.icon-text .icon:not(:last-child){margin-right:.25em}.icon-text .icon:not(:first-child){margin-left:.25em}div.icon-text{display:flex}.image,#documenter .docs-sidebar .docs-logo>img{display:block;position:relative}.image img,#documenter .docs-sidebar .docs-logo>img img{display:block;height:auto;width:100%}.image img.is-rounded,#documenter .docs-sidebar .docs-logo>img img.is-rounded{border-radius:9999px}.image.is-fullwidth,#documenter .docs-sidebar .docs-logo>img.is-fullwidth{width:100%}.image.is-square img,#documenter .docs-sidebar .docs-logo>img.is-square img,.image.is-square .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-square .has-ratio,.image.is-1by1 img,#documenter .docs-sidebar .docs-logo>img.is-1by1 img,.image.is-1by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by1 .has-ratio,.image.is-5by4 img,#documenter .docs-sidebar .docs-logo>img.is-5by4 img,.image.is-5by4 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-5by4 .has-ratio,.image.is-4by3 img,#documenter .docs-sidebar .docs-logo>img.is-4by3 img,.image.is-4by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-4by3 .has-ratio,.image.is-3by2 img,#documenter .docs-sidebar .docs-logo>img.is-3by2 img,.image.is-3by2 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by2 .has-ratio,.image.is-5by3 img,#documenter .docs-sidebar .docs-logo>img.is-5by3 img,.image.is-5by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-5by3 .has-ratio,.image.is-16by9 img,#documenter .docs-sidebar .docs-logo>img.is-16by9 img,.image.is-16by9 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-16by9 .has-ratio,.image.is-2by1 img,#documenter .docs-sidebar .docs-logo>img.is-2by1 img,.image.is-2by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-2by1 .has-ratio,.image.is-3by1 img,#documenter .docs-sidebar .docs-logo>img.is-3by1 img,.image.is-3by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by1 .has-ratio,.image.is-4by5 img,#documenter .docs-sidebar .docs-logo>img.is-4by5 img,.image.is-4by5 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-4by5 .has-ratio,.image.is-3by4 img,#documenter .docs-sidebar .docs-logo>img.is-3by4 img,.image.is-3by4 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by4 .has-ratio,.image.is-2by3 img,#documenter .docs-sidebar .docs-logo>img.is-2by3 img,.image.is-2by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-2by3 .has-ratio,.image.is-3by5 img,#documenter .docs-sidebar .docs-logo>img.is-3by5 img,.image.is-3by5 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by5 .has-ratio,.image.is-9by16 img,#documenter .docs-sidebar .docs-logo>img.is-9by16 img,.image.is-9by16 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-9by16 .has-ratio,.image.is-1by2 img,#documenter .docs-sidebar .docs-logo>img.is-1by2 img,.image.is-1by2 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by2 .has-ratio,.image.is-1by3 img,#documenter .docs-sidebar .docs-logo>img.is-1by3 img,.image.is-1by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by3 .has-ratio{height:100%;width:100%}.image.is-square,#documenter .docs-sidebar .docs-logo>img.is-square,.image.is-1by1,#documenter .docs-sidebar .docs-logo>img.is-1by1{padding-top:100%}.image.is-5by4,#documenter .docs-sidebar .docs-logo>img.is-5by4{padding-top:80%}.image.is-4by3,#documenter .docs-sidebar .docs-logo>img.is-4by3{padding-top:75%}.image.is-3by2,#documenter .docs-sidebar .docs-logo>img.is-3by2{padding-top:66.6666%}.image.is-5by3,#documenter .docs-sidebar .docs-logo>img.is-5by3{padding-top:60%}.image.is-16by9,#documenter .docs-sidebar .docs-logo>img.is-16by9{padding-top:56.25%}.image.is-2by1,#documenter .docs-sidebar .docs-logo>img.is-2by1{padding-top:50%}.image.is-3by1,#documenter .docs-sidebar .docs-logo>img.is-3by1{padding-top:33.3333%}.image.is-4by5,#documenter .docs-sidebar .docs-logo>img.is-4by5{padding-top:125%}.image.is-3by4,#documenter .docs-sidebar .docs-logo>img.is-3by4{padding-top:133.3333%}.image.is-2by3,#documenter .docs-sidebar .docs-logo>img.is-2by3{padding-top:150%}.image.is-3by5,#documenter .docs-sidebar .docs-logo>img.is-3by5{padding-top:166.6666%}.image.is-9by16,#documenter .docs-sidebar .docs-logo>img.is-9by16{padding-top:177.7777%}.image.is-1by2,#documenter .docs-sidebar .docs-logo>img.is-1by2{padding-top:200%}.image.is-1by3,#documenter .docs-sidebar .docs-logo>img.is-1by3{padding-top:300%}.image.is-16x16,#documenter .docs-sidebar .docs-logo>img.is-16x16{height:16px;width:16px}.image.is-24x24,#documenter .docs-sidebar .docs-logo>img.is-24x24{height:24px;width:24px}.image.is-32x32,#documenter .docs-sidebar .docs-logo>img.is-32x32{height:32px;width:32px}.image.is-48x48,#documenter .docs-sidebar .docs-logo>img.is-48x48{height:48px;width:48px}.image.is-64x64,#documenter .docs-sidebar .docs-logo>img.is-64x64{height:64px;width:64px}.image.is-96x96,#documenter .docs-sidebar .docs-logo>img.is-96x96{height:96px;width:96px}.image.is-128x128,#documenter .docs-sidebar .docs-logo>img.is-128x128{height:128px;width:128px}.notification{background-color:#f5f5f5;border-radius:4px;position:relative;padding:1.25rem 2.5rem 1.25rem 1.5rem}.notification a:not(.button):not(.dropdown-item){color:currentColor;text-decoration:underline}.notification strong{color:currentColor}.notification code,.notification pre{background:#fff}.notification pre code{background:transparent}.notification>.delete{right:.5rem;position:absolute;top:0.5rem}.notification .title,.notification .subtitle,.notification .content{color:currentColor}.notification.is-white{background-color:#fff;color:#0a0a0a}.notification.is-black{background-color:#0a0a0a;color:#fff}.notification.is-light{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.notification.is-dark,.content kbd.notification{background-color:#363636;color:#fff}.notification.is-primary,.docstring>section>a.notification.docs-sourcelink{background-color:#4eb5de;color:#fff}.notification.is-primary.is-light,.docstring>section>a.notification.is-light.docs-sourcelink{background-color:#eef8fc;color:#1a6d8e}.notification.is-link{background-color:#2e63b8;color:#fff}.notification.is-link.is-light{background-color:#eff3fb;color:#3169c4}.notification.is-info{background-color:#209cee;color:#fff}.notification.is-info.is-light{background-color:#ecf7fe;color:#0e72b4}.notification.is-success{background-color:#22c35b;color:#fff}.notification.is-success.is-light{background-color:#eefcf3;color:#198f43}.notification.is-warning{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.notification.is-warning.is-light{background-color:#fffbeb;color:#947600}.notification.is-danger{background-color:#da0b00;color:#fff}.notification.is-danger.is-light{background-color:#ffeceb;color:#f50c00}.progress{-moz-appearance:none;-webkit-appearance:none;border:none;border-radius:9999px;display:block;height:1rem;overflow:hidden;padding:0;width:100%}.progress::-webkit-progress-bar{background-color:#ededed}.progress::-webkit-progress-value{background-color:#222}.progress::-moz-progress-bar{background-color:#222}.progress::-ms-fill{background-color:#222;border:none}.progress.is-white::-webkit-progress-value{background-color:#fff}.progress.is-white::-moz-progress-bar{background-color:#fff}.progress.is-white::-ms-fill{background-color:#fff}.progress.is-white:indeterminate{background-image:linear-gradient(to right, #fff 30%, #ededed 30%)}.progress.is-black::-webkit-progress-value{background-color:#0a0a0a}.progress.is-black::-moz-progress-bar{background-color:#0a0a0a}.progress.is-black::-ms-fill{background-color:#0a0a0a}.progress.is-black:indeterminate{background-image:linear-gradient(to right, #0a0a0a 30%, #ededed 30%)}.progress.is-light::-webkit-progress-value{background-color:#f5f5f5}.progress.is-light::-moz-progress-bar{background-color:#f5f5f5}.progress.is-light::-ms-fill{background-color:#f5f5f5}.progress.is-light:indeterminate{background-image:linear-gradient(to right, #f5f5f5 30%, #ededed 30%)}.progress.is-dark::-webkit-progress-value,.content kbd.progress::-webkit-progress-value{background-color:#363636}.progress.is-dark::-moz-progress-bar,.content kbd.progress::-moz-progress-bar{background-color:#363636}.progress.is-dark::-ms-fill,.content kbd.progress::-ms-fill{background-color:#363636}.progress.is-dark:indeterminate,.content kbd.progress:indeterminate{background-image:linear-gradient(to right, #363636 30%, #ededed 30%)}.progress.is-primary::-webkit-progress-value,.docstring>section>a.progress.docs-sourcelink::-webkit-progress-value{background-color:#4eb5de}.progress.is-primary::-moz-progress-bar,.docstring>section>a.progress.docs-sourcelink::-moz-progress-bar{background-color:#4eb5de}.progress.is-primary::-ms-fill,.docstring>section>a.progress.docs-sourcelink::-ms-fill{background-color:#4eb5de}.progress.is-primary:indeterminate,.docstring>section>a.progress.docs-sourcelink:indeterminate{background-image:linear-gradient(to right, #4eb5de 30%, #ededed 30%)}.progress.is-link::-webkit-progress-value{background-color:#2e63b8}.progress.is-link::-moz-progress-bar{background-color:#2e63b8}.progress.is-link::-ms-fill{background-color:#2e63b8}.progress.is-link:indeterminate{background-image:linear-gradient(to right, #2e63b8 30%, #ededed 30%)}.progress.is-info::-webkit-progress-value{background-color:#209cee}.progress.is-info::-moz-progress-bar{background-color:#209cee}.progress.is-info::-ms-fill{background-color:#209cee}.progress.is-info:indeterminate{background-image:linear-gradient(to right, #209cee 30%, #ededed 30%)}.progress.is-success::-webkit-progress-value{background-color:#22c35b}.progress.is-success::-moz-progress-bar{background-color:#22c35b}.progress.is-success::-ms-fill{background-color:#22c35b}.progress.is-success:indeterminate{background-image:linear-gradient(to right, #22c35b 30%, #ededed 30%)}.progress.is-warning::-webkit-progress-value{background-color:#ffdd57}.progress.is-warning::-moz-progress-bar{background-color:#ffdd57}.progress.is-warning::-ms-fill{background-color:#ffdd57}.progress.is-warning:indeterminate{background-image:linear-gradient(to right, #ffdd57 30%, #ededed 30%)}.progress.is-danger::-webkit-progress-value{background-color:#da0b00}.progress.is-danger::-moz-progress-bar{background-color:#da0b00}.progress.is-danger::-ms-fill{background-color:#da0b00}.progress.is-danger:indeterminate{background-image:linear-gradient(to right, #da0b00 30%, #ededed 30%)}.progress:indeterminate{animation-duration:1.5s;animation-iteration-count:infinite;animation-name:moveIndeterminate;animation-timing-function:linear;background-color:#ededed;background-image:linear-gradient(to right, #222 30%, #ededed 30%);background-position:top left;background-repeat:no-repeat;background-size:150% 150%}.progress:indeterminate::-webkit-progress-bar{background-color:transparent}.progress:indeterminate::-moz-progress-bar{background-color:transparent}.progress:indeterminate::-ms-fill{animation-name:none}.progress.is-small,#documenter .docs-sidebar form.docs-search>input.progress{height:.75rem}.progress.is-medium{height:1.25rem}.progress.is-large{height:1.5rem}@keyframes moveIndeterminate{from{background-position:200% 0}to{background-position:-200% 0}}.table{background-color:#fff;color:#222}.table td,.table th{border:1px solid #dbdbdb;border-width:0 0 1px;padding:0.5em 0.75em;vertical-align:top}.table td.is-white,.table th.is-white{background-color:#fff;border-color:#fff;color:#0a0a0a}.table td.is-black,.table th.is-black{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}.table td.is-light,.table th.is-light{background-color:#f5f5f5;border-color:#f5f5f5;color:rgba(0,0,0,0.7)}.table td.is-dark,.table th.is-dark{background-color:#363636;border-color:#363636;color:#fff}.table td.is-primary,.table th.is-primary{background-color:#4eb5de;border-color:#4eb5de;color:#fff}.table td.is-link,.table th.is-link{background-color:#2e63b8;border-color:#2e63b8;color:#fff}.table td.is-info,.table th.is-info{background-color:#209cee;border-color:#209cee;color:#fff}.table td.is-success,.table th.is-success{background-color:#22c35b;border-color:#22c35b;color:#fff}.table td.is-warning,.table th.is-warning{background-color:#ffdd57;border-color:#ffdd57;color:rgba(0,0,0,0.7)}.table td.is-danger,.table th.is-danger{background-color:#da0b00;border-color:#da0b00;color:#fff}.table td.is-narrow,.table th.is-narrow{white-space:nowrap;width:1%}.table td.is-selected,.table th.is-selected{background-color:#4eb5de;color:#fff}.table td.is-selected a,.table td.is-selected strong,.table th.is-selected a,.table th.is-selected strong{color:currentColor}.table td.is-vcentered,.table th.is-vcentered{vertical-align:middle}.table th{color:#222}.table th:not([align]){text-align:left}.table tr.is-selected{background-color:#4eb5de;color:#fff}.table tr.is-selected a,.table tr.is-selected strong{color:currentColor}.table tr.is-selected td,.table tr.is-selected th{border-color:#fff;color:currentColor}.table thead{background-color:rgba(0,0,0,0)}.table thead td,.table thead th{border-width:0 0 2px;color:#222}.table tfoot{background-color:rgba(0,0,0,0)}.table tfoot td,.table tfoot th{border-width:2px 0 0;color:#222}.table tbody{background-color:rgba(0,0,0,0)}.table tbody tr:last-child td,.table tbody tr:last-child th{border-bottom-width:0}.table.is-bordered td,.table.is-bordered th{border-width:1px}.table.is-bordered tr:last-child td,.table.is-bordered tr:last-child th{border-bottom-width:1px}.table.is-fullwidth{width:100%}.table.is-hoverable tbody tr:not(.is-selected):hover{background-color:#fafafa}.table.is-hoverable.is-striped tbody tr:not(.is-selected):hover{background-color:#fafafa}.table.is-hoverable.is-striped tbody tr:not(.is-selected):hover:nth-child(even){background-color:#f5f5f5}.table.is-narrow td,.table.is-narrow th{padding:0.25em 0.5em}.table.is-striped tbody tr:not(.is-selected):nth-child(even){background-color:#fafafa}.table-container{-webkit-overflow-scrolling:touch;overflow:auto;overflow-y:hidden;max-width:100%}.tags{align-items:center;display:flex;flex-wrap:wrap;justify-content:flex-start}.tags .tag,.tags .content kbd,.content .tags kbd,.tags .docstring>section>a.docs-sourcelink{margin-bottom:0.5rem}.tags .tag:not(:last-child),.tags .content kbd:not(:last-child),.content .tags kbd:not(:last-child),.tags .docstring>section>a.docs-sourcelink:not(:last-child){margin-right:.5rem}.tags:last-child{margin-bottom:-0.5rem}.tags:not(:last-child){margin-bottom:1rem}.tags.are-medium .tag:not(.is-normal):not(.is-large),.tags.are-medium .content kbd:not(.is-normal):not(.is-large),.content .tags.are-medium kbd:not(.is-normal):not(.is-large),.tags.are-medium .docstring>section>a.docs-sourcelink:not(.is-normal):not(.is-large){font-size:1rem}.tags.are-large .tag:not(.is-normal):not(.is-medium),.tags.are-large .content kbd:not(.is-normal):not(.is-medium),.content .tags.are-large kbd:not(.is-normal):not(.is-medium),.tags.are-large .docstring>section>a.docs-sourcelink:not(.is-normal):not(.is-medium){font-size:1.25rem}.tags.is-centered{justify-content:center}.tags.is-centered .tag,.tags.is-centered .content kbd,.content .tags.is-centered kbd,.tags.is-centered .docstring>section>a.docs-sourcelink{margin-right:0.25rem;margin-left:0.25rem}.tags.is-right{justify-content:flex-end}.tags.is-right .tag:not(:first-child),.tags.is-right .content kbd:not(:first-child),.content .tags.is-right kbd:not(:first-child),.tags.is-right .docstring>section>a.docs-sourcelink:not(:first-child){margin-left:0.5rem}.tags.is-right .tag:not(:last-child),.tags.is-right .content kbd:not(:last-child),.content .tags.is-right kbd:not(:last-child),.tags.is-right .docstring>section>a.docs-sourcelink:not(:last-child){margin-right:0}.tags.has-addons .tag,.tags.has-addons .content kbd,.content .tags.has-addons kbd,.tags.has-addons .docstring>section>a.docs-sourcelink{margin-right:0}.tags.has-addons .tag:not(:first-child),.tags.has-addons .content kbd:not(:first-child),.content .tags.has-addons kbd:not(:first-child),.tags.has-addons .docstring>section>a.docs-sourcelink:not(:first-child){margin-left:0;border-top-left-radius:0;border-bottom-left-radius:0}.tags.has-addons .tag:not(:last-child),.tags.has-addons .content kbd:not(:last-child),.content .tags.has-addons kbd:not(:last-child),.tags.has-addons .docstring>section>a.docs-sourcelink:not(:last-child){border-top-right-radius:0;border-bottom-right-radius:0}.tag:not(body),.content kbd:not(body),.docstring>section>a.docs-sourcelink:not(body){align-items:center;background-color:#f5f5f5;border-radius:4px;color:#222;display:inline-flex;font-size:.75rem;height:2em;justify-content:center;line-height:1.5;padding-left:0.75em;padding-right:0.75em;white-space:nowrap}.tag:not(body) .delete,.content kbd:not(body) .delete,.docstring>section>a.docs-sourcelink:not(body) .delete{margin-left:.25rem;margin-right:-.375rem}.tag.is-white:not(body),.content kbd.is-white:not(body),.docstring>section>a.docs-sourcelink.is-white:not(body){background-color:#fff;color:#0a0a0a}.tag.is-black:not(body),.content kbd.is-black:not(body),.docstring>section>a.docs-sourcelink.is-black:not(body){background-color:#0a0a0a;color:#fff}.tag.is-light:not(body),.content kbd.is-light:not(body),.docstring>section>a.docs-sourcelink.is-light:not(body){background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.tag.is-dark:not(body),.content kbd:not(body),.docstring>section>a.docs-sourcelink.is-dark:not(body),.content .docstring>section>kbd:not(body){background-color:#363636;color:#fff}.tag.is-primary:not(body),.content kbd.is-primary:not(body),.docstring>section>a.docs-sourcelink:not(body){background-color:#4eb5de;color:#fff}.tag.is-primary.is-light:not(body),.content kbd.is-primary.is-light:not(body),.docstring>section>a.docs-sourcelink.is-light:not(body){background-color:#eef8fc;color:#1a6d8e}.tag.is-link:not(body),.content kbd.is-link:not(body),.docstring>section>a.docs-sourcelink.is-link:not(body){background-color:#2e63b8;color:#fff}.tag.is-link.is-light:not(body),.content kbd.is-link.is-light:not(body),.docstring>section>a.docs-sourcelink.is-link.is-light:not(body){background-color:#eff3fb;color:#3169c4}.tag.is-info:not(body),.content kbd.is-info:not(body),.docstring>section>a.docs-sourcelink.is-info:not(body){background-color:#209cee;color:#fff}.tag.is-info.is-light:not(body),.content kbd.is-info.is-light:not(body),.docstring>section>a.docs-sourcelink.is-info.is-light:not(body){background-color:#ecf7fe;color:#0e72b4}.tag.is-success:not(body),.content kbd.is-success:not(body),.docstring>section>a.docs-sourcelink.is-success:not(body){background-color:#22c35b;color:#fff}.tag.is-success.is-light:not(body),.content kbd.is-success.is-light:not(body),.docstring>section>a.docs-sourcelink.is-success.is-light:not(body){background-color:#eefcf3;color:#198f43}.tag.is-warning:not(body),.content kbd.is-warning:not(body),.docstring>section>a.docs-sourcelink.is-warning:not(body){background-color:#ffdd57;color:rgba(0,0,0,0.7)}.tag.is-warning.is-light:not(body),.content kbd.is-warning.is-light:not(body),.docstring>section>a.docs-sourcelink.is-warning.is-light:not(body){background-color:#fffbeb;color:#947600}.tag.is-danger:not(body),.content kbd.is-danger:not(body),.docstring>section>a.docs-sourcelink.is-danger:not(body){background-color:#da0b00;color:#fff}.tag.is-danger.is-light:not(body),.content kbd.is-danger.is-light:not(body),.docstring>section>a.docs-sourcelink.is-danger.is-light:not(body){background-color:#ffeceb;color:#f50c00}.tag.is-normal:not(body),.content kbd.is-normal:not(body),.docstring>section>a.docs-sourcelink.is-normal:not(body){font-size:.75rem}.tag.is-medium:not(body),.content kbd.is-medium:not(body),.docstring>section>a.docs-sourcelink.is-medium:not(body){font-size:1rem}.tag.is-large:not(body),.content kbd.is-large:not(body),.docstring>section>a.docs-sourcelink.is-large:not(body){font-size:1.25rem}.tag:not(body) .icon:first-child:not(:last-child),.content kbd:not(body) .icon:first-child:not(:last-child),.docstring>section>a.docs-sourcelink:not(body) .icon:first-child:not(:last-child){margin-left:-.375em;margin-right:.1875em}.tag:not(body) .icon:last-child:not(:first-child),.content kbd:not(body) .icon:last-child:not(:first-child),.docstring>section>a.docs-sourcelink:not(body) .icon:last-child:not(:first-child){margin-left:.1875em;margin-right:-.375em}.tag:not(body) .icon:first-child:last-child,.content kbd:not(body) .icon:first-child:last-child,.docstring>section>a.docs-sourcelink:not(body) .icon:first-child:last-child{margin-left:-.375em;margin-right:-.375em}.tag.is-delete:not(body),.content kbd.is-delete:not(body),.docstring>section>a.docs-sourcelink.is-delete:not(body){margin-left:1px;padding:0;position:relative;width:2em}.tag.is-delete:not(body)::before,.content kbd.is-delete:not(body)::before,.docstring>section>a.docs-sourcelink.is-delete:not(body)::before,.tag.is-delete:not(body)::after,.content kbd.is-delete:not(body)::after,.docstring>section>a.docs-sourcelink.is-delete:not(body)::after{background-color:currentColor;content:"";display:block;left:50%;position:absolute;top:50%;transform:translateX(-50%) translateY(-50%) rotate(45deg);transform-origin:center center}.tag.is-delete:not(body)::before,.content kbd.is-delete:not(body)::before,.docstring>section>a.docs-sourcelink.is-delete:not(body)::before{height:1px;width:50%}.tag.is-delete:not(body)::after,.content kbd.is-delete:not(body)::after,.docstring>section>a.docs-sourcelink.is-delete:not(body)::after{height:50%;width:1px}.tag.is-delete:not(body):hover,.content kbd.is-delete:not(body):hover,.docstring>section>a.docs-sourcelink.is-delete:not(body):hover,.tag.is-delete:not(body):focus,.content kbd.is-delete:not(body):focus,.docstring>section>a.docs-sourcelink.is-delete:not(body):focus{background-color:#e8e8e8}.tag.is-delete:not(body):active,.content kbd.is-delete:not(body):active,.docstring>section>a.docs-sourcelink.is-delete:not(body):active{background-color:#dbdbdb}.tag.is-rounded:not(body),#documenter .docs-sidebar form.docs-search>input:not(body),.content kbd.is-rounded:not(body),#documenter .docs-sidebar .content form.docs-search>input:not(body),.docstring>section>a.docs-sourcelink.is-rounded:not(body){border-radius:9999px}a.tag:hover,.docstring>section>a.docs-sourcelink:hover{text-decoration:underline}.title,.subtitle{word-break:break-word}.title em,.title span,.subtitle em,.subtitle span{font-weight:inherit}.title sub,.subtitle sub{font-size:.75em}.title sup,.subtitle sup{font-size:.75em}.title .tag,.title .content kbd,.content .title kbd,.title .docstring>section>a.docs-sourcelink,.subtitle .tag,.subtitle .content kbd,.content .subtitle kbd,.subtitle .docstring>section>a.docs-sourcelink{vertical-align:middle}.title{color:#222;font-size:2rem;font-weight:600;line-height:1.125}.title strong{color:inherit;font-weight:inherit}.title:not(.is-spaced)+.subtitle{margin-top:-1.25rem}.title.is-1{font-size:3rem}.title.is-2{font-size:2.5rem}.title.is-3{font-size:2rem}.title.is-4{font-size:1.5rem}.title.is-5{font-size:1.25rem}.title.is-6{font-size:1rem}.title.is-7{font-size:.75rem}.subtitle{color:#222;font-size:1.25rem;font-weight:400;line-height:1.25}.subtitle strong{color:#222;font-weight:600}.subtitle:not(.is-spaced)+.title{margin-top:-1.25rem}.subtitle.is-1{font-size:3rem}.subtitle.is-2{font-size:2.5rem}.subtitle.is-3{font-size:2rem}.subtitle.is-4{font-size:1.5rem}.subtitle.is-5{font-size:1.25rem}.subtitle.is-6{font-size:1rem}.subtitle.is-7{font-size:.75rem}.heading{display:block;font-size:11px;letter-spacing:1px;margin-bottom:5px;text-transform:uppercase}.number{align-items:center;background-color:#f5f5f5;border-radius:9999px;display:inline-flex;font-size:1.25rem;height:2em;justify-content:center;margin-right:1.5rem;min-width:2.5em;padding:0.25rem 0.5rem;text-align:center;vertical-align:top}.select select,.textarea,.input,#documenter .docs-sidebar form.docs-search>input{background-color:#fff;border-color:#dbdbdb;border-radius:4px;color:#222}.select select::-moz-placeholder,.textarea::-moz-placeholder,.input::-moz-placeholder,#documenter .docs-sidebar form.docs-search>input::-moz-placeholder{color:#707070}.select select::-webkit-input-placeholder,.textarea::-webkit-input-placeholder,.input::-webkit-input-placeholder,#documenter .docs-sidebar form.docs-search>input::-webkit-input-placeholder{color:#707070}.select select:-moz-placeholder,.textarea:-moz-placeholder,.input:-moz-placeholder,#documenter .docs-sidebar form.docs-search>input:-moz-placeholder{color:#707070}.select select:-ms-input-placeholder,.textarea:-ms-input-placeholder,.input:-ms-input-placeholder,#documenter .docs-sidebar form.docs-search>input:-ms-input-placeholder{color:#707070}.select select:hover,.textarea:hover,.input:hover,#documenter .docs-sidebar form.docs-search>input:hover,.select select.is-hovered,.is-hovered.textarea,.is-hovered.input,#documenter .docs-sidebar form.docs-search>input.is-hovered{border-color:#b5b5b5}.select select:focus,.textarea:focus,.input:focus,#documenter .docs-sidebar form.docs-search>input:focus,.select select.is-focused,.is-focused.textarea,.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.select select:active,.textarea:active,.input:active,#documenter .docs-sidebar form.docs-search>input:active,.select select.is-active,.is-active.textarea,.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{border-color:#2e63b8;box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.select select[disabled],.textarea[disabled],.input[disabled],#documenter .docs-sidebar form.docs-search>input[disabled],fieldset[disabled] .select select,.select fieldset[disabled] select,fieldset[disabled] .textarea,fieldset[disabled] .input,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input{background-color:#f5f5f5;border-color:#f5f5f5;box-shadow:none;color:#6b6b6b}.select select[disabled]::-moz-placeholder,.textarea[disabled]::-moz-placeholder,.input[disabled]::-moz-placeholder,#documenter .docs-sidebar form.docs-search>input[disabled]::-moz-placeholder,fieldset[disabled] .select select::-moz-placeholder,.select fieldset[disabled] select::-moz-placeholder,fieldset[disabled] .textarea::-moz-placeholder,fieldset[disabled] .input::-moz-placeholder,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input::-moz-placeholder,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input::-moz-placeholder{color:rgba(107,107,107,0.3)}.select select[disabled]::-webkit-input-placeholder,.textarea[disabled]::-webkit-input-placeholder,.input[disabled]::-webkit-input-placeholder,#documenter .docs-sidebar form.docs-search>input[disabled]::-webkit-input-placeholder,fieldset[disabled] .select select::-webkit-input-placeholder,.select fieldset[disabled] select::-webkit-input-placeholder,fieldset[disabled] .textarea::-webkit-input-placeholder,fieldset[disabled] .input::-webkit-input-placeholder,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input::-webkit-input-placeholder,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input::-webkit-input-placeholder{color:rgba(107,107,107,0.3)}.select select[disabled]:-moz-placeholder,.textarea[disabled]:-moz-placeholder,.input[disabled]:-moz-placeholder,#documenter .docs-sidebar form.docs-search>input[disabled]:-moz-placeholder,fieldset[disabled] .select select:-moz-placeholder,.select fieldset[disabled] select:-moz-placeholder,fieldset[disabled] .textarea:-moz-placeholder,fieldset[disabled] .input:-moz-placeholder,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input:-moz-placeholder,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input:-moz-placeholder{color:rgba(107,107,107,0.3)}.select select[disabled]:-ms-input-placeholder,.textarea[disabled]:-ms-input-placeholder,.input[disabled]:-ms-input-placeholder,#documenter .docs-sidebar form.docs-search>input[disabled]:-ms-input-placeholder,fieldset[disabled] .select select:-ms-input-placeholder,.select fieldset[disabled] select:-ms-input-placeholder,fieldset[disabled] .textarea:-ms-input-placeholder,fieldset[disabled] .input:-ms-input-placeholder,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input:-ms-input-placeholder,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input:-ms-input-placeholder{color:rgba(107,107,107,0.3)}.textarea,.input,#documenter .docs-sidebar form.docs-search>input{box-shadow:inset 0 0.0625em 0.125em rgba(10,10,10,0.05);max-width:100%;width:100%}.textarea[readonly],.input[readonly],#documenter .docs-sidebar form.docs-search>input[readonly]{box-shadow:none}.is-white.textarea,.is-white.input,#documenter .docs-sidebar form.docs-search>input.is-white{border-color:#fff}.is-white.textarea:focus,.is-white.input:focus,#documenter .docs-sidebar form.docs-search>input.is-white:focus,.is-white.is-focused.textarea,.is-white.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-white.textarea:active,.is-white.input:active,#documenter .docs-sidebar form.docs-search>input.is-white:active,.is-white.is-active.textarea,.is-white.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}.is-black.textarea,.is-black.input,#documenter .docs-sidebar form.docs-search>input.is-black{border-color:#0a0a0a}.is-black.textarea:focus,.is-black.input:focus,#documenter .docs-sidebar form.docs-search>input.is-black:focus,.is-black.is-focused.textarea,.is-black.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-black.textarea:active,.is-black.input:active,#documenter .docs-sidebar form.docs-search>input.is-black:active,.is-black.is-active.textarea,.is-black.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}.is-light.textarea,.is-light.input,#documenter .docs-sidebar form.docs-search>input.is-light{border-color:#f5f5f5}.is-light.textarea:focus,.is-light.input:focus,#documenter .docs-sidebar form.docs-search>input.is-light:focus,.is-light.is-focused.textarea,.is-light.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-light.textarea:active,.is-light.input:active,#documenter .docs-sidebar form.docs-search>input.is-light:active,.is-light.is-active.textarea,.is-light.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(245,245,245,0.25)}.is-dark.textarea,.content kbd.textarea,.is-dark.input,#documenter .docs-sidebar form.docs-search>input.is-dark,.content kbd.input{border-color:#363636}.is-dark.textarea:focus,.content kbd.textarea:focus,.is-dark.input:focus,#documenter .docs-sidebar form.docs-search>input.is-dark:focus,.content kbd.input:focus,.is-dark.is-focused.textarea,.content kbd.is-focused.textarea,.is-dark.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.content kbd.is-focused.input,#documenter .docs-sidebar .content form.docs-search>input.is-focused,.is-dark.textarea:active,.content kbd.textarea:active,.is-dark.input:active,#documenter .docs-sidebar form.docs-search>input.is-dark:active,.content kbd.input:active,.is-dark.is-active.textarea,.content kbd.is-active.textarea,.is-dark.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active,.content kbd.is-active.input,#documenter .docs-sidebar .content form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(54,54,54,0.25)}.is-primary.textarea,.docstring>section>a.textarea.docs-sourcelink,.is-primary.input,#documenter .docs-sidebar form.docs-search>input.is-primary,.docstring>section>a.input.docs-sourcelink{border-color:#4eb5de}.is-primary.textarea:focus,.docstring>section>a.textarea.docs-sourcelink:focus,.is-primary.input:focus,#documenter .docs-sidebar form.docs-search>input.is-primary:focus,.docstring>section>a.input.docs-sourcelink:focus,.is-primary.is-focused.textarea,.docstring>section>a.is-focused.textarea.docs-sourcelink,.is-primary.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.docstring>section>a.is-focused.input.docs-sourcelink,.is-primary.textarea:active,.docstring>section>a.textarea.docs-sourcelink:active,.is-primary.input:active,#documenter .docs-sidebar form.docs-search>input.is-primary:active,.docstring>section>a.input.docs-sourcelink:active,.is-primary.is-active.textarea,.docstring>section>a.is-active.textarea.docs-sourcelink,.is-primary.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active,.docstring>section>a.is-active.input.docs-sourcelink{box-shadow:0 0 0 0.125em rgba(78,181,222,0.25)}.is-link.textarea,.is-link.input,#documenter .docs-sidebar form.docs-search>input.is-link{border-color:#2e63b8}.is-link.textarea:focus,.is-link.input:focus,#documenter .docs-sidebar form.docs-search>input.is-link:focus,.is-link.is-focused.textarea,.is-link.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-link.textarea:active,.is-link.input:active,#documenter .docs-sidebar form.docs-search>input.is-link:active,.is-link.is-active.textarea,.is-link.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.is-info.textarea,.is-info.input,#documenter .docs-sidebar form.docs-search>input.is-info{border-color:#209cee}.is-info.textarea:focus,.is-info.input:focus,#documenter .docs-sidebar form.docs-search>input.is-info:focus,.is-info.is-focused.textarea,.is-info.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-info.textarea:active,.is-info.input:active,#documenter .docs-sidebar form.docs-search>input.is-info:active,.is-info.is-active.textarea,.is-info.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(32,156,238,0.25)}.is-success.textarea,.is-success.input,#documenter .docs-sidebar form.docs-search>input.is-success{border-color:#22c35b}.is-success.textarea:focus,.is-success.input:focus,#documenter .docs-sidebar form.docs-search>input.is-success:focus,.is-success.is-focused.textarea,.is-success.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-success.textarea:active,.is-success.input:active,#documenter .docs-sidebar form.docs-search>input.is-success:active,.is-success.is-active.textarea,.is-success.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(34,195,91,0.25)}.is-warning.textarea,.is-warning.input,#documenter .docs-sidebar form.docs-search>input.is-warning{border-color:#ffdd57}.is-warning.textarea:focus,.is-warning.input:focus,#documenter .docs-sidebar form.docs-search>input.is-warning:focus,.is-warning.is-focused.textarea,.is-warning.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-warning.textarea:active,.is-warning.input:active,#documenter .docs-sidebar form.docs-search>input.is-warning:active,.is-warning.is-active.textarea,.is-warning.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(255,221,87,0.25)}.is-danger.textarea,.is-danger.input,#documenter .docs-sidebar form.docs-search>input.is-danger{border-color:#da0b00}.is-danger.textarea:focus,.is-danger.input:focus,#documenter .docs-sidebar form.docs-search>input.is-danger:focus,.is-danger.is-focused.textarea,.is-danger.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-danger.textarea:active,.is-danger.input:active,#documenter .docs-sidebar form.docs-search>input.is-danger:active,.is-danger.is-active.textarea,.is-danger.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(218,11,0,0.25)}.is-small.textarea,.is-small.input,#documenter .docs-sidebar form.docs-search>input{border-radius:2px;font-size:.75rem}.is-medium.textarea,.is-medium.input,#documenter .docs-sidebar form.docs-search>input.is-medium{font-size:1.25rem}.is-large.textarea,.is-large.input,#documenter .docs-sidebar form.docs-search>input.is-large{font-size:1.5rem}.is-fullwidth.textarea,.is-fullwidth.input,#documenter .docs-sidebar form.docs-search>input.is-fullwidth{display:block;width:100%}.is-inline.textarea,.is-inline.input,#documenter .docs-sidebar form.docs-search>input.is-inline{display:inline;width:auto}.input.is-rounded,#documenter .docs-sidebar form.docs-search>input{border-radius:9999px;padding-left:calc(calc(0.75em - 1px) + 0.375em);padding-right:calc(calc(0.75em - 1px) + 0.375em)}.input.is-static,#documenter .docs-sidebar form.docs-search>input.is-static{background-color:transparent;border-color:transparent;box-shadow:none;padding-left:0;padding-right:0}.textarea{display:block;max-width:100%;min-width:100%;padding:calc(0.75em - 1px);resize:vertical}.textarea:not([rows]){max-height:40em;min-height:8em}.textarea[rows]{height:initial}.textarea.has-fixed-size{resize:none}.radio,.checkbox{cursor:pointer;display:inline-block;line-height:1.25;position:relative}.radio input,.checkbox input{cursor:pointer}.radio:hover,.checkbox:hover{color:#222}.radio[disabled],.checkbox[disabled],fieldset[disabled] .radio,fieldset[disabled] .checkbox,.radio input[disabled],.checkbox input[disabled]{color:#6b6b6b;cursor:not-allowed}.radio+.radio{margin-left:.5em}.select{display:inline-block;max-width:100%;position:relative;vertical-align:top}.select:not(.is-multiple){height:2.5em}.select:not(.is-multiple):not(.is-loading)::after{border-color:#2e63b8;right:1.125em;z-index:4}.select.is-rounded select,#documenter .docs-sidebar form.docs-search>input.select select{border-radius:9999px;padding-left:1em}.select select{cursor:pointer;display:block;font-size:1em;max-width:100%;outline:none}.select select::-ms-expand{display:none}.select select[disabled]:hover,fieldset[disabled] .select select:hover{border-color:#f5f5f5}.select select:not([multiple]){padding-right:2.5em}.select select[multiple]{height:auto;padding:0}.select select[multiple] option{padding:0.5em 1em}.select:not(.is-multiple):not(.is-loading):hover::after{border-color:#222}.select.is-white:not(:hover)::after{border-color:#fff}.select.is-white select{border-color:#fff}.select.is-white select:hover,.select.is-white select.is-hovered{border-color:#f2f2f2}.select.is-white select:focus,.select.is-white select.is-focused,.select.is-white select:active,.select.is-white select.is-active{box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}.select.is-black:not(:hover)::after{border-color:#0a0a0a}.select.is-black select{border-color:#0a0a0a}.select.is-black select:hover,.select.is-black select.is-hovered{border-color:#000}.select.is-black select:focus,.select.is-black select.is-focused,.select.is-black select:active,.select.is-black select.is-active{box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}.select.is-light:not(:hover)::after{border-color:#f5f5f5}.select.is-light select{border-color:#f5f5f5}.select.is-light select:hover,.select.is-light select.is-hovered{border-color:#e8e8e8}.select.is-light select:focus,.select.is-light select.is-focused,.select.is-light select:active,.select.is-light select.is-active{box-shadow:0 0 0 0.125em rgba(245,245,245,0.25)}.select.is-dark:not(:hover)::after,.content kbd.select:not(:hover)::after{border-color:#363636}.select.is-dark select,.content kbd.select select{border-color:#363636}.select.is-dark select:hover,.content kbd.select select:hover,.select.is-dark select.is-hovered,.content kbd.select select.is-hovered{border-color:#292929}.select.is-dark select:focus,.content kbd.select select:focus,.select.is-dark select.is-focused,.content kbd.select select.is-focused,.select.is-dark select:active,.content kbd.select select:active,.select.is-dark select.is-active,.content kbd.select select.is-active{box-shadow:0 0 0 0.125em rgba(54,54,54,0.25)}.select.is-primary:not(:hover)::after,.docstring>section>a.select.docs-sourcelink:not(:hover)::after{border-color:#4eb5de}.select.is-primary select,.docstring>section>a.select.docs-sourcelink select{border-color:#4eb5de}.select.is-primary select:hover,.docstring>section>a.select.docs-sourcelink select:hover,.select.is-primary select.is-hovered,.docstring>section>a.select.docs-sourcelink select.is-hovered{border-color:#39acda}.select.is-primary select:focus,.docstring>section>a.select.docs-sourcelink select:focus,.select.is-primary select.is-focused,.docstring>section>a.select.docs-sourcelink select.is-focused,.select.is-primary select:active,.docstring>section>a.select.docs-sourcelink select:active,.select.is-primary select.is-active,.docstring>section>a.select.docs-sourcelink select.is-active{box-shadow:0 0 0 0.125em rgba(78,181,222,0.25)}.select.is-link:not(:hover)::after{border-color:#2e63b8}.select.is-link select{border-color:#2e63b8}.select.is-link select:hover,.select.is-link select.is-hovered{border-color:#2958a4}.select.is-link select:focus,.select.is-link select.is-focused,.select.is-link select:active,.select.is-link select.is-active{box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.select.is-info:not(:hover)::after{border-color:#209cee}.select.is-info select{border-color:#209cee}.select.is-info select:hover,.select.is-info select.is-hovered{border-color:#1190e3}.select.is-info select:focus,.select.is-info select.is-focused,.select.is-info select:active,.select.is-info select.is-active{box-shadow:0 0 0 0.125em rgba(32,156,238,0.25)}.select.is-success:not(:hover)::after{border-color:#22c35b}.select.is-success select{border-color:#22c35b}.select.is-success select:hover,.select.is-success select.is-hovered{border-color:#1ead51}.select.is-success select:focus,.select.is-success select.is-focused,.select.is-success select:active,.select.is-success select.is-active{box-shadow:0 0 0 0.125em rgba(34,195,91,0.25)}.select.is-warning:not(:hover)::after{border-color:#ffdd57}.select.is-warning select{border-color:#ffdd57}.select.is-warning select:hover,.select.is-warning select.is-hovered{border-color:#ffd83e}.select.is-warning select:focus,.select.is-warning select.is-focused,.select.is-warning select:active,.select.is-warning select.is-active{box-shadow:0 0 0 0.125em rgba(255,221,87,0.25)}.select.is-danger:not(:hover)::after{border-color:#da0b00}.select.is-danger select{border-color:#da0b00}.select.is-danger select:hover,.select.is-danger select.is-hovered{border-color:#c10a00}.select.is-danger select:focus,.select.is-danger select.is-focused,.select.is-danger select:active,.select.is-danger select.is-active{box-shadow:0 0 0 0.125em rgba(218,11,0,0.25)}.select.is-small,#documenter .docs-sidebar form.docs-search>input.select{border-radius:2px;font-size:.75rem}.select.is-medium{font-size:1.25rem}.select.is-large{font-size:1.5rem}.select.is-disabled::after{border-color:#6b6b6b !important;opacity:0.5}.select.is-fullwidth{width:100%}.select.is-fullwidth select{width:100%}.select.is-loading::after{margin-top:0;position:absolute;right:.625em;top:0.625em;transform:none}.select.is-loading.is-small:after,#documenter .docs-sidebar form.docs-search>input.is-loading:after{font-size:.75rem}.select.is-loading.is-medium:after{font-size:1.25rem}.select.is-loading.is-large:after{font-size:1.5rem}.file{align-items:stretch;display:flex;justify-content:flex-start;position:relative}.file.is-white .file-cta{background-color:#fff;border-color:transparent;color:#0a0a0a}.file.is-white:hover .file-cta,.file.is-white.is-hovered .file-cta{background-color:#f9f9f9;border-color:transparent;color:#0a0a0a}.file.is-white:focus .file-cta,.file.is-white.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(255,255,255,0.25);color:#0a0a0a}.file.is-white:active .file-cta,.file.is-white.is-active .file-cta{background-color:#f2f2f2;border-color:transparent;color:#0a0a0a}.file.is-black .file-cta{background-color:#0a0a0a;border-color:transparent;color:#fff}.file.is-black:hover .file-cta,.file.is-black.is-hovered .file-cta{background-color:#040404;border-color:transparent;color:#fff}.file.is-black:focus .file-cta,.file.is-black.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(10,10,10,0.25);color:#fff}.file.is-black:active .file-cta,.file.is-black.is-active .file-cta{background-color:#000;border-color:transparent;color:#fff}.file.is-light .file-cta{background-color:#f5f5f5;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-light:hover .file-cta,.file.is-light.is-hovered .file-cta{background-color:#eee;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-light:focus .file-cta,.file.is-light.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(245,245,245,0.25);color:rgba(0,0,0,0.7)}.file.is-light:active .file-cta,.file.is-light.is-active .file-cta{background-color:#e8e8e8;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-dark .file-cta,.content kbd.file .file-cta{background-color:#363636;border-color:transparent;color:#fff}.file.is-dark:hover .file-cta,.content kbd.file:hover .file-cta,.file.is-dark.is-hovered .file-cta,.content kbd.file.is-hovered .file-cta{background-color:#2f2f2f;border-color:transparent;color:#fff}.file.is-dark:focus .file-cta,.content kbd.file:focus .file-cta,.file.is-dark.is-focused .file-cta,.content kbd.file.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(54,54,54,0.25);color:#fff}.file.is-dark:active .file-cta,.content kbd.file:active .file-cta,.file.is-dark.is-active .file-cta,.content kbd.file.is-active .file-cta{background-color:#292929;border-color:transparent;color:#fff}.file.is-primary .file-cta,.docstring>section>a.file.docs-sourcelink .file-cta{background-color:#4eb5de;border-color:transparent;color:#fff}.file.is-primary:hover .file-cta,.docstring>section>a.file.docs-sourcelink:hover .file-cta,.file.is-primary.is-hovered .file-cta,.docstring>section>a.file.is-hovered.docs-sourcelink .file-cta{background-color:#43b1dc;border-color:transparent;color:#fff}.file.is-primary:focus .file-cta,.docstring>section>a.file.docs-sourcelink:focus .file-cta,.file.is-primary.is-focused .file-cta,.docstring>section>a.file.is-focused.docs-sourcelink .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(78,181,222,0.25);color:#fff}.file.is-primary:active .file-cta,.docstring>section>a.file.docs-sourcelink:active .file-cta,.file.is-primary.is-active .file-cta,.docstring>section>a.file.is-active.docs-sourcelink .file-cta{background-color:#39acda;border-color:transparent;color:#fff}.file.is-link .file-cta{background-color:#2e63b8;border-color:transparent;color:#fff}.file.is-link:hover .file-cta,.file.is-link.is-hovered .file-cta{background-color:#2b5eae;border-color:transparent;color:#fff}.file.is-link:focus .file-cta,.file.is-link.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(46,99,184,0.25);color:#fff}.file.is-link:active .file-cta,.file.is-link.is-active .file-cta{background-color:#2958a4;border-color:transparent;color:#fff}.file.is-info .file-cta{background-color:#209cee;border-color:transparent;color:#fff}.file.is-info:hover .file-cta,.file.is-info.is-hovered .file-cta{background-color:#1497ed;border-color:transparent;color:#fff}.file.is-info:focus .file-cta,.file.is-info.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(32,156,238,0.25);color:#fff}.file.is-info:active .file-cta,.file.is-info.is-active .file-cta{background-color:#1190e3;border-color:transparent;color:#fff}.file.is-success .file-cta{background-color:#22c35b;border-color:transparent;color:#fff}.file.is-success:hover .file-cta,.file.is-success.is-hovered .file-cta{background-color:#20b856;border-color:transparent;color:#fff}.file.is-success:focus .file-cta,.file.is-success.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(34,195,91,0.25);color:#fff}.file.is-success:active .file-cta,.file.is-success.is-active .file-cta{background-color:#1ead51;border-color:transparent;color:#fff}.file.is-warning .file-cta{background-color:#ffdd57;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-warning:hover .file-cta,.file.is-warning.is-hovered .file-cta{background-color:#ffda4a;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-warning:focus .file-cta,.file.is-warning.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(255,221,87,0.25);color:rgba(0,0,0,0.7)}.file.is-warning:active .file-cta,.file.is-warning.is-active .file-cta{background-color:#ffd83e;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-danger .file-cta{background-color:#da0b00;border-color:transparent;color:#fff}.file.is-danger:hover .file-cta,.file.is-danger.is-hovered .file-cta{background-color:#cd0a00;border-color:transparent;color:#fff}.file.is-danger:focus .file-cta,.file.is-danger.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(218,11,0,0.25);color:#fff}.file.is-danger:active .file-cta,.file.is-danger.is-active .file-cta{background-color:#c10a00;border-color:transparent;color:#fff}.file.is-small,#documenter .docs-sidebar form.docs-search>input.file{font-size:.75rem}.file.is-normal{font-size:1rem}.file.is-medium{font-size:1.25rem}.file.is-medium .file-icon .fa{font-size:21px}.file.is-large{font-size:1.5rem}.file.is-large .file-icon .fa{font-size:28px}.file.has-name .file-cta{border-bottom-right-radius:0;border-top-right-radius:0}.file.has-name .file-name{border-bottom-left-radius:0;border-top-left-radius:0}.file.has-name.is-empty .file-cta{border-radius:4px}.file.has-name.is-empty .file-name{display:none}.file.is-boxed .file-label{flex-direction:column}.file.is-boxed .file-cta{flex-direction:column;height:auto;padding:1em 3em}.file.is-boxed .file-name{border-width:0 1px 1px}.file.is-boxed .file-icon{height:1.5em;width:1.5em}.file.is-boxed .file-icon .fa{font-size:21px}.file.is-boxed.is-small .file-icon .fa,#documenter .docs-sidebar form.docs-search>input.is-boxed .file-icon .fa{font-size:14px}.file.is-boxed.is-medium .file-icon .fa{font-size:28px}.file.is-boxed.is-large .file-icon .fa{font-size:35px}.file.is-boxed.has-name .file-cta{border-radius:4px 4px 0 0}.file.is-boxed.has-name .file-name{border-radius:0 0 4px 4px;border-width:0 1px 1px}.file.is-centered{justify-content:center}.file.is-fullwidth .file-label{width:100%}.file.is-fullwidth .file-name{flex-grow:1;max-width:none}.file.is-right{justify-content:flex-end}.file.is-right .file-cta{border-radius:0 4px 4px 0}.file.is-right .file-name{border-radius:4px 0 0 4px;border-width:1px 0 1px 1px;order:-1}.file-label{align-items:stretch;display:flex;cursor:pointer;justify-content:flex-start;overflow:hidden;position:relative}.file-label:hover .file-cta{background-color:#eee;color:#222}.file-label:hover .file-name{border-color:#d5d5d5}.file-label:active .file-cta{background-color:#e8e8e8;color:#222}.file-label:active .file-name{border-color:#cfcfcf}.file-input{height:100%;left:0;opacity:0;outline:none;position:absolute;top:0;width:100%}.file-cta,.file-name{border-color:#dbdbdb;border-radius:4px;font-size:1em;padding-left:1em;padding-right:1em;white-space:nowrap}.file-cta{background-color:#f5f5f5;color:#222}.file-name{border-color:#dbdbdb;border-style:solid;border-width:1px 1px 1px 0;display:block;max-width:16em;overflow:hidden;text-align:inherit;text-overflow:ellipsis}.file-icon{align-items:center;display:flex;height:1em;justify-content:center;margin-right:.5em;width:1em}.file-icon .fa{font-size:14px}.label{color:#222;display:block;font-size:1rem;font-weight:700}.label:not(:last-child){margin-bottom:0.5em}.label.is-small,#documenter .docs-sidebar form.docs-search>input.label{font-size:.75rem}.label.is-medium{font-size:1.25rem}.label.is-large{font-size:1.5rem}.help{display:block;font-size:.75rem;margin-top:0.25rem}.help.is-white{color:#fff}.help.is-black{color:#0a0a0a}.help.is-light{color:#f5f5f5}.help.is-dark,.content kbd.help{color:#363636}.help.is-primary,.docstring>section>a.help.docs-sourcelink{color:#4eb5de}.help.is-link{color:#2e63b8}.help.is-info{color:#209cee}.help.is-success{color:#22c35b}.help.is-warning{color:#ffdd57}.help.is-danger{color:#da0b00}.field:not(:last-child){margin-bottom:0.75rem}.field.has-addons{display:flex;justify-content:flex-start}.field.has-addons .control:not(:last-child){margin-right:-1px}.field.has-addons .control:not(:first-child):not(:last-child) .button,.field.has-addons .control:not(:first-child):not(:last-child) .input,.field.has-addons .control:not(:first-child):not(:last-child) #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .field.has-addons .control:not(:first-child):not(:last-child) form.docs-search>input,.field.has-addons .control:not(:first-child):not(:last-child) .select select{border-radius:0}.field.has-addons .control:first-child:not(:only-child) .button,.field.has-addons .control:first-child:not(:only-child) .input,.field.has-addons .control:first-child:not(:only-child) #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .field.has-addons .control:first-child:not(:only-child) form.docs-search>input,.field.has-addons .control:first-child:not(:only-child) .select select{border-bottom-right-radius:0;border-top-right-radius:0}.field.has-addons .control:last-child:not(:only-child) .button,.field.has-addons .control:last-child:not(:only-child) .input,.field.has-addons .control:last-child:not(:only-child) #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .field.has-addons .control:last-child:not(:only-child) form.docs-search>input,.field.has-addons .control:last-child:not(:only-child) .select select{border-bottom-left-radius:0;border-top-left-radius:0}.field.has-addons .control .button:not([disabled]):hover,.field.has-addons .control .button.is-hovered:not([disabled]),.field.has-addons .control .input:not([disabled]):hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):hover,.field.has-addons .control .input.is-hovered:not([disabled]),.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-hovered:not([disabled]),#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-hovered:not([disabled]),.field.has-addons .control .select select:not([disabled]):hover,.field.has-addons .control .select select.is-hovered:not([disabled]){z-index:2}.field.has-addons .control .button:not([disabled]):focus,.field.has-addons .control .button.is-focused:not([disabled]),.field.has-addons .control .button:not([disabled]):active,.field.has-addons .control .button.is-active:not([disabled]),.field.has-addons .control .input:not([disabled]):focus,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):focus,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):focus,.field.has-addons .control .input.is-focused:not([disabled]),.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-focused:not([disabled]),#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-focused:not([disabled]),.field.has-addons .control .input:not([disabled]):active,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):active,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):active,.field.has-addons .control .input.is-active:not([disabled]),.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-active:not([disabled]),#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-active:not([disabled]),.field.has-addons .control .select select:not([disabled]):focus,.field.has-addons .control .select select.is-focused:not([disabled]),.field.has-addons .control .select select:not([disabled]):active,.field.has-addons .control .select select.is-active:not([disabled]){z-index:3}.field.has-addons .control .button:not([disabled]):focus:hover,.field.has-addons .control .button.is-focused:not([disabled]):hover,.field.has-addons .control .button:not([disabled]):active:hover,.field.has-addons .control .button.is-active:not([disabled]):hover,.field.has-addons .control .input:not([disabled]):focus:hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):focus:hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):focus:hover,.field.has-addons .control .input.is-focused:not([disabled]):hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-focused:not([disabled]):hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-focused:not([disabled]):hover,.field.has-addons .control .input:not([disabled]):active:hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):active:hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):active:hover,.field.has-addons .control .input.is-active:not([disabled]):hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-active:not([disabled]):hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-active:not([disabled]):hover,.field.has-addons .control .select select:not([disabled]):focus:hover,.field.has-addons .control .select select.is-focused:not([disabled]):hover,.field.has-addons .control .select select:not([disabled]):active:hover,.field.has-addons .control .select select.is-active:not([disabled]):hover{z-index:4}.field.has-addons .control.is-expanded{flex-grow:1;flex-shrink:1}.field.has-addons.has-addons-centered{justify-content:center}.field.has-addons.has-addons-right{justify-content:flex-end}.field.has-addons.has-addons-fullwidth .control{flex-grow:1;flex-shrink:0}.field.is-grouped{display:flex;justify-content:flex-start}.field.is-grouped>.control{flex-shrink:0}.field.is-grouped>.control:not(:last-child){margin-bottom:0;margin-right:.75rem}.field.is-grouped>.control.is-expanded{flex-grow:1;flex-shrink:1}.field.is-grouped.is-grouped-centered{justify-content:center}.field.is-grouped.is-grouped-right{justify-content:flex-end}.field.is-grouped.is-grouped-multiline{flex-wrap:wrap}.field.is-grouped.is-grouped-multiline>.control:last-child,.field.is-grouped.is-grouped-multiline>.control:not(:last-child){margin-bottom:0.75rem}.field.is-grouped.is-grouped-multiline:last-child{margin-bottom:-0.75rem}.field.is-grouped.is-grouped-multiline:not(:last-child){margin-bottom:0}@media screen and (min-width: 769px),print{.field.is-horizontal{display:flex}}.field-label .label{font-size:inherit}@media screen and (max-width: 768px){.field-label{margin-bottom:0.5rem}}@media screen and (min-width: 769px),print{.field-label{flex-basis:0;flex-grow:1;flex-shrink:0;margin-right:1.5rem;text-align:right}.field-label.is-small,#documenter .docs-sidebar form.docs-search>input.field-label{font-size:.75rem;padding-top:0.375em}.field-label.is-normal{padding-top:0.375em}.field-label.is-medium{font-size:1.25rem;padding-top:0.375em}.field-label.is-large{font-size:1.5rem;padding-top:0.375em}}.field-body .field .field{margin-bottom:0}@media screen and (min-width: 769px),print{.field-body{display:flex;flex-basis:0;flex-grow:5;flex-shrink:1}.field-body .field{margin-bottom:0}.field-body>.field{flex-shrink:1}.field-body>.field:not(.is-narrow){flex-grow:1}.field-body>.field:not(:last-child){margin-right:.75rem}}.control{box-sizing:border-box;clear:both;font-size:1rem;position:relative;text-align:inherit}.control.has-icons-left .input:focus~.icon,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input:focus~.icon,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input:focus~.icon,.control.has-icons-left .select:focus~.icon,.control.has-icons-right .input:focus~.icon,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input:focus~.icon,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input:focus~.icon,.control.has-icons-right .select:focus~.icon{color:#222}.control.has-icons-left .input.is-small~.icon,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input~.icon,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input~.icon,.control.has-icons-left .select.is-small~.icon,.control.has-icons-right .input.is-small~.icon,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input~.icon,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input~.icon,.control.has-icons-right .select.is-small~.icon{font-size:.75rem}.control.has-icons-left .input.is-medium~.icon,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input.is-medium~.icon,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input.is-medium~.icon,.control.has-icons-left .select.is-medium~.icon,.control.has-icons-right .input.is-medium~.icon,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input.is-medium~.icon,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input.is-medium~.icon,.control.has-icons-right .select.is-medium~.icon{font-size:1.25rem}.control.has-icons-left .input.is-large~.icon,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input.is-large~.icon,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input.is-large~.icon,.control.has-icons-left .select.is-large~.icon,.control.has-icons-right .input.is-large~.icon,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input.is-large~.icon,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input.is-large~.icon,.control.has-icons-right .select.is-large~.icon{font-size:1.5rem}.control.has-icons-left .icon,.control.has-icons-right .icon{color:#dbdbdb;height:2.5em;pointer-events:none;position:absolute;top:0;width:2.5em;z-index:4}.control.has-icons-left .input,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input,.control.has-icons-left .select select{padding-left:2.5em}.control.has-icons-left .icon.is-left{left:0}.control.has-icons-right .input,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input,.control.has-icons-right .select select{padding-right:2.5em}.control.has-icons-right .icon.is-right{right:0}.control.is-loading::after{position:absolute !important;right:.625em;top:0.625em;z-index:4}.control.is-loading.is-small:after,#documenter .docs-sidebar form.docs-search>input.is-loading:after{font-size:.75rem}.control.is-loading.is-medium:after{font-size:1.25rem}.control.is-loading.is-large:after{font-size:1.5rem}.breadcrumb{font-size:1rem;white-space:nowrap}.breadcrumb a{align-items:center;color:#2e63b8;display:flex;justify-content:center;padding:0 .75em}.breadcrumb a:hover{color:#363636}.breadcrumb li{align-items:center;display:flex}.breadcrumb li:first-child a{padding-left:0}.breadcrumb li.is-active a{color:#222;cursor:default;pointer-events:none}.breadcrumb li+li::before{color:#b5b5b5;content:"\0002f"}.breadcrumb ul,.breadcrumb ol{align-items:flex-start;display:flex;flex-wrap:wrap;justify-content:flex-start}.breadcrumb .icon:first-child{margin-right:.5em}.breadcrumb .icon:last-child{margin-left:.5em}.breadcrumb.is-centered ol,.breadcrumb.is-centered ul{justify-content:center}.breadcrumb.is-right ol,.breadcrumb.is-right ul{justify-content:flex-end}.breadcrumb.is-small,#documenter .docs-sidebar form.docs-search>input.breadcrumb{font-size:.75rem}.breadcrumb.is-medium{font-size:1.25rem}.breadcrumb.is-large{font-size:1.5rem}.breadcrumb.has-arrow-separator li+li::before{content:"\02192"}.breadcrumb.has-bullet-separator li+li::before{content:"\02022"}.breadcrumb.has-dot-separator li+li::before{content:"\000b7"}.breadcrumb.has-succeeds-separator li+li::before{content:"\0227B"}.card{background-color:#fff;border-radius:.25rem;box-shadow:#bbb;color:#222;max-width:100%;position:relative}.card-footer:first-child,.card-content:first-child,.card-header:first-child{border-top-left-radius:.25rem;border-top-right-radius:.25rem}.card-footer:last-child,.card-content:last-child,.card-header:last-child{border-bottom-left-radius:.25rem;border-bottom-right-radius:.25rem}.card-header{background-color:rgba(0,0,0,0);align-items:stretch;box-shadow:0 0.125em 0.25em rgba(10,10,10,0.1);display:flex}.card-header-title{align-items:center;color:#222;display:flex;flex-grow:1;font-weight:700;padding:0.75rem 1rem}.card-header-title.is-centered{justify-content:center}.card-header-icon{-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;color:currentColor;font-family:inherit;font-size:1em;margin:0;padding:0;align-items:center;cursor:pointer;display:flex;justify-content:center;padding:0.75rem 1rem}.card-image{display:block;position:relative}.card-image:first-child img{border-top-left-radius:.25rem;border-top-right-radius:.25rem}.card-image:last-child img{border-bottom-left-radius:.25rem;border-bottom-right-radius:.25rem}.card-content{background-color:rgba(0,0,0,0);padding:1.5rem}.card-footer{background-color:rgba(0,0,0,0);border-top:1px solid #ededed;align-items:stretch;display:flex}.card-footer-item{align-items:center;display:flex;flex-basis:0;flex-grow:1;flex-shrink:0;justify-content:center;padding:.75rem}.card-footer-item:not(:last-child){border-right:1px solid #ededed}.card .media:not(:last-child){margin-bottom:1.5rem}.dropdown{display:inline-flex;position:relative;vertical-align:top}.dropdown.is-active .dropdown-menu,.dropdown.is-hoverable:hover .dropdown-menu{display:block}.dropdown.is-right .dropdown-menu{left:auto;right:0}.dropdown.is-up .dropdown-menu{bottom:100%;padding-bottom:4px;padding-top:initial;top:auto}.dropdown-menu{display:none;left:0;min-width:12rem;padding-top:4px;position:absolute;top:100%;z-index:20}.dropdown-content{background-color:#fff;border-radius:4px;box-shadow:#bbb;padding-bottom:.5rem;padding-top:.5rem}.dropdown-item{color:#222;display:block;font-size:0.875rem;line-height:1.5;padding:0.375rem 1rem;position:relative}a.dropdown-item,button.dropdown-item{padding-right:3rem;text-align:inherit;white-space:nowrap;width:100%}a.dropdown-item:hover,button.dropdown-item:hover{background-color:#f5f5f5;color:#0a0a0a}a.dropdown-item.is-active,button.dropdown-item.is-active{background-color:#2e63b8;color:#fff}.dropdown-divider{background-color:#ededed;border:none;display:block;height:1px;margin:0.5rem 0}.level{align-items:center;justify-content:space-between}.level code{border-radius:4px}.level img{display:inline-block;vertical-align:top}.level.is-mobile{display:flex}.level.is-mobile .level-left,.level.is-mobile .level-right{display:flex}.level.is-mobile .level-left+.level-right{margin-top:0}.level.is-mobile .level-item:not(:last-child){margin-bottom:0;margin-right:.75rem}.level.is-mobile .level-item:not(.is-narrow){flex-grow:1}@media screen and (min-width: 769px),print{.level{display:flex}.level>.level-item:not(.is-narrow){flex-grow:1}}.level-item{align-items:center;display:flex;flex-basis:auto;flex-grow:0;flex-shrink:0;justify-content:center}.level-item .title,.level-item .subtitle{margin-bottom:0}@media screen and (max-width: 768px){.level-item:not(:last-child){margin-bottom:.75rem}}.level-left,.level-right{flex-basis:auto;flex-grow:0;flex-shrink:0}.level-left .level-item.is-flexible,.level-right .level-item.is-flexible{flex-grow:1}@media screen and (min-width: 769px),print{.level-left .level-item:not(:last-child),.level-right .level-item:not(:last-child){margin-right:.75rem}}.level-left{align-items:center;justify-content:flex-start}@media screen and (max-width: 768px){.level-left+.level-right{margin-top:1.5rem}}@media screen and (min-width: 769px),print{.level-left{display:flex}}.level-right{align-items:center;justify-content:flex-end}@media screen and (min-width: 769px),print{.level-right{display:flex}}.media{align-items:flex-start;display:flex;text-align:inherit}.media .content:not(:last-child){margin-bottom:.75rem}.media .media{border-top:1px solid rgba(219,219,219,0.5);display:flex;padding-top:.75rem}.media .media .content:not(:last-child),.media .media .control:not(:last-child){margin-bottom:.5rem}.media .media .media{padding-top:.5rem}.media .media .media+.media{margin-top:.5rem}.media+.media{border-top:1px solid rgba(219,219,219,0.5);margin-top:1rem;padding-top:1rem}.media.is-large+.media{margin-top:1.5rem;padding-top:1.5rem}.media-left,.media-right{flex-basis:auto;flex-grow:0;flex-shrink:0}.media-left{margin-right:1rem}.media-right{margin-left:1rem}.media-content{flex-basis:auto;flex-grow:1;flex-shrink:1;text-align:inherit}@media screen and (max-width: 768px){.media-content{overflow-x:auto}}.menu{font-size:1rem}.menu.is-small,#documenter .docs-sidebar form.docs-search>input.menu{font-size:.75rem}.menu.is-medium{font-size:1.25rem}.menu.is-large{font-size:1.5rem}.menu-list{line-height:1.25}.menu-list a{border-radius:2px;color:#222;display:block;padding:0.5em 0.75em}.menu-list a:hover{background-color:#f5f5f5;color:#222}.menu-list a.is-active{background-color:#2e63b8;color:#fff}.menu-list li ul{border-left:1px solid #dbdbdb;margin:.75em;padding-left:.75em}.menu-label{color:#6b6b6b;font-size:.75em;letter-spacing:.1em;text-transform:uppercase}.menu-label:not(:first-child){margin-top:1em}.menu-label:not(:last-child){margin-bottom:1em}.message{background-color:#f5f5f5;border-radius:4px;font-size:1rem}.message strong{color:currentColor}.message a:not(.button):not(.tag):not(.dropdown-item){color:currentColor;text-decoration:underline}.message.is-small,#documenter .docs-sidebar form.docs-search>input.message{font-size:.75rem}.message.is-medium{font-size:1.25rem}.message.is-large{font-size:1.5rem}.message.is-white{background-color:#fff}.message.is-white .message-header{background-color:#fff;color:#0a0a0a}.message.is-white .message-body{border-color:#fff}.message.is-black{background-color:#fafafa}.message.is-black .message-header{background-color:#0a0a0a;color:#fff}.message.is-black .message-body{border-color:#0a0a0a}.message.is-light{background-color:#fafafa}.message.is-light .message-header{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.message.is-light .message-body{border-color:#f5f5f5}.message.is-dark,.content kbd.message{background-color:#fafafa}.message.is-dark .message-header,.content kbd.message .message-header{background-color:#363636;color:#fff}.message.is-dark .message-body,.content kbd.message .message-body{border-color:#363636}.message.is-primary,.docstring>section>a.message.docs-sourcelink{background-color:#eef8fc}.message.is-primary .message-header,.docstring>section>a.message.docs-sourcelink .message-header{background-color:#4eb5de;color:#fff}.message.is-primary .message-body,.docstring>section>a.message.docs-sourcelink .message-body{border-color:#4eb5de;color:#1a6d8e}.message.is-link{background-color:#eff3fb}.message.is-link .message-header{background-color:#2e63b8;color:#fff}.message.is-link .message-body{border-color:#2e63b8;color:#3169c4}.message.is-info{background-color:#ecf7fe}.message.is-info .message-header{background-color:#209cee;color:#fff}.message.is-info .message-body{border-color:#209cee;color:#0e72b4}.message.is-success{background-color:#eefcf3}.message.is-success .message-header{background-color:#22c35b;color:#fff}.message.is-success .message-body{border-color:#22c35b;color:#198f43}.message.is-warning{background-color:#fffbeb}.message.is-warning .message-header{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.message.is-warning .message-body{border-color:#ffdd57;color:#947600}.message.is-danger{background-color:#ffeceb}.message.is-danger .message-header{background-color:#da0b00;color:#fff}.message.is-danger .message-body{border-color:#da0b00;color:#f50c00}.message-header{align-items:center;background-color:#222;border-radius:4px 4px 0 0;color:#fff;display:flex;font-weight:700;justify-content:space-between;line-height:1.25;padding:0.75em 1em;position:relative}.message-header .delete{flex-grow:0;flex-shrink:0;margin-left:.75em}.message-header+.message-body{border-width:0;border-top-left-radius:0;border-top-right-radius:0}.message-body{border-color:#dbdbdb;border-radius:4px;border-style:solid;border-width:0 0 0 4px;color:#222;padding:1.25em 1.5em}.message-body code,.message-body pre{background-color:#fff}.message-body pre code{background-color:rgba(0,0,0,0)}.modal{align-items:center;display:none;flex-direction:column;justify-content:center;overflow:hidden;position:fixed;z-index:40}.modal.is-active{display:flex}.modal-background{background-color:rgba(10,10,10,0.86)}.modal-content,.modal-card{margin:0 20px;max-height:calc(100vh - 160px);overflow:auto;position:relative;width:100%}@media screen and (min-width: 769px){.modal-content,.modal-card{margin:0 auto;max-height:calc(100vh - 40px);width:640px}}.modal-close{background:none;height:40px;position:fixed;right:20px;top:20px;width:40px}.modal-card{display:flex;flex-direction:column;max-height:calc(100vh - 40px);overflow:hidden;-ms-overflow-y:visible}.modal-card-head,.modal-card-foot{align-items:center;background-color:#f5f5f5;display:flex;flex-shrink:0;justify-content:flex-start;padding:20px;position:relative}.modal-card-head{border-bottom:1px solid #dbdbdb;border-top-left-radius:6px;border-top-right-radius:6px}.modal-card-title{color:#222;flex-grow:1;flex-shrink:0;font-size:1.5rem;line-height:1}.modal-card-foot{border-bottom-left-radius:6px;border-bottom-right-radius:6px;border-top:1px solid #dbdbdb}.modal-card-foot .button:not(:last-child){margin-right:.5em}.modal-card-body{-webkit-overflow-scrolling:touch;background-color:#fff;flex-grow:1;flex-shrink:1;overflow:auto;padding:20px}.navbar{background-color:#fff;min-height:3.25rem;position:relative;z-index:30}.navbar.is-white{background-color:#fff;color:#0a0a0a}.navbar.is-white .navbar-brand>.navbar-item,.navbar.is-white .navbar-brand .navbar-link{color:#0a0a0a}.navbar.is-white .navbar-brand>a.navbar-item:focus,.navbar.is-white .navbar-brand>a.navbar-item:hover,.navbar.is-white .navbar-brand>a.navbar-item.is-active,.navbar.is-white .navbar-brand .navbar-link:focus,.navbar.is-white .navbar-brand .navbar-link:hover,.navbar.is-white .navbar-brand .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}.navbar.is-white .navbar-brand .navbar-link::after{border-color:#0a0a0a}.navbar.is-white .navbar-burger{color:#0a0a0a}@media screen and (min-width: 1056px){.navbar.is-white .navbar-start>.navbar-item,.navbar.is-white .navbar-start .navbar-link,.navbar.is-white .navbar-end>.navbar-item,.navbar.is-white .navbar-end .navbar-link{color:#0a0a0a}.navbar.is-white .navbar-start>a.navbar-item:focus,.navbar.is-white .navbar-start>a.navbar-item:hover,.navbar.is-white .navbar-start>a.navbar-item.is-active,.navbar.is-white .navbar-start .navbar-link:focus,.navbar.is-white .navbar-start .navbar-link:hover,.navbar.is-white .navbar-start .navbar-link.is-active,.navbar.is-white .navbar-end>a.navbar-item:focus,.navbar.is-white .navbar-end>a.navbar-item:hover,.navbar.is-white .navbar-end>a.navbar-item.is-active,.navbar.is-white .navbar-end .navbar-link:focus,.navbar.is-white .navbar-end .navbar-link:hover,.navbar.is-white .navbar-end .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}.navbar.is-white .navbar-start .navbar-link::after,.navbar.is-white .navbar-end .navbar-link::after{border-color:#0a0a0a}.navbar.is-white .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-white .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-white .navbar-item.has-dropdown.is-active .navbar-link{background-color:#f2f2f2;color:#0a0a0a}.navbar.is-white .navbar-dropdown a.navbar-item.is-active{background-color:#fff;color:#0a0a0a}}.navbar.is-black{background-color:#0a0a0a;color:#fff}.navbar.is-black .navbar-brand>.navbar-item,.navbar.is-black .navbar-brand .navbar-link{color:#fff}.navbar.is-black .navbar-brand>a.navbar-item:focus,.navbar.is-black .navbar-brand>a.navbar-item:hover,.navbar.is-black .navbar-brand>a.navbar-item.is-active,.navbar.is-black .navbar-brand .navbar-link:focus,.navbar.is-black .navbar-brand .navbar-link:hover,.navbar.is-black .navbar-brand .navbar-link.is-active{background-color:#000;color:#fff}.navbar.is-black .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-black .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-black .navbar-start>.navbar-item,.navbar.is-black .navbar-start .navbar-link,.navbar.is-black .navbar-end>.navbar-item,.navbar.is-black .navbar-end .navbar-link{color:#fff}.navbar.is-black .navbar-start>a.navbar-item:focus,.navbar.is-black .navbar-start>a.navbar-item:hover,.navbar.is-black .navbar-start>a.navbar-item.is-active,.navbar.is-black .navbar-start .navbar-link:focus,.navbar.is-black .navbar-start .navbar-link:hover,.navbar.is-black .navbar-start .navbar-link.is-active,.navbar.is-black .navbar-end>a.navbar-item:focus,.navbar.is-black .navbar-end>a.navbar-item:hover,.navbar.is-black .navbar-end>a.navbar-item.is-active,.navbar.is-black .navbar-end .navbar-link:focus,.navbar.is-black .navbar-end .navbar-link:hover,.navbar.is-black .navbar-end .navbar-link.is-active{background-color:#000;color:#fff}.navbar.is-black .navbar-start .navbar-link::after,.navbar.is-black .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-black .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-black .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-black .navbar-item.has-dropdown.is-active .navbar-link{background-color:#000;color:#fff}.navbar.is-black .navbar-dropdown a.navbar-item.is-active{background-color:#0a0a0a;color:#fff}}.navbar.is-light{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-brand>.navbar-item,.navbar.is-light .navbar-brand .navbar-link{color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-brand>a.navbar-item:focus,.navbar.is-light .navbar-brand>a.navbar-item:hover,.navbar.is-light .navbar-brand>a.navbar-item.is-active,.navbar.is-light .navbar-brand .navbar-link:focus,.navbar.is-light .navbar-brand .navbar-link:hover,.navbar.is-light .navbar-brand .navbar-link.is-active{background-color:#e8e8e8;color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-brand .navbar-link::after{border-color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-burger{color:rgba(0,0,0,0.7)}@media screen and (min-width: 1056px){.navbar.is-light .navbar-start>.navbar-item,.navbar.is-light .navbar-start .navbar-link,.navbar.is-light .navbar-end>.navbar-item,.navbar.is-light .navbar-end .navbar-link{color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-start>a.navbar-item:focus,.navbar.is-light .navbar-start>a.navbar-item:hover,.navbar.is-light .navbar-start>a.navbar-item.is-active,.navbar.is-light .navbar-start .navbar-link:focus,.navbar.is-light .navbar-start .navbar-link:hover,.navbar.is-light .navbar-start .navbar-link.is-active,.navbar.is-light .navbar-end>a.navbar-item:focus,.navbar.is-light .navbar-end>a.navbar-item:hover,.navbar.is-light .navbar-end>a.navbar-item.is-active,.navbar.is-light .navbar-end .navbar-link:focus,.navbar.is-light .navbar-end .navbar-link:hover,.navbar.is-light .navbar-end .navbar-link.is-active{background-color:#e8e8e8;color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-start .navbar-link::after,.navbar.is-light .navbar-end .navbar-link::after{border-color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-light .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-light .navbar-item.has-dropdown.is-active .navbar-link{background-color:#e8e8e8;color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-dropdown a.navbar-item.is-active{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}}.navbar.is-dark,.content kbd.navbar{background-color:#363636;color:#fff}.navbar.is-dark .navbar-brand>.navbar-item,.content kbd.navbar .navbar-brand>.navbar-item,.navbar.is-dark .navbar-brand .navbar-link,.content kbd.navbar .navbar-brand .navbar-link{color:#fff}.navbar.is-dark .navbar-brand>a.navbar-item:focus,.content kbd.navbar .navbar-brand>a.navbar-item:focus,.navbar.is-dark .navbar-brand>a.navbar-item:hover,.content kbd.navbar .navbar-brand>a.navbar-item:hover,.navbar.is-dark .navbar-brand>a.navbar-item.is-active,.content kbd.navbar .navbar-brand>a.navbar-item.is-active,.navbar.is-dark .navbar-brand .navbar-link:focus,.content kbd.navbar .navbar-brand .navbar-link:focus,.navbar.is-dark .navbar-brand .navbar-link:hover,.content kbd.navbar .navbar-brand .navbar-link:hover,.navbar.is-dark .navbar-brand .navbar-link.is-active,.content kbd.navbar .navbar-brand .navbar-link.is-active{background-color:#292929;color:#fff}.navbar.is-dark .navbar-brand .navbar-link::after,.content kbd.navbar .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-dark .navbar-burger,.content kbd.navbar .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-dark .navbar-start>.navbar-item,.content kbd.navbar .navbar-start>.navbar-item,.navbar.is-dark .navbar-start .navbar-link,.content kbd.navbar .navbar-start .navbar-link,.navbar.is-dark .navbar-end>.navbar-item,.content kbd.navbar .navbar-end>.navbar-item,.navbar.is-dark .navbar-end .navbar-link,.content kbd.navbar .navbar-end .navbar-link{color:#fff}.navbar.is-dark .navbar-start>a.navbar-item:focus,.content kbd.navbar .navbar-start>a.navbar-item:focus,.navbar.is-dark .navbar-start>a.navbar-item:hover,.content kbd.navbar .navbar-start>a.navbar-item:hover,.navbar.is-dark .navbar-start>a.navbar-item.is-active,.content kbd.navbar .navbar-start>a.navbar-item.is-active,.navbar.is-dark .navbar-start .navbar-link:focus,.content kbd.navbar .navbar-start .navbar-link:focus,.navbar.is-dark .navbar-start .navbar-link:hover,.content kbd.navbar .navbar-start .navbar-link:hover,.navbar.is-dark .navbar-start .navbar-link.is-active,.content kbd.navbar .navbar-start .navbar-link.is-active,.navbar.is-dark .navbar-end>a.navbar-item:focus,.content kbd.navbar .navbar-end>a.navbar-item:focus,.navbar.is-dark .navbar-end>a.navbar-item:hover,.content kbd.navbar .navbar-end>a.navbar-item:hover,.navbar.is-dark .navbar-end>a.navbar-item.is-active,.content kbd.navbar .navbar-end>a.navbar-item.is-active,.navbar.is-dark .navbar-end .navbar-link:focus,.content kbd.navbar .navbar-end .navbar-link:focus,.navbar.is-dark .navbar-end .navbar-link:hover,.content kbd.navbar .navbar-end .navbar-link:hover,.navbar.is-dark .navbar-end .navbar-link.is-active,.content kbd.navbar .navbar-end .navbar-link.is-active{background-color:#292929;color:#fff}.navbar.is-dark .navbar-start .navbar-link::after,.content kbd.navbar .navbar-start .navbar-link::after,.navbar.is-dark .navbar-end .navbar-link::after,.content kbd.navbar .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-dark .navbar-item.has-dropdown:focus .navbar-link,.content kbd.navbar .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-dark .navbar-item.has-dropdown:hover .navbar-link,.content kbd.navbar .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-dark .navbar-item.has-dropdown.is-active .navbar-link,.content kbd.navbar .navbar-item.has-dropdown.is-active .navbar-link{background-color:#292929;color:#fff}.navbar.is-dark .navbar-dropdown a.navbar-item.is-active,.content kbd.navbar .navbar-dropdown a.navbar-item.is-active{background-color:#363636;color:#fff}}.navbar.is-primary,.docstring>section>a.navbar.docs-sourcelink{background-color:#4eb5de;color:#fff}.navbar.is-primary .navbar-brand>.navbar-item,.docstring>section>a.navbar.docs-sourcelink .navbar-brand>.navbar-item,.navbar.is-primary .navbar-brand .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link{color:#fff}.navbar.is-primary .navbar-brand>a.navbar-item:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item:focus,.navbar.is-primary .navbar-brand>a.navbar-item:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item:hover,.navbar.is-primary .navbar-brand>a.navbar-item.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item.is-active,.navbar.is-primary .navbar-brand .navbar-link:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link:focus,.navbar.is-primary .navbar-brand .navbar-link:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link:hover,.navbar.is-primary .navbar-brand .navbar-link.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link.is-active{background-color:#39acda;color:#fff}.navbar.is-primary .navbar-brand .navbar-link::after,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-primary .navbar-burger,.docstring>section>a.navbar.docs-sourcelink .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-primary .navbar-start>.navbar-item,.docstring>section>a.navbar.docs-sourcelink .navbar-start>.navbar-item,.navbar.is-primary .navbar-start .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link,.navbar.is-primary .navbar-end>.navbar-item,.docstring>section>a.navbar.docs-sourcelink .navbar-end>.navbar-item,.navbar.is-primary .navbar-end .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link{color:#fff}.navbar.is-primary .navbar-start>a.navbar-item:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item:focus,.navbar.is-primary .navbar-start>a.navbar-item:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item:hover,.navbar.is-primary .navbar-start>a.navbar-item.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item.is-active,.navbar.is-primary .navbar-start .navbar-link:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link:focus,.navbar.is-primary .navbar-start .navbar-link:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link:hover,.navbar.is-primary .navbar-start .navbar-link.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link.is-active,.navbar.is-primary .navbar-end>a.navbar-item:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item:focus,.navbar.is-primary .navbar-end>a.navbar-item:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item:hover,.navbar.is-primary .navbar-end>a.navbar-item.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item.is-active,.navbar.is-primary .navbar-end .navbar-link:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link:focus,.navbar.is-primary .navbar-end .navbar-link:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link:hover,.navbar.is-primary .navbar-end .navbar-link.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link.is-active{background-color:#39acda;color:#fff}.navbar.is-primary .navbar-start .navbar-link::after,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link::after,.navbar.is-primary .navbar-end .navbar-link::after,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-primary .navbar-item.has-dropdown:focus .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-primary .navbar-item.has-dropdown:hover .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-primary .navbar-item.has-dropdown.is-active .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown.is-active .navbar-link{background-color:#39acda;color:#fff}.navbar.is-primary .navbar-dropdown a.navbar-item.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-dropdown a.navbar-item.is-active{background-color:#4eb5de;color:#fff}}.navbar.is-link{background-color:#2e63b8;color:#fff}.navbar.is-link .navbar-brand>.navbar-item,.navbar.is-link .navbar-brand .navbar-link{color:#fff}.navbar.is-link .navbar-brand>a.navbar-item:focus,.navbar.is-link .navbar-brand>a.navbar-item:hover,.navbar.is-link .navbar-brand>a.navbar-item.is-active,.navbar.is-link .navbar-brand .navbar-link:focus,.navbar.is-link .navbar-brand .navbar-link:hover,.navbar.is-link .navbar-brand .navbar-link.is-active{background-color:#2958a4;color:#fff}.navbar.is-link .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-link .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-link .navbar-start>.navbar-item,.navbar.is-link .navbar-start .navbar-link,.navbar.is-link .navbar-end>.navbar-item,.navbar.is-link .navbar-end .navbar-link{color:#fff}.navbar.is-link .navbar-start>a.navbar-item:focus,.navbar.is-link .navbar-start>a.navbar-item:hover,.navbar.is-link .navbar-start>a.navbar-item.is-active,.navbar.is-link .navbar-start .navbar-link:focus,.navbar.is-link .navbar-start .navbar-link:hover,.navbar.is-link .navbar-start .navbar-link.is-active,.navbar.is-link .navbar-end>a.navbar-item:focus,.navbar.is-link .navbar-end>a.navbar-item:hover,.navbar.is-link .navbar-end>a.navbar-item.is-active,.navbar.is-link .navbar-end .navbar-link:focus,.navbar.is-link .navbar-end .navbar-link:hover,.navbar.is-link .navbar-end .navbar-link.is-active{background-color:#2958a4;color:#fff}.navbar.is-link .navbar-start .navbar-link::after,.navbar.is-link .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-link .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-link .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-link .navbar-item.has-dropdown.is-active .navbar-link{background-color:#2958a4;color:#fff}.navbar.is-link .navbar-dropdown a.navbar-item.is-active{background-color:#2e63b8;color:#fff}}.navbar.is-info{background-color:#209cee;color:#fff}.navbar.is-info .navbar-brand>.navbar-item,.navbar.is-info .navbar-brand .navbar-link{color:#fff}.navbar.is-info .navbar-brand>a.navbar-item:focus,.navbar.is-info .navbar-brand>a.navbar-item:hover,.navbar.is-info .navbar-brand>a.navbar-item.is-active,.navbar.is-info .navbar-brand .navbar-link:focus,.navbar.is-info .navbar-brand .navbar-link:hover,.navbar.is-info .navbar-brand .navbar-link.is-active{background-color:#1190e3;color:#fff}.navbar.is-info .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-info .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-info .navbar-start>.navbar-item,.navbar.is-info .navbar-start .navbar-link,.navbar.is-info .navbar-end>.navbar-item,.navbar.is-info .navbar-end .navbar-link{color:#fff}.navbar.is-info .navbar-start>a.navbar-item:focus,.navbar.is-info .navbar-start>a.navbar-item:hover,.navbar.is-info .navbar-start>a.navbar-item.is-active,.navbar.is-info .navbar-start .navbar-link:focus,.navbar.is-info .navbar-start .navbar-link:hover,.navbar.is-info .navbar-start .navbar-link.is-active,.navbar.is-info .navbar-end>a.navbar-item:focus,.navbar.is-info .navbar-end>a.navbar-item:hover,.navbar.is-info .navbar-end>a.navbar-item.is-active,.navbar.is-info .navbar-end .navbar-link:focus,.navbar.is-info .navbar-end .navbar-link:hover,.navbar.is-info .navbar-end .navbar-link.is-active{background-color:#1190e3;color:#fff}.navbar.is-info .navbar-start .navbar-link::after,.navbar.is-info .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-info .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-info .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-info .navbar-item.has-dropdown.is-active .navbar-link{background-color:#1190e3;color:#fff}.navbar.is-info .navbar-dropdown a.navbar-item.is-active{background-color:#209cee;color:#fff}}.navbar.is-success{background-color:#22c35b;color:#fff}.navbar.is-success .navbar-brand>.navbar-item,.navbar.is-success .navbar-brand .navbar-link{color:#fff}.navbar.is-success .navbar-brand>a.navbar-item:focus,.navbar.is-success .navbar-brand>a.navbar-item:hover,.navbar.is-success .navbar-brand>a.navbar-item.is-active,.navbar.is-success .navbar-brand .navbar-link:focus,.navbar.is-success .navbar-brand .navbar-link:hover,.navbar.is-success .navbar-brand .navbar-link.is-active{background-color:#1ead51;color:#fff}.navbar.is-success .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-success .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-success .navbar-start>.navbar-item,.navbar.is-success .navbar-start .navbar-link,.navbar.is-success .navbar-end>.navbar-item,.navbar.is-success .navbar-end .navbar-link{color:#fff}.navbar.is-success .navbar-start>a.navbar-item:focus,.navbar.is-success .navbar-start>a.navbar-item:hover,.navbar.is-success .navbar-start>a.navbar-item.is-active,.navbar.is-success .navbar-start .navbar-link:focus,.navbar.is-success .navbar-start .navbar-link:hover,.navbar.is-success .navbar-start .navbar-link.is-active,.navbar.is-success .navbar-end>a.navbar-item:focus,.navbar.is-success .navbar-end>a.navbar-item:hover,.navbar.is-success .navbar-end>a.navbar-item.is-active,.navbar.is-success .navbar-end .navbar-link:focus,.navbar.is-success .navbar-end .navbar-link:hover,.navbar.is-success .navbar-end .navbar-link.is-active{background-color:#1ead51;color:#fff}.navbar.is-success .navbar-start .navbar-link::after,.navbar.is-success .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-success .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-success .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-success .navbar-item.has-dropdown.is-active .navbar-link{background-color:#1ead51;color:#fff}.navbar.is-success .navbar-dropdown a.navbar-item.is-active{background-color:#22c35b;color:#fff}}.navbar.is-warning{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-brand>.navbar-item,.navbar.is-warning .navbar-brand .navbar-link{color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-brand>a.navbar-item:focus,.navbar.is-warning .navbar-brand>a.navbar-item:hover,.navbar.is-warning .navbar-brand>a.navbar-item.is-active,.navbar.is-warning .navbar-brand .navbar-link:focus,.navbar.is-warning .navbar-brand .navbar-link:hover,.navbar.is-warning .navbar-brand .navbar-link.is-active{background-color:#ffd83e;color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-brand .navbar-link::after{border-color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-burger{color:rgba(0,0,0,0.7)}@media screen and (min-width: 1056px){.navbar.is-warning .navbar-start>.navbar-item,.navbar.is-warning .navbar-start .navbar-link,.navbar.is-warning .navbar-end>.navbar-item,.navbar.is-warning .navbar-end .navbar-link{color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-start>a.navbar-item:focus,.navbar.is-warning .navbar-start>a.navbar-item:hover,.navbar.is-warning .navbar-start>a.navbar-item.is-active,.navbar.is-warning .navbar-start .navbar-link:focus,.navbar.is-warning .navbar-start .navbar-link:hover,.navbar.is-warning .navbar-start .navbar-link.is-active,.navbar.is-warning .navbar-end>a.navbar-item:focus,.navbar.is-warning .navbar-end>a.navbar-item:hover,.navbar.is-warning .navbar-end>a.navbar-item.is-active,.navbar.is-warning .navbar-end .navbar-link:focus,.navbar.is-warning .navbar-end .navbar-link:hover,.navbar.is-warning .navbar-end .navbar-link.is-active{background-color:#ffd83e;color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-start .navbar-link::after,.navbar.is-warning .navbar-end .navbar-link::after{border-color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-warning .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-warning .navbar-item.has-dropdown.is-active .navbar-link{background-color:#ffd83e;color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-dropdown a.navbar-item.is-active{background-color:#ffdd57;color:rgba(0,0,0,0.7)}}.navbar.is-danger{background-color:#da0b00;color:#fff}.navbar.is-danger .navbar-brand>.navbar-item,.navbar.is-danger .navbar-brand .navbar-link{color:#fff}.navbar.is-danger .navbar-brand>a.navbar-item:focus,.navbar.is-danger .navbar-brand>a.navbar-item:hover,.navbar.is-danger .navbar-brand>a.navbar-item.is-active,.navbar.is-danger .navbar-brand .navbar-link:focus,.navbar.is-danger .navbar-brand .navbar-link:hover,.navbar.is-danger .navbar-brand .navbar-link.is-active{background-color:#c10a00;color:#fff}.navbar.is-danger .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-danger .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-danger .navbar-start>.navbar-item,.navbar.is-danger .navbar-start .navbar-link,.navbar.is-danger .navbar-end>.navbar-item,.navbar.is-danger .navbar-end .navbar-link{color:#fff}.navbar.is-danger .navbar-start>a.navbar-item:focus,.navbar.is-danger .navbar-start>a.navbar-item:hover,.navbar.is-danger .navbar-start>a.navbar-item.is-active,.navbar.is-danger .navbar-start .navbar-link:focus,.navbar.is-danger .navbar-start .navbar-link:hover,.navbar.is-danger .navbar-start .navbar-link.is-active,.navbar.is-danger .navbar-end>a.navbar-item:focus,.navbar.is-danger .navbar-end>a.navbar-item:hover,.navbar.is-danger .navbar-end>a.navbar-item.is-active,.navbar.is-danger .navbar-end .navbar-link:focus,.navbar.is-danger .navbar-end .navbar-link:hover,.navbar.is-danger .navbar-end .navbar-link.is-active{background-color:#c10a00;color:#fff}.navbar.is-danger .navbar-start .navbar-link::after,.navbar.is-danger .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-danger .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-danger .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-danger .navbar-item.has-dropdown.is-active .navbar-link{background-color:#c10a00;color:#fff}.navbar.is-danger .navbar-dropdown a.navbar-item.is-active{background-color:#da0b00;color:#fff}}.navbar>.container{align-items:stretch;display:flex;min-height:3.25rem;width:100%}.navbar.has-shadow{box-shadow:0 2px 0 0 #f5f5f5}.navbar.is-fixed-bottom,.navbar.is-fixed-top{left:0;position:fixed;right:0;z-index:30}.navbar.is-fixed-bottom{bottom:0}.navbar.is-fixed-bottom.has-shadow{box-shadow:0 -2px 0 0 #f5f5f5}.navbar.is-fixed-top{top:0}html.has-navbar-fixed-top,body.has-navbar-fixed-top{padding-top:3.25rem}html.has-navbar-fixed-bottom,body.has-navbar-fixed-bottom{padding-bottom:3.25rem}.navbar-brand,.navbar-tabs{align-items:stretch;display:flex;flex-shrink:0;min-height:3.25rem}.navbar-brand a.navbar-item:focus,.navbar-brand a.navbar-item:hover{background-color:transparent}.navbar-tabs{-webkit-overflow-scrolling:touch;max-width:100vw;overflow-x:auto;overflow-y:hidden}.navbar-burger{color:#222;-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;cursor:pointer;display:block;height:3.25rem;position:relative;width:3.25rem;margin-left:auto}.navbar-burger span{background-color:currentColor;display:block;height:1px;left:calc(50% - 8px);position:absolute;transform-origin:center;transition-duration:86ms;transition-property:background-color, opacity, transform;transition-timing-function:ease-out;width:16px}.navbar-burger span:nth-child(1){top:calc(50% - 6px)}.navbar-burger span:nth-child(2){top:calc(50% - 1px)}.navbar-burger span:nth-child(3){top:calc(50% + 4px)}.navbar-burger:hover{background-color:rgba(0,0,0,0.05)}.navbar-burger.is-active span:nth-child(1){transform:translateY(5px) rotate(45deg)}.navbar-burger.is-active span:nth-child(2){opacity:0}.navbar-burger.is-active span:nth-child(3){transform:translateY(-5px) rotate(-45deg)}.navbar-menu{display:none}.navbar-item,.navbar-link{color:#222;display:block;line-height:1.5;padding:0.5rem 0.75rem;position:relative}.navbar-item .icon:only-child,.navbar-link .icon:only-child{margin-left:-0.25rem;margin-right:-0.25rem}a.navbar-item,.navbar-link{cursor:pointer}a.navbar-item:focus,a.navbar-item:focus-within,a.navbar-item:hover,a.navbar-item.is-active,.navbar-link:focus,.navbar-link:focus-within,.navbar-link:hover,.navbar-link.is-active{background-color:#fafafa;color:#2e63b8}.navbar-item{flex-grow:0;flex-shrink:0}.navbar-item img{max-height:1.75rem}.navbar-item.has-dropdown{padding:0}.navbar-item.is-expanded{flex-grow:1;flex-shrink:1}.navbar-item.is-tab{border-bottom:1px solid transparent;min-height:3.25rem;padding-bottom:calc(0.5rem - 1px)}.navbar-item.is-tab:focus,.navbar-item.is-tab:hover{background-color:rgba(0,0,0,0);border-bottom-color:#2e63b8}.navbar-item.is-tab.is-active{background-color:rgba(0,0,0,0);border-bottom-color:#2e63b8;border-bottom-style:solid;border-bottom-width:3px;color:#2e63b8;padding-bottom:calc(0.5rem - 3px)}.navbar-content{flex-grow:1;flex-shrink:1}.navbar-link:not(.is-arrowless){padding-right:2.5em}.navbar-link:not(.is-arrowless)::after{border-color:#2e63b8;margin-top:-0.375em;right:1.125em}.navbar-dropdown{font-size:0.875rem;padding-bottom:0.5rem;padding-top:0.5rem}.navbar-dropdown .navbar-item{padding-left:1.5rem;padding-right:1.5rem}.navbar-divider{background-color:#f5f5f5;border:none;display:none;height:2px;margin:0.5rem 0}@media screen and (max-width: 1055px){.navbar>.container{display:block}.navbar-brand .navbar-item,.navbar-tabs .navbar-item{align-items:center;display:flex}.navbar-link::after{display:none}.navbar-menu{background-color:#fff;box-shadow:0 8px 16px rgba(10,10,10,0.1);padding:0.5rem 0}.navbar-menu.is-active{display:block}.navbar.is-fixed-bottom-touch,.navbar.is-fixed-top-touch{left:0;position:fixed;right:0;z-index:30}.navbar.is-fixed-bottom-touch{bottom:0}.navbar.is-fixed-bottom-touch.has-shadow{box-shadow:0 -2px 3px rgba(10,10,10,0.1)}.navbar.is-fixed-top-touch{top:0}.navbar.is-fixed-top .navbar-menu,.navbar.is-fixed-top-touch .navbar-menu{-webkit-overflow-scrolling:touch;max-height:calc(100vh - 3.25rem);overflow:auto}html.has-navbar-fixed-top-touch,body.has-navbar-fixed-top-touch{padding-top:3.25rem}html.has-navbar-fixed-bottom-touch,body.has-navbar-fixed-bottom-touch{padding-bottom:3.25rem}}@media screen and (min-width: 1056px){.navbar,.navbar-menu,.navbar-start,.navbar-end{align-items:stretch;display:flex}.navbar{min-height:3.25rem}.navbar.is-spaced{padding:1rem 2rem}.navbar.is-spaced .navbar-start,.navbar.is-spaced .navbar-end{align-items:center}.navbar.is-spaced a.navbar-item,.navbar.is-spaced .navbar-link{border-radius:4px}.navbar.is-transparent a.navbar-item:focus,.navbar.is-transparent a.navbar-item:hover,.navbar.is-transparent a.navbar-item.is-active,.navbar.is-transparent .navbar-link:focus,.navbar.is-transparent .navbar-link:hover,.navbar.is-transparent .navbar-link.is-active{background-color:transparent !important}.navbar.is-transparent .navbar-item.has-dropdown.is-active .navbar-link,.navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:focus .navbar-link,.navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:focus-within .navbar-link,.navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:hover .navbar-link{background-color:transparent !important}.navbar.is-transparent .navbar-dropdown a.navbar-item:focus,.navbar.is-transparent .navbar-dropdown a.navbar-item:hover{background-color:#f5f5f5;color:#0a0a0a}.navbar.is-transparent .navbar-dropdown a.navbar-item.is-active{background-color:#f5f5f5;color:#2e63b8}.navbar-burger{display:none}.navbar-item,.navbar-link{align-items:center;display:flex}.navbar-item.has-dropdown{align-items:stretch}.navbar-item.has-dropdown-up .navbar-link::after{transform:rotate(135deg) translate(0.25em, -0.25em)}.navbar-item.has-dropdown-up .navbar-dropdown{border-bottom:2px solid #dbdbdb;border-radius:6px 6px 0 0;border-top:none;bottom:100%;box-shadow:0 -8px 8px rgba(10,10,10,0.1);top:auto}.navbar-item.is-active .navbar-dropdown,.navbar-item.is-hoverable:focus .navbar-dropdown,.navbar-item.is-hoverable:focus-within .navbar-dropdown,.navbar-item.is-hoverable:hover .navbar-dropdown{display:block}.navbar.is-spaced .navbar-item.is-active .navbar-dropdown,.navbar-item.is-active .navbar-dropdown.is-boxed,.navbar.is-spaced .navbar-item.is-hoverable:focus .navbar-dropdown,.navbar-item.is-hoverable:focus .navbar-dropdown.is-boxed,.navbar.is-spaced .navbar-item.is-hoverable:focus-within .navbar-dropdown,.navbar-item.is-hoverable:focus-within .navbar-dropdown.is-boxed,.navbar.is-spaced .navbar-item.is-hoverable:hover .navbar-dropdown,.navbar-item.is-hoverable:hover .navbar-dropdown.is-boxed{opacity:1;pointer-events:auto;transform:translateY(0)}.navbar-menu{flex-grow:1;flex-shrink:0}.navbar-start{justify-content:flex-start;margin-right:auto}.navbar-end{justify-content:flex-end;margin-left:auto}.navbar-dropdown{background-color:#fff;border-bottom-left-radius:6px;border-bottom-right-radius:6px;border-top:2px solid #dbdbdb;box-shadow:0 8px 8px rgba(10,10,10,0.1);display:none;font-size:0.875rem;left:0;min-width:100%;position:absolute;top:100%;z-index:20}.navbar-dropdown .navbar-item{padding:0.375rem 1rem;white-space:nowrap}.navbar-dropdown a.navbar-item{padding-right:3rem}.navbar-dropdown a.navbar-item:focus,.navbar-dropdown a.navbar-item:hover{background-color:#f5f5f5;color:#0a0a0a}.navbar-dropdown a.navbar-item.is-active{background-color:#f5f5f5;color:#2e63b8}.navbar.is-spaced .navbar-dropdown,.navbar-dropdown.is-boxed{border-radius:6px;border-top:none;box-shadow:0 8px 8px rgba(10,10,10,0.1), 0 0 0 1px rgba(10,10,10,0.1);display:block;opacity:0;pointer-events:none;top:calc(100% + (-4px));transform:translateY(-5px);transition-duration:86ms;transition-property:opacity, transform}.navbar-dropdown.is-right{left:auto;right:0}.navbar-divider{display:block}.navbar>.container .navbar-brand,.container>.navbar .navbar-brand{margin-left:-.75rem}.navbar>.container .navbar-menu,.container>.navbar .navbar-menu{margin-right:-.75rem}.navbar.is-fixed-bottom-desktop,.navbar.is-fixed-top-desktop{left:0;position:fixed;right:0;z-index:30}.navbar.is-fixed-bottom-desktop{bottom:0}.navbar.is-fixed-bottom-desktop.has-shadow{box-shadow:0 -2px 3px rgba(10,10,10,0.1)}.navbar.is-fixed-top-desktop{top:0}html.has-navbar-fixed-top-desktop,body.has-navbar-fixed-top-desktop{padding-top:3.25rem}html.has-navbar-fixed-bottom-desktop,body.has-navbar-fixed-bottom-desktop{padding-bottom:3.25rem}html.has-spaced-navbar-fixed-top,body.has-spaced-navbar-fixed-top{padding-top:5.25rem}html.has-spaced-navbar-fixed-bottom,body.has-spaced-navbar-fixed-bottom{padding-bottom:5.25rem}a.navbar-item.is-active,.navbar-link.is-active{color:#0a0a0a}a.navbar-item.is-active:not(:focus):not(:hover),.navbar-link.is-active:not(:focus):not(:hover){background-color:rgba(0,0,0,0)}.navbar-item.has-dropdown:focus .navbar-link,.navbar-item.has-dropdown:hover .navbar-link,.navbar-item.has-dropdown.is-active .navbar-link{background-color:#fafafa}}.hero.is-fullheight-with-navbar{min-height:calc(100vh - 3.25rem)}.pagination{font-size:1rem;margin:-.25rem}.pagination.is-small,#documenter .docs-sidebar form.docs-search>input.pagination{font-size:.75rem}.pagination.is-medium{font-size:1.25rem}.pagination.is-large{font-size:1.5rem}.pagination.is-rounded .pagination-previous,#documenter .docs-sidebar form.docs-search>input.pagination .pagination-previous,.pagination.is-rounded .pagination-next,#documenter .docs-sidebar form.docs-search>input.pagination .pagination-next{padding-left:1em;padding-right:1em;border-radius:9999px}.pagination.is-rounded .pagination-link,#documenter .docs-sidebar form.docs-search>input.pagination .pagination-link{border-radius:9999px}.pagination,.pagination-list{align-items:center;display:flex;justify-content:center;text-align:center}.pagination-previous,.pagination-next,.pagination-link,.pagination-ellipsis{font-size:1em;justify-content:center;margin:.25rem;padding-left:.5em;padding-right:.5em;text-align:center}.pagination-previous,.pagination-next,.pagination-link{border-color:#dbdbdb;color:#222;min-width:2.5em}.pagination-previous:hover,.pagination-next:hover,.pagination-link:hover{border-color:#b5b5b5;color:#363636}.pagination-previous:focus,.pagination-next:focus,.pagination-link:focus{border-color:#3c5dcd}.pagination-previous:active,.pagination-next:active,.pagination-link:active{box-shadow:inset 0 1px 2px rgba(10,10,10,0.2)}.pagination-previous[disabled],.pagination-previous.is-disabled,.pagination-next[disabled],.pagination-next.is-disabled,.pagination-link[disabled],.pagination-link.is-disabled{background-color:#dbdbdb;border-color:#dbdbdb;box-shadow:none;color:#6b6b6b;opacity:0.5}.pagination-previous,.pagination-next{padding-left:.75em;padding-right:.75em;white-space:nowrap}.pagination-link.is-current{background-color:#2e63b8;border-color:#2e63b8;color:#fff}.pagination-ellipsis{color:#b5b5b5;pointer-events:none}.pagination-list{flex-wrap:wrap}.pagination-list li{list-style:none}@media screen and (max-width: 768px){.pagination{flex-wrap:wrap}.pagination-previous,.pagination-next{flex-grow:1;flex-shrink:1}.pagination-list li{flex-grow:1;flex-shrink:1}}@media screen and (min-width: 769px),print{.pagination-list{flex-grow:1;flex-shrink:1;justify-content:flex-start;order:1}.pagination-previous,.pagination-next,.pagination-link,.pagination-ellipsis{margin-bottom:0;margin-top:0}.pagination-previous{order:2}.pagination-next{order:3}.pagination{justify-content:space-between;margin-bottom:0;margin-top:0}.pagination.is-centered .pagination-previous{order:1}.pagination.is-centered .pagination-list{justify-content:center;order:2}.pagination.is-centered .pagination-next{order:3}.pagination.is-right .pagination-previous{order:1}.pagination.is-right .pagination-next{order:2}.pagination.is-right .pagination-list{justify-content:flex-end;order:3}}.panel{border-radius:6px;box-shadow:#bbb;font-size:1rem}.panel:not(:last-child){margin-bottom:1.5rem}.panel.is-white .panel-heading{background-color:#fff;color:#0a0a0a}.panel.is-white .panel-tabs a.is-active{border-bottom-color:#fff}.panel.is-white .panel-block.is-active .panel-icon{color:#fff}.panel.is-black .panel-heading{background-color:#0a0a0a;color:#fff}.panel.is-black .panel-tabs a.is-active{border-bottom-color:#0a0a0a}.panel.is-black .panel-block.is-active .panel-icon{color:#0a0a0a}.panel.is-light .panel-heading{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.panel.is-light .panel-tabs a.is-active{border-bottom-color:#f5f5f5}.panel.is-light .panel-block.is-active .panel-icon{color:#f5f5f5}.panel.is-dark .panel-heading,.content kbd.panel .panel-heading{background-color:#363636;color:#fff}.panel.is-dark .panel-tabs a.is-active,.content kbd.panel .panel-tabs a.is-active{border-bottom-color:#363636}.panel.is-dark .panel-block.is-active .panel-icon,.content kbd.panel .panel-block.is-active .panel-icon{color:#363636}.panel.is-primary .panel-heading,.docstring>section>a.panel.docs-sourcelink .panel-heading{background-color:#4eb5de;color:#fff}.panel.is-primary .panel-tabs a.is-active,.docstring>section>a.panel.docs-sourcelink .panel-tabs a.is-active{border-bottom-color:#4eb5de}.panel.is-primary .panel-block.is-active .panel-icon,.docstring>section>a.panel.docs-sourcelink .panel-block.is-active .panel-icon{color:#4eb5de}.panel.is-link .panel-heading{background-color:#2e63b8;color:#fff}.panel.is-link .panel-tabs a.is-active{border-bottom-color:#2e63b8}.panel.is-link .panel-block.is-active .panel-icon{color:#2e63b8}.panel.is-info .panel-heading{background-color:#209cee;color:#fff}.panel.is-info .panel-tabs a.is-active{border-bottom-color:#209cee}.panel.is-info .panel-block.is-active .panel-icon{color:#209cee}.panel.is-success .panel-heading{background-color:#22c35b;color:#fff}.panel.is-success .panel-tabs a.is-active{border-bottom-color:#22c35b}.panel.is-success .panel-block.is-active .panel-icon{color:#22c35b}.panel.is-warning .panel-heading{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.panel.is-warning .panel-tabs a.is-active{border-bottom-color:#ffdd57}.panel.is-warning .panel-block.is-active .panel-icon{color:#ffdd57}.panel.is-danger .panel-heading{background-color:#da0b00;color:#fff}.panel.is-danger .panel-tabs a.is-active{border-bottom-color:#da0b00}.panel.is-danger .panel-block.is-active .panel-icon{color:#da0b00}.panel-tabs:not(:last-child),.panel-block:not(:last-child){border-bottom:1px solid #ededed}.panel-heading{background-color:#ededed;border-radius:6px 6px 0 0;color:#222;font-size:1.25em;font-weight:700;line-height:1.25;padding:0.75em 1em}.panel-tabs{align-items:flex-end;display:flex;font-size:.875em;justify-content:center}.panel-tabs a{border-bottom:1px solid #dbdbdb;margin-bottom:-1px;padding:0.5em}.panel-tabs a.is-active{border-bottom-color:#4a4a4a;color:#363636}.panel-list a{color:#222}.panel-list a:hover{color:#2e63b8}.panel-block{align-items:center;color:#222;display:flex;justify-content:flex-start;padding:0.5em 0.75em}.panel-block input[type="checkbox"]{margin-right:.75em}.panel-block>.control{flex-grow:1;flex-shrink:1;width:100%}.panel-block.is-wrapped{flex-wrap:wrap}.panel-block.is-active{border-left-color:#2e63b8;color:#363636}.panel-block.is-active .panel-icon{color:#2e63b8}.panel-block:last-child{border-bottom-left-radius:6px;border-bottom-right-radius:6px}a.panel-block,label.panel-block{cursor:pointer}a.panel-block:hover,label.panel-block:hover{background-color:#f5f5f5}.panel-icon{display:inline-block;font-size:14px;height:1em;line-height:1em;text-align:center;vertical-align:top;width:1em;color:#6b6b6b;margin-right:.75em}.panel-icon .fa{font-size:inherit;line-height:inherit}.tabs{-webkit-overflow-scrolling:touch;align-items:stretch;display:flex;font-size:1rem;justify-content:space-between;overflow:hidden;overflow-x:auto;white-space:nowrap}.tabs a{align-items:center;border-bottom-color:#dbdbdb;border-bottom-style:solid;border-bottom-width:1px;color:#222;display:flex;justify-content:center;margin-bottom:-1px;padding:0.5em 1em;vertical-align:top}.tabs a:hover{border-bottom-color:#222;color:#222}.tabs li{display:block}.tabs li.is-active a{border-bottom-color:#2e63b8;color:#2e63b8}.tabs ul{align-items:center;border-bottom-color:#dbdbdb;border-bottom-style:solid;border-bottom-width:1px;display:flex;flex-grow:1;flex-shrink:0;justify-content:flex-start}.tabs ul.is-left{padding-right:0.75em}.tabs ul.is-center{flex:none;justify-content:center;padding-left:0.75em;padding-right:0.75em}.tabs ul.is-right{justify-content:flex-end;padding-left:0.75em}.tabs .icon:first-child{margin-right:.5em}.tabs .icon:last-child{margin-left:.5em}.tabs.is-centered ul{justify-content:center}.tabs.is-right ul{justify-content:flex-end}.tabs.is-boxed a{border:1px solid transparent;border-radius:4px 4px 0 0}.tabs.is-boxed a:hover{background-color:#f5f5f5;border-bottom-color:#dbdbdb}.tabs.is-boxed li.is-active a{background-color:#fff;border-color:#dbdbdb;border-bottom-color:rgba(0,0,0,0) !important}.tabs.is-fullwidth li{flex-grow:1;flex-shrink:0}.tabs.is-toggle a{border-color:#dbdbdb;border-style:solid;border-width:1px;margin-bottom:0;position:relative}.tabs.is-toggle a:hover{background-color:#f5f5f5;border-color:#b5b5b5;z-index:2}.tabs.is-toggle li+li{margin-left:-1px}.tabs.is-toggle li:first-child a{border-top-left-radius:4px;border-bottom-left-radius:4px}.tabs.is-toggle li:last-child a{border-top-right-radius:4px;border-bottom-right-radius:4px}.tabs.is-toggle li.is-active a{background-color:#2e63b8;border-color:#2e63b8;color:#fff;z-index:1}.tabs.is-toggle ul{border-bottom:none}.tabs.is-toggle.is-toggle-rounded li:first-child a{border-bottom-left-radius:9999px;border-top-left-radius:9999px;padding-left:1.25em}.tabs.is-toggle.is-toggle-rounded li:last-child a{border-bottom-right-radius:9999px;border-top-right-radius:9999px;padding-right:1.25em}.tabs.is-small,#documenter .docs-sidebar form.docs-search>input.tabs{font-size:.75rem}.tabs.is-medium{font-size:1.25rem}.tabs.is-large{font-size:1.5rem}.column{display:block;flex-basis:0;flex-grow:1;flex-shrink:1;padding:.75rem}.columns.is-mobile>.column.is-narrow{flex:none;width:unset}.columns.is-mobile>.column.is-full{flex:none;width:100%}.columns.is-mobile>.column.is-three-quarters{flex:none;width:75%}.columns.is-mobile>.column.is-two-thirds{flex:none;width:66.6666%}.columns.is-mobile>.column.is-half{flex:none;width:50%}.columns.is-mobile>.column.is-one-third{flex:none;width:33.3333%}.columns.is-mobile>.column.is-one-quarter{flex:none;width:25%}.columns.is-mobile>.column.is-one-fifth{flex:none;width:20%}.columns.is-mobile>.column.is-two-fifths{flex:none;width:40%}.columns.is-mobile>.column.is-three-fifths{flex:none;width:60%}.columns.is-mobile>.column.is-four-fifths{flex:none;width:80%}.columns.is-mobile>.column.is-offset-three-quarters{margin-left:75%}.columns.is-mobile>.column.is-offset-two-thirds{margin-left:66.6666%}.columns.is-mobile>.column.is-offset-half{margin-left:50%}.columns.is-mobile>.column.is-offset-one-third{margin-left:33.3333%}.columns.is-mobile>.column.is-offset-one-quarter{margin-left:25%}.columns.is-mobile>.column.is-offset-one-fifth{margin-left:20%}.columns.is-mobile>.column.is-offset-two-fifths{margin-left:40%}.columns.is-mobile>.column.is-offset-three-fifths{margin-left:60%}.columns.is-mobile>.column.is-offset-four-fifths{margin-left:80%}.columns.is-mobile>.column.is-0{flex:none;width:0%}.columns.is-mobile>.column.is-offset-0{margin-left:0%}.columns.is-mobile>.column.is-1{flex:none;width:8.33333337%}.columns.is-mobile>.column.is-offset-1{margin-left:8.33333337%}.columns.is-mobile>.column.is-2{flex:none;width:16.66666674%}.columns.is-mobile>.column.is-offset-2{margin-left:16.66666674%}.columns.is-mobile>.column.is-3{flex:none;width:25%}.columns.is-mobile>.column.is-offset-3{margin-left:25%}.columns.is-mobile>.column.is-4{flex:none;width:33.33333337%}.columns.is-mobile>.column.is-offset-4{margin-left:33.33333337%}.columns.is-mobile>.column.is-5{flex:none;width:41.66666674%}.columns.is-mobile>.column.is-offset-5{margin-left:41.66666674%}.columns.is-mobile>.column.is-6{flex:none;width:50%}.columns.is-mobile>.column.is-offset-6{margin-left:50%}.columns.is-mobile>.column.is-7{flex:none;width:58.33333337%}.columns.is-mobile>.column.is-offset-7{margin-left:58.33333337%}.columns.is-mobile>.column.is-8{flex:none;width:66.66666674%}.columns.is-mobile>.column.is-offset-8{margin-left:66.66666674%}.columns.is-mobile>.column.is-9{flex:none;width:75%}.columns.is-mobile>.column.is-offset-9{margin-left:75%}.columns.is-mobile>.column.is-10{flex:none;width:83.33333337%}.columns.is-mobile>.column.is-offset-10{margin-left:83.33333337%}.columns.is-mobile>.column.is-11{flex:none;width:91.66666674%}.columns.is-mobile>.column.is-offset-11{margin-left:91.66666674%}.columns.is-mobile>.column.is-12{flex:none;width:100%}.columns.is-mobile>.column.is-offset-12{margin-left:100%}@media screen and (max-width: 768px){.column.is-narrow-mobile{flex:none;width:unset}.column.is-full-mobile{flex:none;width:100%}.column.is-three-quarters-mobile{flex:none;width:75%}.column.is-two-thirds-mobile{flex:none;width:66.6666%}.column.is-half-mobile{flex:none;width:50%}.column.is-one-third-mobile{flex:none;width:33.3333%}.column.is-one-quarter-mobile{flex:none;width:25%}.column.is-one-fifth-mobile{flex:none;width:20%}.column.is-two-fifths-mobile{flex:none;width:40%}.column.is-three-fifths-mobile{flex:none;width:60%}.column.is-four-fifths-mobile{flex:none;width:80%}.column.is-offset-three-quarters-mobile{margin-left:75%}.column.is-offset-two-thirds-mobile{margin-left:66.6666%}.column.is-offset-half-mobile{margin-left:50%}.column.is-offset-one-third-mobile{margin-left:33.3333%}.column.is-offset-one-quarter-mobile{margin-left:25%}.column.is-offset-one-fifth-mobile{margin-left:20%}.column.is-offset-two-fifths-mobile{margin-left:40%}.column.is-offset-three-fifths-mobile{margin-left:60%}.column.is-offset-four-fifths-mobile{margin-left:80%}.column.is-0-mobile{flex:none;width:0%}.column.is-offset-0-mobile{margin-left:0%}.column.is-1-mobile{flex:none;width:8.33333337%}.column.is-offset-1-mobile{margin-left:8.33333337%}.column.is-2-mobile{flex:none;width:16.66666674%}.column.is-offset-2-mobile{margin-left:16.66666674%}.column.is-3-mobile{flex:none;width:25%}.column.is-offset-3-mobile{margin-left:25%}.column.is-4-mobile{flex:none;width:33.33333337%}.column.is-offset-4-mobile{margin-left:33.33333337%}.column.is-5-mobile{flex:none;width:41.66666674%}.column.is-offset-5-mobile{margin-left:41.66666674%}.column.is-6-mobile{flex:none;width:50%}.column.is-offset-6-mobile{margin-left:50%}.column.is-7-mobile{flex:none;width:58.33333337%}.column.is-offset-7-mobile{margin-left:58.33333337%}.column.is-8-mobile{flex:none;width:66.66666674%}.column.is-offset-8-mobile{margin-left:66.66666674%}.column.is-9-mobile{flex:none;width:75%}.column.is-offset-9-mobile{margin-left:75%}.column.is-10-mobile{flex:none;width:83.33333337%}.column.is-offset-10-mobile{margin-left:83.33333337%}.column.is-11-mobile{flex:none;width:91.66666674%}.column.is-offset-11-mobile{margin-left:91.66666674%}.column.is-12-mobile{flex:none;width:100%}.column.is-offset-12-mobile{margin-left:100%}}@media screen and (min-width: 769px),print{.column.is-narrow,.column.is-narrow-tablet{flex:none;width:unset}.column.is-full,.column.is-full-tablet{flex:none;width:100%}.column.is-three-quarters,.column.is-three-quarters-tablet{flex:none;width:75%}.column.is-two-thirds,.column.is-two-thirds-tablet{flex:none;width:66.6666%}.column.is-half,.column.is-half-tablet{flex:none;width:50%}.column.is-one-third,.column.is-one-third-tablet{flex:none;width:33.3333%}.column.is-one-quarter,.column.is-one-quarter-tablet{flex:none;width:25%}.column.is-one-fifth,.column.is-one-fifth-tablet{flex:none;width:20%}.column.is-two-fifths,.column.is-two-fifths-tablet{flex:none;width:40%}.column.is-three-fifths,.column.is-three-fifths-tablet{flex:none;width:60%}.column.is-four-fifths,.column.is-four-fifths-tablet{flex:none;width:80%}.column.is-offset-three-quarters,.column.is-offset-three-quarters-tablet{margin-left:75%}.column.is-offset-two-thirds,.column.is-offset-two-thirds-tablet{margin-left:66.6666%}.column.is-offset-half,.column.is-offset-half-tablet{margin-left:50%}.column.is-offset-one-third,.column.is-offset-one-third-tablet{margin-left:33.3333%}.column.is-offset-one-quarter,.column.is-offset-one-quarter-tablet{margin-left:25%}.column.is-offset-one-fifth,.column.is-offset-one-fifth-tablet{margin-left:20%}.column.is-offset-two-fifths,.column.is-offset-two-fifths-tablet{margin-left:40%}.column.is-offset-three-fifths,.column.is-offset-three-fifths-tablet{margin-left:60%}.column.is-offset-four-fifths,.column.is-offset-four-fifths-tablet{margin-left:80%}.column.is-0,.column.is-0-tablet{flex:none;width:0%}.column.is-offset-0,.column.is-offset-0-tablet{margin-left:0%}.column.is-1,.column.is-1-tablet{flex:none;width:8.33333337%}.column.is-offset-1,.column.is-offset-1-tablet{margin-left:8.33333337%}.column.is-2,.column.is-2-tablet{flex:none;width:16.66666674%}.column.is-offset-2,.column.is-offset-2-tablet{margin-left:16.66666674%}.column.is-3,.column.is-3-tablet{flex:none;width:25%}.column.is-offset-3,.column.is-offset-3-tablet{margin-left:25%}.column.is-4,.column.is-4-tablet{flex:none;width:33.33333337%}.column.is-offset-4,.column.is-offset-4-tablet{margin-left:33.33333337%}.column.is-5,.column.is-5-tablet{flex:none;width:41.66666674%}.column.is-offset-5,.column.is-offset-5-tablet{margin-left:41.66666674%}.column.is-6,.column.is-6-tablet{flex:none;width:50%}.column.is-offset-6,.column.is-offset-6-tablet{margin-left:50%}.column.is-7,.column.is-7-tablet{flex:none;width:58.33333337%}.column.is-offset-7,.column.is-offset-7-tablet{margin-left:58.33333337%}.column.is-8,.column.is-8-tablet{flex:none;width:66.66666674%}.column.is-offset-8,.column.is-offset-8-tablet{margin-left:66.66666674%}.column.is-9,.column.is-9-tablet{flex:none;width:75%}.column.is-offset-9,.column.is-offset-9-tablet{margin-left:75%}.column.is-10,.column.is-10-tablet{flex:none;width:83.33333337%}.column.is-offset-10,.column.is-offset-10-tablet{margin-left:83.33333337%}.column.is-11,.column.is-11-tablet{flex:none;width:91.66666674%}.column.is-offset-11,.column.is-offset-11-tablet{margin-left:91.66666674%}.column.is-12,.column.is-12-tablet{flex:none;width:100%}.column.is-offset-12,.column.is-offset-12-tablet{margin-left:100%}}@media screen and (max-width: 1055px){.column.is-narrow-touch{flex:none;width:unset}.column.is-full-touch{flex:none;width:100%}.column.is-three-quarters-touch{flex:none;width:75%}.column.is-two-thirds-touch{flex:none;width:66.6666%}.column.is-half-touch{flex:none;width:50%}.column.is-one-third-touch{flex:none;width:33.3333%}.column.is-one-quarter-touch{flex:none;width:25%}.column.is-one-fifth-touch{flex:none;width:20%}.column.is-two-fifths-touch{flex:none;width:40%}.column.is-three-fifths-touch{flex:none;width:60%}.column.is-four-fifths-touch{flex:none;width:80%}.column.is-offset-three-quarters-touch{margin-left:75%}.column.is-offset-two-thirds-touch{margin-left:66.6666%}.column.is-offset-half-touch{margin-left:50%}.column.is-offset-one-third-touch{margin-left:33.3333%}.column.is-offset-one-quarter-touch{margin-left:25%}.column.is-offset-one-fifth-touch{margin-left:20%}.column.is-offset-two-fifths-touch{margin-left:40%}.column.is-offset-three-fifths-touch{margin-left:60%}.column.is-offset-four-fifths-touch{margin-left:80%}.column.is-0-touch{flex:none;width:0%}.column.is-offset-0-touch{margin-left:0%}.column.is-1-touch{flex:none;width:8.33333337%}.column.is-offset-1-touch{margin-left:8.33333337%}.column.is-2-touch{flex:none;width:16.66666674%}.column.is-offset-2-touch{margin-left:16.66666674%}.column.is-3-touch{flex:none;width:25%}.column.is-offset-3-touch{margin-left:25%}.column.is-4-touch{flex:none;width:33.33333337%}.column.is-offset-4-touch{margin-left:33.33333337%}.column.is-5-touch{flex:none;width:41.66666674%}.column.is-offset-5-touch{margin-left:41.66666674%}.column.is-6-touch{flex:none;width:50%}.column.is-offset-6-touch{margin-left:50%}.column.is-7-touch{flex:none;width:58.33333337%}.column.is-offset-7-touch{margin-left:58.33333337%}.column.is-8-touch{flex:none;width:66.66666674%}.column.is-offset-8-touch{margin-left:66.66666674%}.column.is-9-touch{flex:none;width:75%}.column.is-offset-9-touch{margin-left:75%}.column.is-10-touch{flex:none;width:83.33333337%}.column.is-offset-10-touch{margin-left:83.33333337%}.column.is-11-touch{flex:none;width:91.66666674%}.column.is-offset-11-touch{margin-left:91.66666674%}.column.is-12-touch{flex:none;width:100%}.column.is-offset-12-touch{margin-left:100%}}@media screen and (min-width: 1056px){.column.is-narrow-desktop{flex:none;width:unset}.column.is-full-desktop{flex:none;width:100%}.column.is-three-quarters-desktop{flex:none;width:75%}.column.is-two-thirds-desktop{flex:none;width:66.6666%}.column.is-half-desktop{flex:none;width:50%}.column.is-one-third-desktop{flex:none;width:33.3333%}.column.is-one-quarter-desktop{flex:none;width:25%}.column.is-one-fifth-desktop{flex:none;width:20%}.column.is-two-fifths-desktop{flex:none;width:40%}.column.is-three-fifths-desktop{flex:none;width:60%}.column.is-four-fifths-desktop{flex:none;width:80%}.column.is-offset-three-quarters-desktop{margin-left:75%}.column.is-offset-two-thirds-desktop{margin-left:66.6666%}.column.is-offset-half-desktop{margin-left:50%}.column.is-offset-one-third-desktop{margin-left:33.3333%}.column.is-offset-one-quarter-desktop{margin-left:25%}.column.is-offset-one-fifth-desktop{margin-left:20%}.column.is-offset-two-fifths-desktop{margin-left:40%}.column.is-offset-three-fifths-desktop{margin-left:60%}.column.is-offset-four-fifths-desktop{margin-left:80%}.column.is-0-desktop{flex:none;width:0%}.column.is-offset-0-desktop{margin-left:0%}.column.is-1-desktop{flex:none;width:8.33333337%}.column.is-offset-1-desktop{margin-left:8.33333337%}.column.is-2-desktop{flex:none;width:16.66666674%}.column.is-offset-2-desktop{margin-left:16.66666674%}.column.is-3-desktop{flex:none;width:25%}.column.is-offset-3-desktop{margin-left:25%}.column.is-4-desktop{flex:none;width:33.33333337%}.column.is-offset-4-desktop{margin-left:33.33333337%}.column.is-5-desktop{flex:none;width:41.66666674%}.column.is-offset-5-desktop{margin-left:41.66666674%}.column.is-6-desktop{flex:none;width:50%}.column.is-offset-6-desktop{margin-left:50%}.column.is-7-desktop{flex:none;width:58.33333337%}.column.is-offset-7-desktop{margin-left:58.33333337%}.column.is-8-desktop{flex:none;width:66.66666674%}.column.is-offset-8-desktop{margin-left:66.66666674%}.column.is-9-desktop{flex:none;width:75%}.column.is-offset-9-desktop{margin-left:75%}.column.is-10-desktop{flex:none;width:83.33333337%}.column.is-offset-10-desktop{margin-left:83.33333337%}.column.is-11-desktop{flex:none;width:91.66666674%}.column.is-offset-11-desktop{margin-left:91.66666674%}.column.is-12-desktop{flex:none;width:100%}.column.is-offset-12-desktop{margin-left:100%}}@media screen and (min-width: 1216px){.column.is-narrow-widescreen{flex:none;width:unset}.column.is-full-widescreen{flex:none;width:100%}.column.is-three-quarters-widescreen{flex:none;width:75%}.column.is-two-thirds-widescreen{flex:none;width:66.6666%}.column.is-half-widescreen{flex:none;width:50%}.column.is-one-third-widescreen{flex:none;width:33.3333%}.column.is-one-quarter-widescreen{flex:none;width:25%}.column.is-one-fifth-widescreen{flex:none;width:20%}.column.is-two-fifths-widescreen{flex:none;width:40%}.column.is-three-fifths-widescreen{flex:none;width:60%}.column.is-four-fifths-widescreen{flex:none;width:80%}.column.is-offset-three-quarters-widescreen{margin-left:75%}.column.is-offset-two-thirds-widescreen{margin-left:66.6666%}.column.is-offset-half-widescreen{margin-left:50%}.column.is-offset-one-third-widescreen{margin-left:33.3333%}.column.is-offset-one-quarter-widescreen{margin-left:25%}.column.is-offset-one-fifth-widescreen{margin-left:20%}.column.is-offset-two-fifths-widescreen{margin-left:40%}.column.is-offset-three-fifths-widescreen{margin-left:60%}.column.is-offset-four-fifths-widescreen{margin-left:80%}.column.is-0-widescreen{flex:none;width:0%}.column.is-offset-0-widescreen{margin-left:0%}.column.is-1-widescreen{flex:none;width:8.33333337%}.column.is-offset-1-widescreen{margin-left:8.33333337%}.column.is-2-widescreen{flex:none;width:16.66666674%}.column.is-offset-2-widescreen{margin-left:16.66666674%}.column.is-3-widescreen{flex:none;width:25%}.column.is-offset-3-widescreen{margin-left:25%}.column.is-4-widescreen{flex:none;width:33.33333337%}.column.is-offset-4-widescreen{margin-left:33.33333337%}.column.is-5-widescreen{flex:none;width:41.66666674%}.column.is-offset-5-widescreen{margin-left:41.66666674%}.column.is-6-widescreen{flex:none;width:50%}.column.is-offset-6-widescreen{margin-left:50%}.column.is-7-widescreen{flex:none;width:58.33333337%}.column.is-offset-7-widescreen{margin-left:58.33333337%}.column.is-8-widescreen{flex:none;width:66.66666674%}.column.is-offset-8-widescreen{margin-left:66.66666674%}.column.is-9-widescreen{flex:none;width:75%}.column.is-offset-9-widescreen{margin-left:75%}.column.is-10-widescreen{flex:none;width:83.33333337%}.column.is-offset-10-widescreen{margin-left:83.33333337%}.column.is-11-widescreen{flex:none;width:91.66666674%}.column.is-offset-11-widescreen{margin-left:91.66666674%}.column.is-12-widescreen{flex:none;width:100%}.column.is-offset-12-widescreen{margin-left:100%}}@media screen and (min-width: 1408px){.column.is-narrow-fullhd{flex:none;width:unset}.column.is-full-fullhd{flex:none;width:100%}.column.is-three-quarters-fullhd{flex:none;width:75%}.column.is-two-thirds-fullhd{flex:none;width:66.6666%}.column.is-half-fullhd{flex:none;width:50%}.column.is-one-third-fullhd{flex:none;width:33.3333%}.column.is-one-quarter-fullhd{flex:none;width:25%}.column.is-one-fifth-fullhd{flex:none;width:20%}.column.is-two-fifths-fullhd{flex:none;width:40%}.column.is-three-fifths-fullhd{flex:none;width:60%}.column.is-four-fifths-fullhd{flex:none;width:80%}.column.is-offset-three-quarters-fullhd{margin-left:75%}.column.is-offset-two-thirds-fullhd{margin-left:66.6666%}.column.is-offset-half-fullhd{margin-left:50%}.column.is-offset-one-third-fullhd{margin-left:33.3333%}.column.is-offset-one-quarter-fullhd{margin-left:25%}.column.is-offset-one-fifth-fullhd{margin-left:20%}.column.is-offset-two-fifths-fullhd{margin-left:40%}.column.is-offset-three-fifths-fullhd{margin-left:60%}.column.is-offset-four-fifths-fullhd{margin-left:80%}.column.is-0-fullhd{flex:none;width:0%}.column.is-offset-0-fullhd{margin-left:0%}.column.is-1-fullhd{flex:none;width:8.33333337%}.column.is-offset-1-fullhd{margin-left:8.33333337%}.column.is-2-fullhd{flex:none;width:16.66666674%}.column.is-offset-2-fullhd{margin-left:16.66666674%}.column.is-3-fullhd{flex:none;width:25%}.column.is-offset-3-fullhd{margin-left:25%}.column.is-4-fullhd{flex:none;width:33.33333337%}.column.is-offset-4-fullhd{margin-left:33.33333337%}.column.is-5-fullhd{flex:none;width:41.66666674%}.column.is-offset-5-fullhd{margin-left:41.66666674%}.column.is-6-fullhd{flex:none;width:50%}.column.is-offset-6-fullhd{margin-left:50%}.column.is-7-fullhd{flex:none;width:58.33333337%}.column.is-offset-7-fullhd{margin-left:58.33333337%}.column.is-8-fullhd{flex:none;width:66.66666674%}.column.is-offset-8-fullhd{margin-left:66.66666674%}.column.is-9-fullhd{flex:none;width:75%}.column.is-offset-9-fullhd{margin-left:75%}.column.is-10-fullhd{flex:none;width:83.33333337%}.column.is-offset-10-fullhd{margin-left:83.33333337%}.column.is-11-fullhd{flex:none;width:91.66666674%}.column.is-offset-11-fullhd{margin-left:91.66666674%}.column.is-12-fullhd{flex:none;width:100%}.column.is-offset-12-fullhd{margin-left:100%}}.columns{margin-left:-.75rem;margin-right:-.75rem;margin-top:-.75rem}.columns:last-child{margin-bottom:-.75rem}.columns:not(:last-child){margin-bottom:calc(1.5rem - .75rem)}.columns.is-centered{justify-content:center}.columns.is-gapless{margin-left:0;margin-right:0;margin-top:0}.columns.is-gapless>.column{margin:0;padding:0 !important}.columns.is-gapless:not(:last-child){margin-bottom:1.5rem}.columns.is-gapless:last-child{margin-bottom:0}.columns.is-mobile{display:flex}.columns.is-multiline{flex-wrap:wrap}.columns.is-vcentered{align-items:center}@media screen and (min-width: 769px),print{.columns:not(.is-desktop){display:flex}}@media screen and (min-width: 1056px){.columns.is-desktop{display:flex}}.columns.is-variable{--columnGap: 0.75rem;margin-left:calc(-1 * var(--columnGap));margin-right:calc(-1 * var(--columnGap))}.columns.is-variable>.column{padding-left:var(--columnGap);padding-right:var(--columnGap)}.columns.is-variable.is-0{--columnGap: 0rem}@media screen and (max-width: 768px){.columns.is-variable.is-0-mobile{--columnGap: 0rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-0-tablet{--columnGap: 0rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-0-tablet-only{--columnGap: 0rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-0-touch{--columnGap: 0rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-0-desktop{--columnGap: 0rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-0-desktop-only{--columnGap: 0rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-0-widescreen{--columnGap: 0rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-0-widescreen-only{--columnGap: 0rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-0-fullhd{--columnGap: 0rem}}.columns.is-variable.is-1{--columnGap: .25rem}@media screen and (max-width: 768px){.columns.is-variable.is-1-mobile{--columnGap: .25rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-1-tablet{--columnGap: .25rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-1-tablet-only{--columnGap: .25rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-1-touch{--columnGap: .25rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-1-desktop{--columnGap: .25rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-1-desktop-only{--columnGap: .25rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-1-widescreen{--columnGap: .25rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-1-widescreen-only{--columnGap: .25rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-1-fullhd{--columnGap: .25rem}}.columns.is-variable.is-2{--columnGap: .5rem}@media screen and (max-width: 768px){.columns.is-variable.is-2-mobile{--columnGap: .5rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-2-tablet{--columnGap: .5rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-2-tablet-only{--columnGap: .5rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-2-touch{--columnGap: .5rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-2-desktop{--columnGap: .5rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-2-desktop-only{--columnGap: .5rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-2-widescreen{--columnGap: .5rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-2-widescreen-only{--columnGap: .5rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-2-fullhd{--columnGap: .5rem}}.columns.is-variable.is-3{--columnGap: .75rem}@media screen and (max-width: 768px){.columns.is-variable.is-3-mobile{--columnGap: .75rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-3-tablet{--columnGap: .75rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-3-tablet-only{--columnGap: .75rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-3-touch{--columnGap: .75rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-3-desktop{--columnGap: .75rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-3-desktop-only{--columnGap: .75rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-3-widescreen{--columnGap: .75rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-3-widescreen-only{--columnGap: .75rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-3-fullhd{--columnGap: .75rem}}.columns.is-variable.is-4{--columnGap: 1rem}@media screen and (max-width: 768px){.columns.is-variable.is-4-mobile{--columnGap: 1rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-4-tablet{--columnGap: 1rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-4-tablet-only{--columnGap: 1rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-4-touch{--columnGap: 1rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-4-desktop{--columnGap: 1rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-4-desktop-only{--columnGap: 1rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-4-widescreen{--columnGap: 1rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-4-widescreen-only{--columnGap: 1rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-4-fullhd{--columnGap: 1rem}}.columns.is-variable.is-5{--columnGap: 1.25rem}@media screen and (max-width: 768px){.columns.is-variable.is-5-mobile{--columnGap: 1.25rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-5-tablet{--columnGap: 1.25rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-5-tablet-only{--columnGap: 1.25rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-5-touch{--columnGap: 1.25rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-5-desktop{--columnGap: 1.25rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-5-desktop-only{--columnGap: 1.25rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-5-widescreen{--columnGap: 1.25rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-5-widescreen-only{--columnGap: 1.25rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-5-fullhd{--columnGap: 1.25rem}}.columns.is-variable.is-6{--columnGap: 1.5rem}@media screen and (max-width: 768px){.columns.is-variable.is-6-mobile{--columnGap: 1.5rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-6-tablet{--columnGap: 1.5rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-6-tablet-only{--columnGap: 1.5rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-6-touch{--columnGap: 1.5rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-6-desktop{--columnGap: 1.5rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-6-desktop-only{--columnGap: 1.5rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-6-widescreen{--columnGap: 1.5rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-6-widescreen-only{--columnGap: 1.5rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-6-fullhd{--columnGap: 1.5rem}}.columns.is-variable.is-7{--columnGap: 1.75rem}@media screen and (max-width: 768px){.columns.is-variable.is-7-mobile{--columnGap: 1.75rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-7-tablet{--columnGap: 1.75rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-7-tablet-only{--columnGap: 1.75rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-7-touch{--columnGap: 1.75rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-7-desktop{--columnGap: 1.75rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-7-desktop-only{--columnGap: 1.75rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-7-widescreen{--columnGap: 1.75rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-7-widescreen-only{--columnGap: 1.75rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-7-fullhd{--columnGap: 1.75rem}}.columns.is-variable.is-8{--columnGap: 2rem}@media screen and (max-width: 768px){.columns.is-variable.is-8-mobile{--columnGap: 2rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-8-tablet{--columnGap: 2rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-8-tablet-only{--columnGap: 2rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-8-touch{--columnGap: 2rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-8-desktop{--columnGap: 2rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-8-desktop-only{--columnGap: 2rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-8-widescreen{--columnGap: 2rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-8-widescreen-only{--columnGap: 2rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-8-fullhd{--columnGap: 2rem}}.tile{align-items:stretch;display:block;flex-basis:0;flex-grow:1;flex-shrink:1;min-height:min-content}.tile.is-ancestor{margin-left:-.75rem;margin-right:-.75rem;margin-top:-.75rem}.tile.is-ancestor:last-child{margin-bottom:-.75rem}.tile.is-ancestor:not(:last-child){margin-bottom:.75rem}.tile.is-child{margin:0 !important}.tile.is-parent{padding:.75rem}.tile.is-vertical{flex-direction:column}.tile.is-vertical>.tile.is-child:not(:last-child){margin-bottom:1.5rem !important}@media screen and (min-width: 769px),print{.tile:not(.is-child){display:flex}.tile.is-1{flex:none;width:8.33333337%}.tile.is-2{flex:none;width:16.66666674%}.tile.is-3{flex:none;width:25%}.tile.is-4{flex:none;width:33.33333337%}.tile.is-5{flex:none;width:41.66666674%}.tile.is-6{flex:none;width:50%}.tile.is-7{flex:none;width:58.33333337%}.tile.is-8{flex:none;width:66.66666674%}.tile.is-9{flex:none;width:75%}.tile.is-10{flex:none;width:83.33333337%}.tile.is-11{flex:none;width:91.66666674%}.tile.is-12{flex:none;width:100%}}.hero{align-items:stretch;display:flex;flex-direction:column;justify-content:space-between}.hero .navbar{background:none}.hero .tabs ul{border-bottom:none}.hero.is-white{background-color:#fff;color:#0a0a0a}.hero.is-white a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-white strong{color:inherit}.hero.is-white .title{color:#0a0a0a}.hero.is-white .subtitle{color:rgba(10,10,10,0.9)}.hero.is-white .subtitle a:not(.button),.hero.is-white .subtitle strong{color:#0a0a0a}@media screen and (max-width: 1055px){.hero.is-white .navbar-menu{background-color:#fff}}.hero.is-white .navbar-item,.hero.is-white .navbar-link{color:rgba(10,10,10,0.7)}.hero.is-white a.navbar-item:hover,.hero.is-white a.navbar-item.is-active,.hero.is-white .navbar-link:hover,.hero.is-white .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}.hero.is-white .tabs a{color:#0a0a0a;opacity:0.9}.hero.is-white .tabs a:hover{opacity:1}.hero.is-white .tabs li.is-active a{color:#fff !important;opacity:1}.hero.is-white .tabs.is-boxed a,.hero.is-white .tabs.is-toggle a{color:#0a0a0a}.hero.is-white .tabs.is-boxed a:hover,.hero.is-white .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-white .tabs.is-boxed li.is-active a,.hero.is-white .tabs.is-boxed li.is-active a:hover,.hero.is-white .tabs.is-toggle li.is-active a,.hero.is-white .tabs.is-toggle li.is-active a:hover{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}.hero.is-white.is-bold{background-image:linear-gradient(141deg, #e8e3e4 0%, #fff 71%, #fff 100%)}@media screen and (max-width: 768px){.hero.is-white.is-bold .navbar-menu{background-image:linear-gradient(141deg, #e8e3e4 0%, #fff 71%, #fff 100%)}}.hero.is-black{background-color:#0a0a0a;color:#fff}.hero.is-black a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-black strong{color:inherit}.hero.is-black .title{color:#fff}.hero.is-black .subtitle{color:rgba(255,255,255,0.9)}.hero.is-black .subtitle a:not(.button),.hero.is-black .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-black .navbar-menu{background-color:#0a0a0a}}.hero.is-black .navbar-item,.hero.is-black .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-black a.navbar-item:hover,.hero.is-black a.navbar-item.is-active,.hero.is-black .navbar-link:hover,.hero.is-black .navbar-link.is-active{background-color:#000;color:#fff}.hero.is-black .tabs a{color:#fff;opacity:0.9}.hero.is-black .tabs a:hover{opacity:1}.hero.is-black .tabs li.is-active a{color:#0a0a0a !important;opacity:1}.hero.is-black .tabs.is-boxed a,.hero.is-black .tabs.is-toggle a{color:#fff}.hero.is-black .tabs.is-boxed a:hover,.hero.is-black .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-black .tabs.is-boxed li.is-active a,.hero.is-black .tabs.is-boxed li.is-active a:hover,.hero.is-black .tabs.is-toggle li.is-active a,.hero.is-black .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#0a0a0a}.hero.is-black.is-bold{background-image:linear-gradient(141deg, #000 0%, #0a0a0a 71%, #181616 100%)}@media screen and (max-width: 768px){.hero.is-black.is-bold .navbar-menu{background-image:linear-gradient(141deg, #000 0%, #0a0a0a 71%, #181616 100%)}}.hero.is-light{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.hero.is-light a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-light strong{color:inherit}.hero.is-light .title{color:rgba(0,0,0,0.7)}.hero.is-light .subtitle{color:rgba(0,0,0,0.9)}.hero.is-light .subtitle a:not(.button),.hero.is-light .subtitle strong{color:rgba(0,0,0,0.7)}@media screen and (max-width: 1055px){.hero.is-light .navbar-menu{background-color:#f5f5f5}}.hero.is-light .navbar-item,.hero.is-light .navbar-link{color:rgba(0,0,0,0.7)}.hero.is-light a.navbar-item:hover,.hero.is-light a.navbar-item.is-active,.hero.is-light .navbar-link:hover,.hero.is-light .navbar-link.is-active{background-color:#e8e8e8;color:rgba(0,0,0,0.7)}.hero.is-light .tabs a{color:rgba(0,0,0,0.7);opacity:0.9}.hero.is-light .tabs a:hover{opacity:1}.hero.is-light .tabs li.is-active a{color:#f5f5f5 !important;opacity:1}.hero.is-light .tabs.is-boxed a,.hero.is-light .tabs.is-toggle a{color:rgba(0,0,0,0.7)}.hero.is-light .tabs.is-boxed a:hover,.hero.is-light .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-light .tabs.is-boxed li.is-active a,.hero.is-light .tabs.is-boxed li.is-active a:hover,.hero.is-light .tabs.is-toggle li.is-active a,.hero.is-light .tabs.is-toggle li.is-active a:hover{background-color:rgba(0,0,0,0.7);border-color:rgba(0,0,0,0.7);color:#f5f5f5}.hero.is-light.is-bold{background-image:linear-gradient(141deg, #dfd8d9 0%, #f5f5f5 71%, #fff 100%)}@media screen and (max-width: 768px){.hero.is-light.is-bold .navbar-menu{background-image:linear-gradient(141deg, #dfd8d9 0%, #f5f5f5 71%, #fff 100%)}}.hero.is-dark,.content kbd.hero{background-color:#363636;color:#fff}.hero.is-dark a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.content kbd.hero a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-dark strong,.content kbd.hero strong{color:inherit}.hero.is-dark .title,.content kbd.hero .title{color:#fff}.hero.is-dark .subtitle,.content kbd.hero .subtitle{color:rgba(255,255,255,0.9)}.hero.is-dark .subtitle a:not(.button),.content kbd.hero .subtitle a:not(.button),.hero.is-dark .subtitle strong,.content kbd.hero .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-dark .navbar-menu,.content kbd.hero .navbar-menu{background-color:#363636}}.hero.is-dark .navbar-item,.content kbd.hero .navbar-item,.hero.is-dark .navbar-link,.content kbd.hero .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-dark a.navbar-item:hover,.content kbd.hero a.navbar-item:hover,.hero.is-dark a.navbar-item.is-active,.content kbd.hero a.navbar-item.is-active,.hero.is-dark .navbar-link:hover,.content kbd.hero .navbar-link:hover,.hero.is-dark .navbar-link.is-active,.content kbd.hero .navbar-link.is-active{background-color:#292929;color:#fff}.hero.is-dark .tabs a,.content kbd.hero .tabs a{color:#fff;opacity:0.9}.hero.is-dark .tabs a:hover,.content kbd.hero .tabs a:hover{opacity:1}.hero.is-dark .tabs li.is-active a,.content kbd.hero .tabs li.is-active a{color:#363636 !important;opacity:1}.hero.is-dark .tabs.is-boxed a,.content kbd.hero .tabs.is-boxed a,.hero.is-dark .tabs.is-toggle a,.content kbd.hero .tabs.is-toggle a{color:#fff}.hero.is-dark .tabs.is-boxed a:hover,.content kbd.hero .tabs.is-boxed a:hover,.hero.is-dark .tabs.is-toggle a:hover,.content kbd.hero .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-dark .tabs.is-boxed li.is-active a,.content kbd.hero .tabs.is-boxed li.is-active a,.hero.is-dark .tabs.is-boxed li.is-active a:hover,.hero.is-dark .tabs.is-toggle li.is-active a,.content kbd.hero .tabs.is-toggle li.is-active a,.hero.is-dark .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#363636}.hero.is-dark.is-bold,.content kbd.hero.is-bold{background-image:linear-gradient(141deg, #1f191a 0%, #363636 71%, #46403f 100%)}@media screen and (max-width: 768px){.hero.is-dark.is-bold .navbar-menu,.content kbd.hero.is-bold .navbar-menu{background-image:linear-gradient(141deg, #1f191a 0%, #363636 71%, #46403f 100%)}}.hero.is-primary,.docstring>section>a.hero.docs-sourcelink{background-color:#4eb5de;color:#fff}.hero.is-primary a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.docstring>section>a.hero.docs-sourcelink a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-primary strong,.docstring>section>a.hero.docs-sourcelink strong{color:inherit}.hero.is-primary .title,.docstring>section>a.hero.docs-sourcelink .title{color:#fff}.hero.is-primary .subtitle,.docstring>section>a.hero.docs-sourcelink .subtitle{color:rgba(255,255,255,0.9)}.hero.is-primary .subtitle a:not(.button),.docstring>section>a.hero.docs-sourcelink .subtitle a:not(.button),.hero.is-primary .subtitle strong,.docstring>section>a.hero.docs-sourcelink .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-primary .navbar-menu,.docstring>section>a.hero.docs-sourcelink .navbar-menu{background-color:#4eb5de}}.hero.is-primary .navbar-item,.docstring>section>a.hero.docs-sourcelink .navbar-item,.hero.is-primary .navbar-link,.docstring>section>a.hero.docs-sourcelink .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-primary a.navbar-item:hover,.docstring>section>a.hero.docs-sourcelink a.navbar-item:hover,.hero.is-primary a.navbar-item.is-active,.docstring>section>a.hero.docs-sourcelink a.navbar-item.is-active,.hero.is-primary .navbar-link:hover,.docstring>section>a.hero.docs-sourcelink .navbar-link:hover,.hero.is-primary .navbar-link.is-active,.docstring>section>a.hero.docs-sourcelink .navbar-link.is-active{background-color:#39acda;color:#fff}.hero.is-primary .tabs a,.docstring>section>a.hero.docs-sourcelink .tabs a{color:#fff;opacity:0.9}.hero.is-primary .tabs a:hover,.docstring>section>a.hero.docs-sourcelink .tabs a:hover{opacity:1}.hero.is-primary .tabs li.is-active a,.docstring>section>a.hero.docs-sourcelink .tabs li.is-active a{color:#4eb5de !important;opacity:1}.hero.is-primary .tabs.is-boxed a,.docstring>section>a.hero.docs-sourcelink .tabs.is-boxed a,.hero.is-primary .tabs.is-toggle a,.docstring>section>a.hero.docs-sourcelink .tabs.is-toggle a{color:#fff}.hero.is-primary .tabs.is-boxed a:hover,.docstring>section>a.hero.docs-sourcelink .tabs.is-boxed a:hover,.hero.is-primary .tabs.is-toggle a:hover,.docstring>section>a.hero.docs-sourcelink .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-primary .tabs.is-boxed li.is-active a,.docstring>section>a.hero.docs-sourcelink .tabs.is-boxed li.is-active a,.hero.is-primary .tabs.is-boxed li.is-active a:hover,.hero.is-primary .tabs.is-toggle li.is-active a,.docstring>section>a.hero.docs-sourcelink .tabs.is-toggle li.is-active a,.hero.is-primary .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#4eb5de}.hero.is-primary.is-bold,.docstring>section>a.hero.is-bold.docs-sourcelink{background-image:linear-gradient(141deg, #1bc7de 0%, #4eb5de 71%, #5fa9e7 100%)}@media screen and (max-width: 768px){.hero.is-primary.is-bold .navbar-menu,.docstring>section>a.hero.is-bold.docs-sourcelink .navbar-menu{background-image:linear-gradient(141deg, #1bc7de 0%, #4eb5de 71%, #5fa9e7 100%)}}.hero.is-link{background-color:#2e63b8;color:#fff}.hero.is-link a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-link strong{color:inherit}.hero.is-link .title{color:#fff}.hero.is-link .subtitle{color:rgba(255,255,255,0.9)}.hero.is-link .subtitle a:not(.button),.hero.is-link .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-link .navbar-menu{background-color:#2e63b8}}.hero.is-link .navbar-item,.hero.is-link .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-link a.navbar-item:hover,.hero.is-link a.navbar-item.is-active,.hero.is-link .navbar-link:hover,.hero.is-link .navbar-link.is-active{background-color:#2958a4;color:#fff}.hero.is-link .tabs a{color:#fff;opacity:0.9}.hero.is-link .tabs a:hover{opacity:1}.hero.is-link .tabs li.is-active a{color:#2e63b8 !important;opacity:1}.hero.is-link .tabs.is-boxed a,.hero.is-link .tabs.is-toggle a{color:#fff}.hero.is-link .tabs.is-boxed a:hover,.hero.is-link .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-link .tabs.is-boxed li.is-active a,.hero.is-link .tabs.is-boxed li.is-active a:hover,.hero.is-link .tabs.is-toggle li.is-active a,.hero.is-link .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#2e63b8}.hero.is-link.is-bold{background-image:linear-gradient(141deg, #1b6098 0%, #2e63b8 71%, #2d51d2 100%)}@media screen and (max-width: 768px){.hero.is-link.is-bold .navbar-menu{background-image:linear-gradient(141deg, #1b6098 0%, #2e63b8 71%, #2d51d2 100%)}}.hero.is-info{background-color:#209cee;color:#fff}.hero.is-info a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-info strong{color:inherit}.hero.is-info .title{color:#fff}.hero.is-info .subtitle{color:rgba(255,255,255,0.9)}.hero.is-info .subtitle a:not(.button),.hero.is-info .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-info .navbar-menu{background-color:#209cee}}.hero.is-info .navbar-item,.hero.is-info .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-info a.navbar-item:hover,.hero.is-info a.navbar-item.is-active,.hero.is-info .navbar-link:hover,.hero.is-info .navbar-link.is-active{background-color:#1190e3;color:#fff}.hero.is-info .tabs a{color:#fff;opacity:0.9}.hero.is-info .tabs a:hover{opacity:1}.hero.is-info .tabs li.is-active a{color:#209cee !important;opacity:1}.hero.is-info .tabs.is-boxed a,.hero.is-info .tabs.is-toggle a{color:#fff}.hero.is-info .tabs.is-boxed a:hover,.hero.is-info .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-info .tabs.is-boxed li.is-active a,.hero.is-info .tabs.is-boxed li.is-active a:hover,.hero.is-info .tabs.is-toggle li.is-active a,.hero.is-info .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#209cee}.hero.is-info.is-bold{background-image:linear-gradient(141deg, #05a6d6 0%, #209cee 71%, #3287f5 100%)}@media screen and (max-width: 768px){.hero.is-info.is-bold .navbar-menu{background-image:linear-gradient(141deg, #05a6d6 0%, #209cee 71%, #3287f5 100%)}}.hero.is-success{background-color:#22c35b;color:#fff}.hero.is-success a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-success strong{color:inherit}.hero.is-success .title{color:#fff}.hero.is-success .subtitle{color:rgba(255,255,255,0.9)}.hero.is-success .subtitle a:not(.button),.hero.is-success .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-success .navbar-menu{background-color:#22c35b}}.hero.is-success .navbar-item,.hero.is-success .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-success a.navbar-item:hover,.hero.is-success a.navbar-item.is-active,.hero.is-success .navbar-link:hover,.hero.is-success .navbar-link.is-active{background-color:#1ead51;color:#fff}.hero.is-success .tabs a{color:#fff;opacity:0.9}.hero.is-success .tabs a:hover{opacity:1}.hero.is-success .tabs li.is-active a{color:#22c35b !important;opacity:1}.hero.is-success .tabs.is-boxed a,.hero.is-success .tabs.is-toggle a{color:#fff}.hero.is-success .tabs.is-boxed a:hover,.hero.is-success .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-success .tabs.is-boxed li.is-active a,.hero.is-success .tabs.is-boxed li.is-active a:hover,.hero.is-success .tabs.is-toggle li.is-active a,.hero.is-success .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#22c35b}.hero.is-success.is-bold{background-image:linear-gradient(141deg, #12a02c 0%, #22c35b 71%, #1fdf83 100%)}@media screen and (max-width: 768px){.hero.is-success.is-bold .navbar-menu{background-image:linear-gradient(141deg, #12a02c 0%, #22c35b 71%, #1fdf83 100%)}}.hero.is-warning{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.hero.is-warning a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-warning strong{color:inherit}.hero.is-warning .title{color:rgba(0,0,0,0.7)}.hero.is-warning .subtitle{color:rgba(0,0,0,0.9)}.hero.is-warning .subtitle a:not(.button),.hero.is-warning .subtitle strong{color:rgba(0,0,0,0.7)}@media screen and (max-width: 1055px){.hero.is-warning .navbar-menu{background-color:#ffdd57}}.hero.is-warning .navbar-item,.hero.is-warning .navbar-link{color:rgba(0,0,0,0.7)}.hero.is-warning a.navbar-item:hover,.hero.is-warning a.navbar-item.is-active,.hero.is-warning .navbar-link:hover,.hero.is-warning .navbar-link.is-active{background-color:#ffd83e;color:rgba(0,0,0,0.7)}.hero.is-warning .tabs a{color:rgba(0,0,0,0.7);opacity:0.9}.hero.is-warning .tabs a:hover{opacity:1}.hero.is-warning .tabs li.is-active a{color:#ffdd57 !important;opacity:1}.hero.is-warning .tabs.is-boxed a,.hero.is-warning .tabs.is-toggle a{color:rgba(0,0,0,0.7)}.hero.is-warning .tabs.is-boxed a:hover,.hero.is-warning .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-warning .tabs.is-boxed li.is-active a,.hero.is-warning .tabs.is-boxed li.is-active a:hover,.hero.is-warning .tabs.is-toggle li.is-active a,.hero.is-warning .tabs.is-toggle li.is-active a:hover{background-color:rgba(0,0,0,0.7);border-color:rgba(0,0,0,0.7);color:#ffdd57}.hero.is-warning.is-bold{background-image:linear-gradient(141deg, #ffae24 0%, #ffdd57 71%, #fffa71 100%)}@media screen and (max-width: 768px){.hero.is-warning.is-bold .navbar-menu{background-image:linear-gradient(141deg, #ffae24 0%, #ffdd57 71%, #fffa71 100%)}}.hero.is-danger{background-color:#da0b00;color:#fff}.hero.is-danger a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-danger strong{color:inherit}.hero.is-danger .title{color:#fff}.hero.is-danger .subtitle{color:rgba(255,255,255,0.9)}.hero.is-danger .subtitle a:not(.button),.hero.is-danger .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-danger .navbar-menu{background-color:#da0b00}}.hero.is-danger .navbar-item,.hero.is-danger .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-danger a.navbar-item:hover,.hero.is-danger a.navbar-item.is-active,.hero.is-danger .navbar-link:hover,.hero.is-danger .navbar-link.is-active{background-color:#c10a00;color:#fff}.hero.is-danger .tabs a{color:#fff;opacity:0.9}.hero.is-danger .tabs a:hover{opacity:1}.hero.is-danger .tabs li.is-active a{color:#da0b00 !important;opacity:1}.hero.is-danger .tabs.is-boxed a,.hero.is-danger .tabs.is-toggle a{color:#fff}.hero.is-danger .tabs.is-boxed a:hover,.hero.is-danger .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-danger .tabs.is-boxed li.is-active a,.hero.is-danger .tabs.is-boxed li.is-active a:hover,.hero.is-danger .tabs.is-toggle li.is-active a,.hero.is-danger .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#da0b00}.hero.is-danger.is-bold{background-image:linear-gradient(141deg, #a70013 0%, #da0b00 71%, #f43500 100%)}@media screen and (max-width: 768px){.hero.is-danger.is-bold .navbar-menu{background-image:linear-gradient(141deg, #a70013 0%, #da0b00 71%, #f43500 100%)}}.hero.is-small .hero-body,#documenter .docs-sidebar form.docs-search>input.hero .hero-body{padding:1.5rem}@media screen and (min-width: 769px),print{.hero.is-medium .hero-body{padding:9rem 4.5rem}}@media screen and (min-width: 769px),print{.hero.is-large .hero-body{padding:18rem 6rem}}.hero.is-halfheight .hero-body,.hero.is-fullheight .hero-body,.hero.is-fullheight-with-navbar .hero-body{align-items:center;display:flex}.hero.is-halfheight .hero-body>.container,.hero.is-fullheight .hero-body>.container,.hero.is-fullheight-with-navbar .hero-body>.container{flex-grow:1;flex-shrink:1}.hero.is-halfheight{min-height:50vh}.hero.is-fullheight{min-height:100vh}.hero-video{overflow:hidden}.hero-video video{left:50%;min-height:100%;min-width:100%;position:absolute;top:50%;transform:translate3d(-50%, -50%, 0)}.hero-video.is-transparent{opacity:0.3}@media screen and (max-width: 768px){.hero-video{display:none}}.hero-buttons{margin-top:1.5rem}@media screen and (max-width: 768px){.hero-buttons .button{display:flex}.hero-buttons .button:not(:last-child){margin-bottom:0.75rem}}@media screen and (min-width: 769px),print{.hero-buttons{display:flex;justify-content:center}.hero-buttons .button:not(:last-child){margin-right:1.5rem}}.hero-head,.hero-foot{flex-grow:0;flex-shrink:0}.hero-body{flex-grow:1;flex-shrink:0;padding:3rem 1.5rem}@media screen and (min-width: 769px),print{.hero-body{padding:3rem 3rem}}.section{padding:3rem 1.5rem}@media screen and (min-width: 1056px){.section{padding:3rem 3rem}.section.is-medium{padding:9rem 4.5rem}.section.is-large{padding:18rem 6rem}}.footer{background-color:#fafafa;padding:3rem 1.5rem 6rem}h1 .docs-heading-anchor,h1 .docs-heading-anchor:hover,h1 .docs-heading-anchor:visited,h2 .docs-heading-anchor,h2 .docs-heading-anchor:hover,h2 .docs-heading-anchor:visited,h3 .docs-heading-anchor,h3 .docs-heading-anchor:hover,h3 .docs-heading-anchor:visited,h4 .docs-heading-anchor,h4 .docs-heading-anchor:hover,h4 .docs-heading-anchor:visited,h5 .docs-heading-anchor,h5 .docs-heading-anchor:hover,h5 .docs-heading-anchor:visited,h6 .docs-heading-anchor,h6 .docs-heading-anchor:hover,h6 .docs-heading-anchor:visited{color:#222}h1 .docs-heading-anchor-permalink,h2 .docs-heading-anchor-permalink,h3 .docs-heading-anchor-permalink,h4 .docs-heading-anchor-permalink,h5 .docs-heading-anchor-permalink,h6 .docs-heading-anchor-permalink{visibility:hidden;vertical-align:middle;margin-left:0.5em;font-size:0.7rem}h1 .docs-heading-anchor-permalink::before,h2 .docs-heading-anchor-permalink::before,h3 .docs-heading-anchor-permalink::before,h4 .docs-heading-anchor-permalink::before,h5 .docs-heading-anchor-permalink::before,h6 .docs-heading-anchor-permalink::before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f0c1"}h1:hover .docs-heading-anchor-permalink,h2:hover .docs-heading-anchor-permalink,h3:hover .docs-heading-anchor-permalink,h4:hover .docs-heading-anchor-permalink,h5:hover .docs-heading-anchor-permalink,h6:hover .docs-heading-anchor-permalink{visibility:visible}.docs-dark-only{display:none !important}pre{position:relative;overflow:hidden}pre code,pre code.hljs{padding:0 .75rem !important;overflow:auto;display:block}pre code:first-of-type,pre code.hljs:first-of-type{padding-top:0.5rem !important}pre code:last-of-type,pre code.hljs:last-of-type{padding-bottom:0.5rem !important}pre .copy-button{opacity:0.2;transition:opacity 0.2s;position:absolute;right:0em;top:0em;padding:0.5em;width:2.5em;height:2.5em;background:transparent;border:none;font-family:"Font Awesome 6 Free";color:#222;cursor:pointer;text-align:center}pre .copy-button:focus,pre .copy-button:hover{opacity:1;background:rgba(34,34,34,0.1);color:#2e63b8}pre .copy-button.success{color:#259a12;opacity:1}pre .copy-button.error{color:#cb3c33;opacity:1}pre:hover .copy-button{opacity:1}.admonition{background-color:#b5b5b5;border-style:solid;border-width:1px;border-color:#363636;border-radius:4px;font-size:1rem}.admonition strong{color:currentColor}.admonition.is-small,#documenter .docs-sidebar form.docs-search>input.admonition{font-size:.75rem}.admonition.is-medium{font-size:1.25rem}.admonition.is-large{font-size:1.5rem}.admonition.is-default{background-color:#b5b5b5;border-color:#363636}.admonition.is-default>.admonition-header{background-color:#363636;color:#fff}.admonition.is-default>.admonition-body{color:#fff}.admonition.is-info{background-color:#def0fc;border-color:#209cee}.admonition.is-info>.admonition-header{background-color:#209cee;color:#fff}.admonition.is-info>.admonition-body{color:rgba(0,0,0,0.7)}.admonition.is-success{background-color:#bdf4d1;border-color:#22c35b}.admonition.is-success>.admonition-header{background-color:#22c35b;color:#fff}.admonition.is-success>.admonition-body{color:rgba(0,0,0,0.7)}.admonition.is-warning{background-color:#fff3c5;border-color:#ffdd57}.admonition.is-warning>.admonition-header{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.admonition.is-warning>.admonition-body{color:rgba(0,0,0,0.7)}.admonition.is-danger{background-color:#ffaba7;border-color:#da0b00}.admonition.is-danger>.admonition-header{background-color:#da0b00;color:#fff}.admonition.is-danger>.admonition-body{color:rgba(0,0,0,0.7)}.admonition.is-compat{background-color:#bdeff5;border-color:#1db5c9}.admonition.is-compat>.admonition-header{background-color:#1db5c9;color:#fff}.admonition.is-compat>.admonition-body{color:rgba(0,0,0,0.7)}.admonition-header{color:#fff;background-color:#363636;align-items:center;font-weight:700;justify-content:space-between;line-height:1.25;padding:0.5rem .75rem;position:relative}.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;margin-right:.75rem;content:"\f06a"}details.admonition.is-details>.admonition-header{list-style:none}details.admonition.is-details>.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f055"}details.admonition.is-details[open]>.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f056"}.admonition-body{color:#222;padding:0.5rem .75rem}.admonition-body pre{background-color:#f5f5f5}.admonition-body code{background-color:rgba(0,0,0,0.05)}.docstring{margin-bottom:1em;background-color:rgba(0,0,0,0);border:1px solid #dbdbdb;box-shadow:2px 2px 3px rgba(10,10,10,0.1);max-width:100%}.docstring>header{cursor:pointer;display:flex;flex-grow:1;align-items:stretch;padding:0.5rem .75rem;background-color:#f5f5f5;box-shadow:0 0.125em 0.25em rgba(10,10,10,0.1);box-shadow:none;border-bottom:1px solid #dbdbdb}.docstring>header code{background-color:transparent}.docstring>header .docstring-article-toggle-button{min-width:1.1rem;padding:0.2rem 0.2rem 0.2rem 0}.docstring>header .docstring-binding{margin-right:0.3em}.docstring>header .docstring-category{margin-left:0.3em}.docstring>section{position:relative;padding:.75rem .75rem;border-bottom:1px solid #dbdbdb}.docstring>section:last-child{border-bottom:none}.docstring>section>a.docs-sourcelink{transition:opacity 0.3s;opacity:0;position:absolute;right:.375rem;bottom:.375rem}.docstring>section>a.docs-sourcelink:focus{opacity:1 !important}.docstring:hover>section>a.docs-sourcelink{opacity:0.2}.docstring:focus-within>section>a.docs-sourcelink{opacity:0.2}.docstring>section:hover a.docs-sourcelink{opacity:1}.documenter-example-output{background-color:#fff}.outdated-warning-overlay{position:fixed;top:0;left:0;right:0;box-shadow:0 0 10px rgba(0,0,0,0.3);z-index:999;background-color:#ffaba7;color:rgba(0,0,0,0.7);border-bottom:3px solid #da0b00;padding:10px 35px;text-align:center;font-size:15px}.outdated-warning-overlay .outdated-warning-closer{position:absolute;top:calc(50% - 10px);right:18px;cursor:pointer;width:12px}.outdated-warning-overlay a{color:#2e63b8}.outdated-warning-overlay a:hover{color:#363636}.content pre{border:1px solid #dbdbdb}.content code{font-weight:inherit}.content a code{color:#2e63b8}.content h1 code,.content h2 code,.content h3 code,.content h4 code,.content h5 code,.content h6 code{color:#222}.content table{display:block;width:initial;max-width:100%;overflow-x:auto}.content blockquote>ul:first-child,.content blockquote>ol:first-child,.content .admonition-body>ul:first-child,.content .admonition-body>ol:first-child{margin-top:0}pre,code{font-variant-ligatures:no-contextual}.breadcrumb a.is-disabled{cursor:default;pointer-events:none}.breadcrumb a.is-disabled,.breadcrumb a.is-disabled:hover{color:#222}.hljs{background:initial !important}.katex .katex-mathml{top:0;right:0}.katex-display,mjx-container,.MathJax_Display{margin:0.5em 0 !important}html{-moz-osx-font-smoothing:auto;-webkit-font-smoothing:auto}li.no-marker{list-style:none}#documenter .docs-main>article{overflow-wrap:break-word}#documenter .docs-main>article .math-container{overflow-x:auto;overflow-y:hidden}@media screen and (min-width: 1056px){#documenter .docs-main{max-width:52rem;margin-left:20rem;padding-right:1rem}}@media screen and (max-width: 1055px){#documenter .docs-main{width:100%}#documenter .docs-main>article{max-width:52rem;margin-left:auto;margin-right:auto;margin-bottom:1rem;padding:0 1rem}#documenter .docs-main>header,#documenter .docs-main>nav{max-width:100%;width:100%;margin:0}}#documenter .docs-main header.docs-navbar{background-color:#fff;border-bottom:1px solid #dbdbdb;z-index:2;min-height:4rem;margin-bottom:1rem;display:flex}#documenter .docs-main header.docs-navbar .breadcrumb{flex-grow:1}#documenter .docs-main header.docs-navbar .docs-sidebar-button{display:block;font-size:1.5rem;padding-bottom:0.1rem;margin-right:1rem}#documenter .docs-main header.docs-navbar .docs-right{display:flex;white-space:nowrap;gap:1rem;align-items:center}#documenter .docs-main header.docs-navbar .docs-right .docs-icon,#documenter .docs-main header.docs-navbar .docs-right .docs-label{display:inline-block}#documenter .docs-main header.docs-navbar .docs-right .docs-label{padding:0;margin-left:0.3em}@media screen and (max-width: 1055px){#documenter .docs-main header.docs-navbar .docs-right .docs-navbar-link{margin-left:0.4rem;margin-right:0.4rem}}#documenter .docs-main header.docs-navbar>*{margin:auto 0}@media screen and (max-width: 1055px){#documenter .docs-main header.docs-navbar{position:sticky;top:0;padding:0 1rem;transition-property:top, box-shadow;-webkit-transition-property:top, box-shadow;transition-duration:0.3s;-webkit-transition-duration:0.3s}#documenter .docs-main header.docs-navbar.headroom--not-top{box-shadow:.2rem 0rem .4rem #bbb;transition-duration:0.7s;-webkit-transition-duration:0.7s}#documenter .docs-main header.docs-navbar.headroom--unpinned.headroom--not-top.headroom--not-bottom{top:-4.5rem;transition-duration:0.7s;-webkit-transition-duration:0.7s}}#documenter .docs-main section.footnotes{border-top:1px solid #dbdbdb}#documenter .docs-main section.footnotes li .tag:first-child,#documenter .docs-main section.footnotes li .docstring>section>a.docs-sourcelink:first-child,#documenter .docs-main section.footnotes li .content kbd:first-child,.content #documenter .docs-main section.footnotes li kbd:first-child{margin-right:1em;margin-bottom:0.4em}#documenter .docs-main .docs-footer{display:flex;flex-wrap:wrap;margin-left:0;margin-right:0;border-top:1px solid #dbdbdb;padding-top:1rem;padding-bottom:1rem}@media screen and (max-width: 1055px){#documenter .docs-main .docs-footer{padding-left:1rem;padding-right:1rem}}#documenter .docs-main .docs-footer .docs-footer-nextpage,#documenter .docs-main .docs-footer .docs-footer-prevpage{flex-grow:1}#documenter .docs-main .docs-footer .docs-footer-nextpage{text-align:right}#documenter .docs-main .docs-footer .flexbox-break{flex-basis:100%;height:0}#documenter .docs-main .docs-footer .footer-message{font-size:0.8em;margin:0.5em auto 0 auto;text-align:center}#documenter .docs-sidebar{display:flex;flex-direction:column;color:#0a0a0a;background-color:#f5f5f5;border-right:1px solid #dbdbdb;padding:0;flex:0 0 18rem;z-index:5;font-size:1rem;position:fixed;left:-18rem;width:18rem;height:100%;transition:left 0.3s}#documenter .docs-sidebar.visible{left:0;box-shadow:.4rem 0rem .8rem #bbb}@media screen and (min-width: 1056px){#documenter .docs-sidebar.visible{box-shadow:none}}@media screen and (min-width: 1056px){#documenter .docs-sidebar{left:0;top:0}}#documenter .docs-sidebar .docs-logo{margin-top:1rem;padding:0 1rem}#documenter .docs-sidebar .docs-logo>img{max-height:6rem;margin:auto}#documenter .docs-sidebar .docs-package-name{flex-shrink:0;font-size:1.5rem;font-weight:700;text-align:center;white-space:nowrap;overflow:hidden;padding:0.5rem 0}#documenter .docs-sidebar .docs-package-name .docs-autofit{max-width:16.2rem}#documenter .docs-sidebar .docs-package-name a,#documenter .docs-sidebar .docs-package-name a:hover{color:#0a0a0a}#documenter .docs-sidebar .docs-version-selector{border-top:1px solid #dbdbdb;display:none;padding:0.5rem}#documenter .docs-sidebar .docs-version-selector.visible{display:flex}#documenter .docs-sidebar ul.docs-menu{flex-grow:1;user-select:none;border-top:1px solid #dbdbdb;padding-bottom:1.5rem}#documenter .docs-sidebar ul.docs-menu>li>.tocitem{font-weight:bold}#documenter .docs-sidebar ul.docs-menu>li li{font-size:.95rem;margin-left:1em;border-left:1px solid #dbdbdb}#documenter .docs-sidebar ul.docs-menu input.collapse-toggle{display:none}#documenter .docs-sidebar ul.docs-menu ul.collapsed{display:none}#documenter .docs-sidebar ul.docs-menu input:checked~ul.collapsed{display:block}#documenter .docs-sidebar ul.docs-menu label.tocitem{display:flex}#documenter .docs-sidebar ul.docs-menu label.tocitem .docs-label{flex-grow:2}#documenter .docs-sidebar ul.docs-menu label.tocitem .docs-chevron{display:inline-block;font-style:normal;font-variant:normal;text-rendering:auto;line-height:1;font-size:.75rem;margin-left:1rem;margin-top:auto;margin-bottom:auto}#documenter .docs-sidebar ul.docs-menu label.tocitem .docs-chevron::before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f054"}#documenter .docs-sidebar ul.docs-menu input:checked~label.tocitem .docs-chevron::before{content:"\f078"}#documenter .docs-sidebar ul.docs-menu .tocitem{display:block;padding:0.5rem 0.5rem}#documenter .docs-sidebar ul.docs-menu .tocitem,#documenter .docs-sidebar ul.docs-menu .tocitem:hover{color:#0a0a0a;background:#f5f5f5}#documenter .docs-sidebar ul.docs-menu a.tocitem:hover,#documenter .docs-sidebar ul.docs-menu label.tocitem:hover{color:#0a0a0a;background-color:#ebebeb}#documenter .docs-sidebar ul.docs-menu li.is-active{border-top:1px solid #dbdbdb;border-bottom:1px solid #dbdbdb;background-color:#fff}#documenter .docs-sidebar ul.docs-menu li.is-active .tocitem,#documenter .docs-sidebar ul.docs-menu li.is-active .tocitem:hover{background-color:#fff;color:#0a0a0a}#documenter .docs-sidebar ul.docs-menu li.is-active ul.internal .tocitem:hover{background-color:#ebebeb;color:#0a0a0a}#documenter .docs-sidebar ul.docs-menu>li.is-active:first-child{border-top:none}#documenter .docs-sidebar ul.docs-menu ul.internal{margin:0 0.5rem 0.5rem;border-top:1px solid #dbdbdb}#documenter .docs-sidebar ul.docs-menu ul.internal li{font-size:.85rem;border-left:none;margin-left:0;margin-top:0.5rem}#documenter .docs-sidebar ul.docs-menu ul.internal .tocitem{width:100%;padding:0}#documenter .docs-sidebar ul.docs-menu ul.internal .tocitem::before{content:"⚬";margin-right:0.4em}#documenter .docs-sidebar form.docs-search{margin:auto;margin-top:0.5rem;margin-bottom:0.5rem}#documenter .docs-sidebar form.docs-search>input{width:14.4rem}#documenter .docs-sidebar #documenter-search-query{color:#707070;width:14.4rem;box-shadow:inset 0 1px 2px rgba(10,10,10,0.1)}@media screen and (min-width: 1056px){#documenter .docs-sidebar ul.docs-menu{overflow-y:auto;-webkit-overflow-scroll:touch}#documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar{width:.3rem;background:none}#documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar-thumb{border-radius:5px 0px 0px 5px;background:#e0e0e0}#documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar-thumb:hover{background:#ccc}}@media screen and (max-width: 1055px){#documenter .docs-sidebar{overflow-y:auto;-webkit-overflow-scroll:touch}#documenter .docs-sidebar::-webkit-scrollbar{width:.3rem;background:none}#documenter .docs-sidebar::-webkit-scrollbar-thumb{border-radius:5px 0px 0px 5px;background:#e0e0e0}#documenter .docs-sidebar::-webkit-scrollbar-thumb:hover{background:#ccc}}kbd.search-modal-key-hints{border-radius:0.25rem;border:1px solid rgba(0,0,0,0.6);box-shadow:0 2px 0 1px rgba(0,0,0,0.6);cursor:default;font-size:0.9rem;line-height:1.5;min-width:0.75rem;text-align:center;padding:0.1rem 0.3rem;position:relative;top:-1px}.search-min-width-50{min-width:50%}.search-min-height-100{min-height:100%}.search-modal-card-body{max-height:calc(100vh - 15rem)}.search-result-link{border-radius:0.7em;transition:all 300ms}.search-result-link:hover,.search-result-link:focus{background-color:rgba(0,128,128,0.1)}.search-result-link .property-search-result-badge,.search-result-link .search-filter{transition:all 300ms}.property-search-result-badge,.search-filter{padding:0.15em 0.5em;font-size:0.8em;font-style:italic;text-transform:none !important;line-height:1.5;color:#f5f5f5;background-color:rgba(51,65,85,0.501961);border-radius:0.6rem}.search-result-link:hover .property-search-result-badge,.search-result-link:hover .search-filter,.search-result-link:focus .property-search-result-badge,.search-result-link:focus .search-filter{color:#f1f5f9;background-color:#333}.search-filter{color:#333;background-color:#f5f5f5;transition:all 300ms}.search-filter:hover,.search-filter:focus{color:#333}.search-filter-selected{color:#f5f5f5;background-color:rgba(139,0,139,0.5)}.search-filter-selected:hover,.search-filter-selected:focus{color:#f5f5f5}.search-result-highlight{background-color:#ffdd57;color:black}.search-divider{border-bottom:1px solid #dbdbdb}.search-result-title{width:85%;color:#333}.search-result-code-title{font-size:0.875rem;font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace}#search-modal .modal-card-body::-webkit-scrollbar,#search-modal .filter-tabs::-webkit-scrollbar{height:10px;width:10px;background-color:transparent}#search-modal .modal-card-body::-webkit-scrollbar-thumb,#search-modal .filter-tabs::-webkit-scrollbar-thumb{background-color:gray;border-radius:1rem}#search-modal .modal-card-body::-webkit-scrollbar-track,#search-modal .filter-tabs::-webkit-scrollbar-track{-webkit-box-shadow:inset 0 0 6px rgba(0,0,0,0.6);background-color:transparent}.w-100{width:100%}.gap-2{gap:0.5rem}.gap-4{gap:1rem}.gap-8{gap:2rem}.ansi span.sgr1{font-weight:bolder}.ansi span.sgr2{font-weight:lighter}.ansi span.sgr3{font-style:italic}.ansi span.sgr4{text-decoration:underline}.ansi span.sgr7{color:#fff;background-color:#222}.ansi span.sgr8{color:transparent}.ansi span.sgr8 span{color:transparent}.ansi span.sgr9{text-decoration:line-through}.ansi span.sgr30{color:#242424}.ansi span.sgr31{color:#a7201f}.ansi span.sgr32{color:#066f00}.ansi span.sgr33{color:#856b00}.ansi span.sgr34{color:#2149b0}.ansi span.sgr35{color:#7d4498}.ansi span.sgr36{color:#007989}.ansi span.sgr37{color:gray}.ansi span.sgr40{background-color:#242424}.ansi span.sgr41{background-color:#a7201f}.ansi span.sgr42{background-color:#066f00}.ansi span.sgr43{background-color:#856b00}.ansi span.sgr44{background-color:#2149b0}.ansi span.sgr45{background-color:#7d4498}.ansi span.sgr46{background-color:#007989}.ansi span.sgr47{background-color:gray}.ansi span.sgr90{color:#616161}.ansi span.sgr91{color:#cb3c33}.ansi span.sgr92{color:#0e8300}.ansi span.sgr93{color:#a98800}.ansi span.sgr94{color:#3c5dcd}.ansi span.sgr95{color:#9256af}.ansi span.sgr96{color:#008fa3}.ansi span.sgr97{color:#f5f5f5}.ansi span.sgr100{background-color:#616161}.ansi span.sgr101{background-color:#cb3c33}.ansi span.sgr102{background-color:#0e8300}.ansi span.sgr103{background-color:#a98800}.ansi span.sgr104{background-color:#3c5dcd}.ansi span.sgr105{background-color:#9256af}.ansi span.sgr106{background-color:#008fa3}.ansi span.sgr107{background-color:#f5f5f5}code.language-julia-repl>span.hljs-meta{color:#066f00;font-weight:bolder}/*!
+ Theme: Default
+ Description: Original highlight.js style
+ Author: (c) Ivan Sagalaev
"
+ ],
+ "metadata": {}
+ }
+ ],
+ "nbformat_minor": 3,
+ "metadata": {
+ "language_info": {
+ "file_extension": ".jl",
+ "mimetype": "application/julia",
+ "name": "julia",
+ "version": "1.9.3"
+ },
+ "kernelspec": {
+ "name": "julia-1.9",
+ "display_name": "Julia 1.9.3",
+ "language": "julia"
+ }
+ },
+ "nbformat": 4
+}
diff --git a/previews/PR176/assets/paraviewcolormap.png b/previews/PR176/assets/paraviewcolormap.png
new file mode 100644
index 000000000..1d1073abe
Binary files /dev/null and b/previews/PR176/assets/paraviewcolormap.png differ
diff --git a/previews/PR176/assets/paraviewimport.png b/previews/PR176/assets/paraviewimport.png
new file mode 100644
index 000000000..dd6d0b1f5
Binary files /dev/null and b/previews/PR176/assets/paraviewimport.png differ
diff --git a/previews/PR176/assets/paraviewrender.jpg b/previews/PR176/assets/paraviewrender.jpg
new file mode 100644
index 000000000..975c20149
Binary files /dev/null and b/previews/PR176/assets/paraviewrender.jpg differ
diff --git a/previews/PR176/assets/paraviewvolume.png b/previews/PR176/assets/paraviewvolume.png
new file mode 100644
index 000000000..eed55ec5e
Binary files /dev/null and b/previews/PR176/assets/paraviewvolume.png differ
diff --git a/previews/PR176/assets/scripts/08_Kagome_AFM.jl b/previews/PR176/assets/scripts/08_Kagome_AFM.jl
new file mode 100644
index 000000000..5fdaecb1d
--- /dev/null
+++ b/previews/PR176/assets/scripts/08_Kagome_AFM.jl
@@ -0,0 +1,36 @@
+using Sunny, GLMakie
+
+a = b = 6.0 # (Å)
+c = 40.0
+latvecs = lattice_vectors(a, b, c, 90, 90, 120)
+crystal = Crystal(latvecs, [[1/2,0,0]], 147; types=["Cr"])
+
+S = 1
+sys = System(crystal, (3,3,1), [SpinInfo(1; S, g=2)], :dipole)
+J = 1.0
+set_exchange!(sys, J, Bond(2,3,[0,0,0]))
+
+q = -[1/3, 1/3, 0]
+axis = [0,0,1]
+set_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[cos(0),sin(0),0])
+set_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[cos(0),sin(0),0])
+set_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[cos(2π/3),sin(2π/3),0])
+plot_spins(sys; ghost_radius=30, orthographic=true)
+
+@assert energy_per_site(sys) ≈ (4/2)*J*S^2*cos(2π/3)
+
+points_rlu = [[-1/2, 0, 0], [0, 0, 0], [1/2, 1/2, 0]]
+density = 100
+path, xticks = reciprocal_space_path(crystal, points_rlu, density);
+
+swt = SpinWaveTheory(sys)
+formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)
+disp, intensity = intensities_bands(swt, path, formula);
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+ylims!(ax, -1e-1, 2.3)
+for i in axes(disp)[2]
+ lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange=(0,1e-2))
+end
+fig
diff --git a/previews/PR176/assets/scripts/15_Ba3NbFe3Si2O14.jl b/previews/PR176/assets/scripts/15_Ba3NbFe3Si2O14.jl
new file mode 100644
index 000000000..a6d7a837d
--- /dev/null
+++ b/previews/PR176/assets/scripts/15_Ba3NbFe3Si2O14.jl
@@ -0,0 +1,61 @@
+using Sunny, GLMakie
+
+a = b = 8.539 # (Å)
+c = 5.2414
+latvecs = lattice_vectors(a, b, c, 90, 90, 120)
+types = ["Fe","Nb","Ba","Si","O","O","O"]
+positions = [[0.24964,0,0.5],[0,0,0],[0.56598,0,0],[2/3,1/3,0.5220],[2/3,1/3,0.2162],[0.5259,0.7024,0.3536],[0.7840,0.9002,0.7760]]
+langasite = Crystal(latvecs, positions, 150; types)
+crystal = subcrystal(langasite, "Fe")
+view_crystal(crystal, 7)
+
+latsize = (1,1,7)
+S = 5/2
+seed = 5
+sys = System(crystal, latsize, [SpinInfo(1; S, g=2)], :dipole; seed)
+
+J₁ = 0.85
+J₂ = 0.24
+J₃ = 0.053
+J₄ = 0.017
+J₅ = 0.24
+set_exchange!(sys, J₁, Bond(3, 2, [1,1,0]))
+set_exchange!(sys, J₄, Bond(1, 1, [0,0,1]))
+set_exchange!(sys, J₂, Bond(1, 3, [0,0,0]))
+
+ϵD = -1
+ϵH = +1
+ϵT = ϵD * ϵH
+
+if ϵT == -1
+ set_exchange!(sys, J₃, Bond(2, 3, [-1,-1,1]))
+ set_exchange!(sys, J₅, Bond(3, 2, [1,1,1]))
+elseif ϵT == 1
+ set_exchange!(sys, J₅, Bond(2, 3, [-1,-1,1]))
+ set_exchange!(sys, J₃, Bond(3, 2, [1,1,1]))
+else
+ throw("Provide a valid chirality")
+end
+
+q = [0, 0, 1/7]
+axis = [0,0,1]
+set_spiral_order_on_sublattice!(sys, 1; q, axis, S0=[1, 0, 0])
+set_spiral_order_on_sublattice!(sys, 2; q, axis, S0=[-1/2, -sqrt(3)/2, 0])
+set_spiral_order_on_sublattice!(sys, 3; q, axis, S0=[-1/2, +sqrt(3)/2, 0])
+
+plot_spins(sys; color=[s[1] for s in sys.dipoles])
+
+points_rlu = [[0,1,-1],[0,1,-1+1],[0,1,-1+2],[0,1,-1+3]];
+density = 100
+path, xticks = reciprocal_space_path(crystal, points_rlu, density);
+
+swt = SpinWaveTheory(sys)
+γ = 0.15 # width in meV
+broadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))
+energies = collect(0:0.01:6) # 0 < ω < 6 (meV).
+is = intensities_broadened(swt, path, energies, broadened_formula);
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+heatmap!(ax, 1:size(is,1), energies, is, colorrange=(0,5))
+fig
diff --git a/previews/PR176/assets/scripts/fei2_classical.jl b/previews/PR176/assets/scripts/fei2_classical.jl
new file mode 100644
index 000000000..a6940adbd
--- /dev/null
+++ b/previews/PR176/assets/scripts/fei2_classical.jl
@@ -0,0 +1,250 @@
+using Sunny, LinearAlgebra, GLMakie
+
+a = b = 4.05012#hide
+c = 6.75214#hide
+latvecs = lattice_vectors(a, b, c, 90, 90, 120)#hide
+positions = [[0,0,0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]]#hide
+types = ["Fe", "I", "I"]#hide
+FeI2 = Crystal(latvecs, positions; types)#hide
+cryst = subcrystal(FeI2, "Fe")#hide
+sys = System(cryst, (4,4,4), [SpinInfo(1,S=1,g=2)], :SUN, seed=2)#hide
+J1pm = -0.236#hide
+J1pmpm = -0.161#hide
+J1zpm = -0.261#hide
+J2pm = 0.026#hide
+J3pm = 0.166#hide
+J′0pm = 0.037#hide
+J′1pm = 0.013#hide
+J′2apm = 0.068#hide
+J1zz = -0.236#hide
+J2zz = 0.113#hide
+J3zz = 0.211#hide
+J′0zz = -0.036#hide
+J′1zz = 0.051#hide
+J′2azz = 0.073#hide
+J1xx = J1pm + J1pmpm#hide
+J1yy = J1pm - J1pmpm#hide
+J1yz = J1zpm#hide
+set_exchange!(sys, [J1xx 0.0 0.0; 0.0 J1yy J1yz; 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))#hide
+set_exchange!(sys, [J2pm 0.0 0.0; 0.0 J2pm 0.0; 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))#hide
+set_exchange!(sys, [J3pm 0.0 0.0; 0.0 J3pm 0.0; 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))#hide
+set_exchange!(sys, [J′0pm 0.0 0.0; 0.0 J′0pm 0.0; 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))#hide
+set_exchange!(sys, [J′1pm 0.0 0.0; 0.0 J′1pm 0.0; 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))#hide
+set_exchange!(sys, [J′2apm 0.0 0.0; 0.0 J′2apm 0.0; 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))#hide
+D = 2.165#hide
+S = spin_operators(sys, 1)#hide
+set_onsite_coupling!(sys, -D*S[3]^2, 1)#hide
+sys
+
+Δt = 0.05/D # Should be inversely proportional to the largest energy scale
+ # in the system. For FeI2, this is the easy-axis anisotropy,
+ # `D = 2.165` (meV). The prefactor 0.05 is relatively small,
+ # and achieves high accuracy.
+kT = 0.2 # Temperature of the thermal bath (meV).
+λ = 0.1 # This value is typically good for Monte Carlo sampling,
+ # independent of system details.
+
+langevin = Langevin(Δt; kT, λ);
+
+randomize_spins!(sys)
+for _ in 1:20_000
+ step!(sys, langevin)
+end
+
+plot_spins(sys; color=[s[3] for s in sys.dipoles])
+
+sys_large = resize_supercell(sys, (16,16,4)) # 16x16x4 copies of the original unit cell
+plot_spins(sys_large; color=[s[3] for s in sys_large.dipoles])
+
+kT = 3.5 * meV_per_K # 3.5K ≈ 0.30 meV
+langevin.kT = kT
+for _ in 1:10_000
+ step!(sys_large, langevin)
+end
+
+sc = dynamical_correlations(sys_large; Δt=2Δt, nω=120, ωmax=7.5)
+
+add_sample!(sc, sys_large) # Accumulate the sample into `sc`
+
+for _ in 1:2
+ for _ in 1:1000 # Enough steps to decorrelate spins
+ step!(sys_large, langevin)
+ end
+ add_sample!(sc, sys_large) # Accumulate the sample into `sc`
+end
+
+sc
+
+formula = intensity_formula(sc, :trace; kT)
+
+qs = [[0, 0, 0], [0.5, 0.5, 0.5]]
+is = intensities_interpolated(sc, qs, formula; interpolation = :round)
+
+ωs = available_energies(sc)
+fig = lines(ωs, is[1,:]; axis=(xlabel="meV", ylabel="Intensity"), label="(0,0,0)")
+lines!(ωs, is[2,:]; label="(π,π,π)")
+axislegend()
+fig
+
+formfactors = [FormFactor("Fe2"; g_lande=3/2)]
+new_formula = intensity_formula(sc, :perp; kT, formfactors = formfactors)
+
+points = [[0, 0, 0], # List of wave vectors that define a path
+ [1, 0, 0],
+ [0, 1, 0],
+ [1/2, 0, 0],
+ [0, 1, 0],
+ [0, 0, 0]]
+density = 40
+path, xticks = reciprocal_space_path(cryst, points, density);
+
+is_interpolated = intensities_interpolated(sc, path, new_formula;
+ interpolation = :linear, # Interpolate between available wave vectors
+);
+# Add artificial broadening
+is_interpolated_broadened = broaden_energy(sc, is, (ω, ω₀)->lorentzian(ω-ω₀, 0.05));
+
+cut_width = 0.3
+density = 15
+paramsList, markers, ranges = reciprocal_space_path_bins(sc,points,density,cut_width);
+
+total_bins = ranges[end][end]
+energy_bins = paramsList[1].numbins[4]
+is_binned = zeros(Float64,total_bins,energy_bins)
+integrated_kernel = integrated_lorentzian(0.05) # Lorentzian broadening
+for k in eachindex(paramsList)
+ bin_data, counts = intensities_binned(sc,paramsList[k], new_formula;
+ integrated_kernel = integrated_kernel
+ )
+ is_binned[ranges[k],:] = bin_data[:,1,1,:] ./ counts[:,1,1,:]
+end
+
+fig = Figure()
+ax_top = Axis(fig[1,1],ylabel = "meV",xticklabelrotation=π/8,xticklabelsize=12;xticks)
+ax_bottom = Axis(fig[2,1],ylabel = "meV",xticks = (markers, string.(points)),xticklabelrotation=π/8,xticklabelsize=12)
+
+heatmap!(ax_top,1:size(is_interpolated,1), ωs, is_interpolated;
+ colorrange=(0.0,0.07),
+)
+
+heatmap!(ax_bottom,1:size(is_binned,1), ωs, is_binned;
+ colorrange=(0.0,0.05),
+)
+
+fig
+
+ωidx = 60
+target_ω = ωs[ωidx]
+
+params = unit_resolution_binning_parameters(sc)
+params.binstart[1:2] .= -1 # Expand plot range slightly
+
+# Set energy integration range
+omega_width = 0.3
+params.binstart[4] = target_ω - (omega_width/2)
+params.binend[4] = target_ω # `binend` should be inside (e.g. at the center) of the range
+params.binwidth[4] = omega_width
+
+integrate_axes!(params, axes = 3) # Integrate out z direction entirely
+
+params
+
+is, counts = intensities_binned(sc,params,new_formula)
+
+fig = Figure()
+ax = Axis(fig[1,1];
+ title="Δω=0.3 meV (Binned)", aspect=true,
+ xlabel = "[H, 0, 0]",
+ ylabel = "[0, K, 0]"
+)
+bcs = axes_bincenters(params)
+hm = heatmap!(ax,bcs[1],bcs[2],is[:,:,1,1] ./ counts[:,:,1,1])
+function add_lines!(ax,params)#hide
+ bes = Sunny.axes_binedges(params)#hide
+ hrange = range(-2,2,length=17)#hide
+ linesegments!(ax,[(Point2f(params.covectors[1,1:3] ⋅ [h,-10,0],params.covectors[2,1:3] ⋅ [h,-10,0]),Point2f(params.covectors[1,1:3] ⋅ [h,10,0],params.covectors[2,1:3] ⋅ [h,10,0])) for h = hrange],linestyle=:dash,color=:black)#hide
+ krange = range(-2,2,length=17)#hide
+ linesegments!(ax,[(Point2f(params.covectors[1,1:3] ⋅ [-10,k,0],params.covectors[2,1:3] ⋅ [-10,k,0]),Point2f(params.covectors[1,1:3] ⋅ [10,k,0],params.covectors[2,1:3] ⋅ [10,k,0])) for k = krange],linestyle=:dash,color=:black)#hide
+ xlims!(ax,bes[1][1],bes[1][end])#hide
+ ylims!(ax,bes[2][1],bes[2][end])#hide
+end#hide
+add_lines!(ax,params)
+Colorbar(fig[1,2], hm);
+fig
+
+latvecs = sys.crystal.latvecs
+metric = latvecs' * I(3) * latvecs
+
+(latvecs * [1/2,1,0]) ⋅ (latvecs * [1,0,0]) == 0
+
+f = Figure()#hide
+ax = Axis(f[1,1])#hide
+arrows!(ax,[Point2f(0,0),Point2f(latvecs[1:2,1] ./ 2)],[Vec2f(latvecs[1:2,1] ./ 2), Vec2f(latvecs[1:2,2])],arrowcolor = :blue,arrowsize = 30.,linewidth = 5.,linecolor = :blue)#hide
+arrows!(ax,[Point2f(0,0)],[Vec2f(latvecs[1:2,:] * [1/2,1,0])],arrowcolor = :red,arrowsize = 30.,linewidth = 5.,linecolor = :red, linestyle = :dash)#hide
+scatter!(ax,[Point2f(latvecs[1:2,:] * [a,b,0]) for a in -1:1, b in -1:1][:],color = :black)#hide
+annotations!(ax,["0","0+b","0+a", "a/2", "b"],[Point2f(0,-0.3),Point2f(latvecs[1:2,2]) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1]) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1] ./ 4) .- Vec2f(0,0.3),Point2f(latvecs[1:2,1] ./ 2) .+ Vec2f(latvecs[1:2,2] ./ 2) .+ Vec2f(0.3,0.3)],color=[:black,:black,:black,:blue,:blue])#hide
+f#hide
+
+params.covectors[2,1:3] = [1/2,1,0] # [1/2,1,0] times [a;b;c] is (a/2 + b)
+params#hide
+
+# Zoom out horizontal axis
+params.binstart[1], params.binend[1] = -2, 2
+
+# Adjust vertical axis bounds to account for
+# length of a/2 + b
+params.binstart[2], params.binend[2] = -2 * sqrt(3/4), 2 * sqrt(3/4)
+
+# Re-compute in the new coordinate system
+is, counts = intensities_binned(sc,params,new_formula)
+
+fig = Figure(; resolution=(1200,500))#hide
+ax_right = Axis(fig[1,3];#hide
+ title="ω≈$(round(target_ω, digits=2)) meV with Δω=0.3 meV (Binned)", aspect=true,#hide
+ xlabel = "[H, -1/2H, 0]"#hide
+)#hide
+bcs = axes_bincenters(params)#hide
+hm_right = heatmap!(ax_right,bcs[1],bcs[2],is[:,:,1,1] ./ counts[:,:,1,1])#hide
+add_lines!(ax_right,params)
+Colorbar(fig[1,4], hm_right);#hide
+
+# New basis matrix
+A = [1 0 0
+ -1/2 1 0
+ 0 0 1]
+
+# Define our grid of wave vectors
+npoints = 60
+as = range(-2, 2, npoints)
+bs = range(-3/√3, 3/√3, npoints)
+qs_ortho = [[a, b, 0] for a in as, b in bs]
+
+# Convert to original RLU system for input to Sunny
+qs = [A * q for q in qs_ortho]
+
+# Use interpolation to get intensities
+is = intensities_interpolated(sc, qs, new_formula; interpolation=:linear)
+
+ax_left = Axis(fig[1,2];#hide
+ title="ω≈$(round(ωs[ωidx], digits=2)) meV (Interpolated)", aspect=true,#hide
+ xlabel = "[H, -1/2H, 0]", ylabel = "[0, K, 0]"#hide
+)#hide
+hm_left = heatmap!(ax_left, as, bs, is[:,:,ωidx])#hide
+add_lines!(ax_left,params)
+Colorbar(fig[1,1], hm_left);#hide
+fig
+
+metric = (latvecs * inv(A'))' * I(3) * (latvecs * inv(A'))
+
+is_static = instant_intensities_interpolated(sc, qs, new_formula; interpolation = :linear)
+
+hm = heatmap(as, bs, is_static;
+ axis=(
+ title="Instantaneous Structure Factor",
+ xlabel = "[H, -1/2H, 0]",
+ ylabel = "[0, K, 0]",
+ aspect=true
+ )
+)
+Colorbar(hm.figure[1,2], hm.plot)
+hm
diff --git a/previews/PR176/assets/scripts/fei2_tutorial.jl b/previews/PR176/assets/scripts/fei2_tutorial.jl
new file mode 100644
index 000000000..5d1b97d8a
--- /dev/null
+++ b/previews/PR176/assets/scripts/fei2_tutorial.jl
@@ -0,0 +1,118 @@
+using Sunny, GLMakie
+
+a = b = 4.05012 # Lattice constants for triangular lattice
+c = 6.75214 # Spacing in the z-direction
+
+latvecs = lattice_vectors(a, b, c, 90, 90, 120) # A 3x3 matrix of lattice vectors that
+ # define the conventional unit cell
+positions = [[0, 0, 0], [1/3, 2/3, 1/4], [2/3, 1/3, 3/4]] # Positions of atoms in fractions
+ # of lattice vectors
+types = ["Fe", "I", "I"]
+FeI2 = Crystal(latvecs, positions; types)
+
+cryst = subcrystal(FeI2, "Fe")
+
+view_crystal(cryst, 8.0)
+
+print_symmetry_table(cryst, 8.0)
+
+sys = System(cryst, (4, 4, 4), [SpinInfo(1, S=1, g=2)], :SUN, seed=2)
+
+J1pm = -0.236
+J1pmpm = -0.161
+J1zpm = -0.261
+J2pm = 0.026
+J3pm = 0.166
+J′0pm = 0.037
+J′1pm = 0.013
+J′2apm = 0.068
+
+J1zz = -0.236
+J2zz = 0.113
+J3zz = 0.211
+J′0zz = -0.036
+J′1zz = 0.051
+J′2azz = 0.073
+
+J1xx = J1pm + J1pmpm
+J1yy = J1pm - J1pmpm
+J1yz = J1zpm
+
+set_exchange!(sys, [J1xx 0.0 0.0;
+ 0.0 J1yy J1yz;
+ 0.0 J1yz J1zz], Bond(1,1,[1,0,0]))
+set_exchange!(sys, [J2pm 0.0 0.0;
+ 0.0 J2pm 0.0;
+ 0.0 0.0 J2zz], Bond(1,1,[1,2,0]))
+set_exchange!(sys, [J3pm 0.0 0.0;
+ 0.0 J3pm 0.0;
+ 0.0 0.0 J3zz], Bond(1,1,[2,0,0]))
+set_exchange!(sys, [J′0pm 0.0 0.0;
+ 0.0 J′0pm 0.0;
+ 0.0 0.0 J′0zz], Bond(1,1,[0,0,1]))
+set_exchange!(sys, [J′1pm 0.0 0.0;
+ 0.0 J′1pm 0.0;
+ 0.0 0.0 J′1zz], Bond(1,1,[1,0,1]))
+set_exchange!(sys, [J′2apm 0.0 0.0;
+ 0.0 J′2apm 0.0;
+ 0.0 0.0 J′2azz], Bond(1,1,[1,2,1]))
+
+D = 2.165
+S = spin_operators(sys, 1)
+set_onsite_coupling!(sys, -D*S[3]^2, 1)
+
+randomize_spins!(sys)
+minimize_energy!(sys)
+
+plot_spins(sys; color=[s[3] for s in sys.dipoles])
+
+print_wrapped_intensities(sys)
+
+suggest_magnetic_supercell([[0, -1/4, 1/4]])
+
+sys_min = reshape_supercell(sys, [1 0 0; 0 2 1; 0 -2 1])
+randomize_spins!(sys_min)
+minimize_energy!(sys_min);
+
+plot_spins(sys_min; color=[s[3] for s in sys_min.dipoles], ghost_radius=12)
+
+swt = SpinWaveTheory(sys_min)
+
+q_points = [[0,0,0], [1,0,0], [0,1,0], [1/2,0,0], [0,1,0], [0,0,0]];
+
+density = 50
+path, xticks = reciprocal_space_path(cryst, q_points, density);
+
+disp = dispersion(swt, path);
+
+formula = intensity_formula(swt, :perp; kernel=delta_function_kernel)
+
+disp, intensity = intensities_bands(swt, path, formula);
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+ylims!(ax, 0.0, 7.5)
+xlims!(ax, 1, size(disp, 1))
+colorrange = extrema(intensity)
+for i in axes(disp)[2]
+ lines!(ax, 1:length(disp[:,i]), disp[:,i]; color=intensity[:,i], colorrange)
+end
+fig
+
+γ = 0.15 # width in meV
+broadened_formula = intensity_formula(swt, :perp; kernel=lorentzian(γ))
+
+energies = collect(0:0.01:10) # 0 < ω < 10 (meV).
+is1 = intensities_broadened(swt, path, energies, broadened_formula);
+
+R = rotation_in_rlu(cryst, [0, 0, 1], 2π/3)
+is2 = intensities_broadened(swt, [R*q for q in path], energies, broadened_formula)
+is3 = intensities_broadened(swt, [R*R*q for q in path], energies, broadened_formula)
+is_averaged = (is1 + is2 + is3) / 3
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Momentum (r.l.u.)", ylabel="Energy (meV)", xticks, xticklabelrotation=π/6)
+heatmap!(ax, 1:size(is_averaged, 1), energies, is_averaged)
+fig
+
+disp, is = dssf(swt, path);
diff --git a/previews/PR176/assets/scripts/ising2d.jl b/previews/PR176/assets/scripts/ising2d.jl
new file mode 100644
index 000000000..6ab158eb6
--- /dev/null
+++ b/previews/PR176/assets/scripts/ising2d.jl
@@ -0,0 +1,24 @@
+using Sunny, GLMakie
+
+a = 1
+latvecs = lattice_vectors(a,a,10a,90,90,90)
+crystal = Crystal(latvecs, [[0,0,0]])
+
+L = 128
+sys = System(crystal, (L,L,1), [SpinInfo(1, S=1, g=1)], :dipole, units=Units.theory, seed=0)
+polarize_spins!(sys, (0,0,1))
+
+set_exchange!(sys, -1.0, Bond(1,1,(1,0,0)))
+
+B = 0
+set_external_field!(sys, (0,0,B))
+
+Tc = 2/log(1+√2)
+
+nsweeps = 4000
+sampler = LocalSampler(kT=Tc, propose=propose_flip)
+for i in 1:nsweeps
+ step!(sys, sampler)
+end
+
+heatmap(reshape([s.z for s in sys.dipoles], (L,L)))
diff --git a/previews/PR176/assets/scripts/out_of_equilibrium.jl b/previews/PR176/assets/scripts/out_of_equilibrium.jl
new file mode 100644
index 000000000..60ce32a36
--- /dev/null
+++ b/previews/PR176/assets/scripts/out_of_equilibrium.jl
@@ -0,0 +1,64 @@
+using Sunny, GLMakie
+
+lat_vecs = lattice_vectors(1.0, 1.0, 2.0, 90, 90, 120)
+basis_vecs = [[0,0,0]]
+cryst = Crystal(lat_vecs, basis_vecs)
+
+L = 40
+dims = (L, L, 1)
+sys = System(cryst, dims, [SpinInfo(1, S=1, g=1)], :SUN; seed=101, units=Units.theory)
+
+J1 = -1 # Nearest-neighbor ferromagnetic
+J2 = (2.0/(1+√5)) # Tune competing exchange to set skyrmion scale length
+Δ = 2.6 # Exchange anisotropy
+
+ex1 = J1 * [1.0 0.0 0.0;
+ 0.0 1.0 0.0;
+ 0.0 0.0 Δ]
+ex2 = J2 * [1.0 0.0 0.0;
+ 0.0 1.0 0.0;
+ 0.0 0.0 Δ]
+set_exchange!(sys, ex1, Bond(1, 1, [1, 0, 0]))
+set_exchange!(sys, ex2, Bond(1, 1, [1, 2, 0]))
+
+h = 15.5
+field = set_external_field!(sys, [0.0 0.0 h])
+
+D = 19.0
+Sz = spin_operators(sys, 1)[3]
+set_onsite_coupling!(sys, D*Sz^2, 1)
+
+randomize_spins!(sys)
+
+Δt = 0.2/D # Integration time step (inverse meV). Typically this will be
+ # inversely proportional to the largest energy scale in the
+ # system. We can use a fairly large time-step here because
+ # accuracy isn't critical.
+kT = 0 # Target equilibrium temperature (meV)
+λ = 0.1 # Magnitude of coupling to thermal bath (dimensionless)
+integrator = Langevin(Δt; kT, λ)
+
+τs = [4., 16, 256] # Times to record snapshots
+frames = [] # Empty array to store snapshots
+for i in eachindex(τs)
+ dur = i == 1 ? τs[1] : τs[i] - τs[i-1] # Determine the length of time to simulate
+ numsteps = round(Int, dur/Δt)
+ for _ in 1:numsteps # Perform the integration
+ step!(sys, integrator)
+ end
+ push!(frames, copy(sys.coherents)) # Save a snapshot spin configuration
+end
+
+include(pkgdir(Sunny, "examples", "extra", "plotting2d.jl"))
+
+function sun_berry_curvature(z₁, z₂, z₃)
+ z₁, z₂, z₃ = normalize.((z₁, z₂, z₃))
+ n₁ = z₁ ⋅ z₂
+ n₂ = z₂ ⋅ z₃
+ n₃ = z₃ ⋅ z₁
+ return angle(n₁ * n₂ * n₃)
+end
+
+plot_triangular_plaquettes(sun_berry_curvature, frames; resolution=(1800,600),
+ offset_spacing=10, texts = ["\tt = "*string(τ) for τ in τs], text_offset = (0.0, 6.0)
+)
diff --git a/previews/PR176/assets/scripts/powder_averaging.jl b/previews/PR176/assets/scripts/powder_averaging.jl
new file mode 100644
index 000000000..5e18d9af5
--- /dev/null
+++ b/previews/PR176/assets/scripts/powder_averaging.jl
@@ -0,0 +1,57 @@
+using Sunny, GLMakie
+
+a = 8.5031 # (Å)
+latvecs = lattice_vectors(a, a, a, 90, 90, 90)
+cryst = Crystal(latvecs, [[0,0,0]], 227, setting="1")
+
+view_crystal(cryst, 8.0)
+
+latsize = (2, 2, 2)
+seed = 0
+S = 3/2
+J = 7.5413*meV_per_K # (~ 0.65 meV)
+sys = System(cryst, latsize, [SpinInfo(1; S, g=2)], :dipole; seed=0)
+set_exchange!(sys, J, Bond(1, 3, [0,0,0]))
+
+randomize_spins!(sys)
+minimize_energy!(sys)
+
+@assert energy_per_site(sys) ≈ -2J*S^2
+
+s0 = sys.dipoles[1,1,1,1]
+plot_spins(sys; color=[s'*s0 for s in sys.dipoles])
+
+shape = cryst.latvecs \ cryst.prim_latvecs
+sys_prim = reshape_supercell(sys, shape)
+@assert energy_per_site(sys_prim) ≈ -2J*S^2
+
+swt = SpinWaveTheory(sys_prim)
+η = 0.4 # (meV)
+kernel = lorentzian(η)
+formfactors = [FormFactor("Co2")]
+formula = intensity_formula(swt, :perp; kernel, formfactors)
+
+qpoints = [[0.0, 0.0, 0.0], [0.5, 0.0, 0.0], [0.5, 0.5, 0.0], [0.0, 0.0, 0.0]]
+path, xticks = reciprocal_space_path(cryst, qpoints, 50)
+energies = collect(0:0.01:6)
+is = intensities_broadened(swt, path, energies, formula)
+
+fig = Figure()
+ax = Axis(fig[1,1]; aspect=1.4, ylabel="ω (meV)", xlabel="𝐪 (r.l.u.)",
+ xticks, xticklabelrotation=π/10)
+heatmap!(ax, 1:size(is, 1), energies, is, colormap=:gnuplot2)
+fig
+
+radii = 0.01:0.02:3 # (1/Å)
+output = zeros(Float64, length(radii), length(energies))
+for (i, radius) in enumerate(radii)
+ n = 300
+ qs = reciprocal_space_shell(cryst, radius, n)
+ is = intensities_broadened(swt, qs, energies, formula)
+ output[i, :] = sum(is, dims=1) / size(is, 1)
+end
+
+fig = Figure()
+ax = Axis(fig[1,1]; xlabel="Q (Å⁻¹)", ylabel="ω (meV)")
+heatmap!(ax, radii, energies, output, colormap=:gnuplot2)
+fig
diff --git a/previews/PR176/assets/themes/documenter-dark.css b/previews/PR176/assets/themes/documenter-dark.css
new file mode 100644
index 000000000..691b83abd
--- /dev/null
+++ b/previews/PR176/assets/themes/documenter-dark.css
@@ -0,0 +1,7 @@
+html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-ellipsis,html.theme--documenter-dark .file-cta,html.theme--documenter-dark .file-name,html.theme--documenter-dark .select select,html.theme--documenter-dark .textarea,html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark .button{-moz-appearance:none;-webkit-appearance:none;align-items:center;border:1px solid transparent;border-radius:.4em;box-shadow:none;display:inline-flex;font-size:1rem;height:2.5em;justify-content:flex-start;line-height:1.5;padding-bottom:calc(0.5em - 1px);padding-left:calc(0.75em - 1px);padding-right:calc(0.75em - 1px);padding-top:calc(0.5em - 1px);position:relative;vertical-align:top}html.theme--documenter-dark .pagination-previous:focus,html.theme--documenter-dark .pagination-next:focus,html.theme--documenter-dark .pagination-link:focus,html.theme--documenter-dark .pagination-ellipsis:focus,html.theme--documenter-dark .file-cta:focus,html.theme--documenter-dark .file-name:focus,html.theme--documenter-dark .select select:focus,html.theme--documenter-dark .textarea:focus,html.theme--documenter-dark .input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:focus,html.theme--documenter-dark .button:focus,html.theme--documenter-dark .is-focused.pagination-previous,html.theme--documenter-dark .is-focused.pagination-next,html.theme--documenter-dark .is-focused.pagination-link,html.theme--documenter-dark .is-focused.pagination-ellipsis,html.theme--documenter-dark .is-focused.file-cta,html.theme--documenter-dark .is-focused.file-name,html.theme--documenter-dark .select select.is-focused,html.theme--documenter-dark .is-focused.textarea,html.theme--documenter-dark .is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-focused.button,html.theme--documenter-dark .pagination-previous:active,html.theme--documenter-dark .pagination-next:active,html.theme--documenter-dark .pagination-link:active,html.theme--documenter-dark .pagination-ellipsis:active,html.theme--documenter-dark .file-cta:active,html.theme--documenter-dark .file-name:active,html.theme--documenter-dark .select select:active,html.theme--documenter-dark .textarea:active,html.theme--documenter-dark .input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:active,html.theme--documenter-dark .button:active,html.theme--documenter-dark .is-active.pagination-previous,html.theme--documenter-dark .is-active.pagination-next,html.theme--documenter-dark .is-active.pagination-link,html.theme--documenter-dark .is-active.pagination-ellipsis,html.theme--documenter-dark .is-active.file-cta,html.theme--documenter-dark .is-active.file-name,html.theme--documenter-dark .select select.is-active,html.theme--documenter-dark .is-active.textarea,html.theme--documenter-dark .is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active,html.theme--documenter-dark .is-active.button{outline:none}html.theme--documenter-dark .pagination-previous[disabled],html.theme--documenter-dark .pagination-next[disabled],html.theme--documenter-dark .pagination-link[disabled],html.theme--documenter-dark .pagination-ellipsis[disabled],html.theme--documenter-dark .file-cta[disabled],html.theme--documenter-dark .file-name[disabled],html.theme--documenter-dark .select select[disabled],html.theme--documenter-dark .textarea[disabled],html.theme--documenter-dark .input[disabled],html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled],html.theme--documenter-dark .button[disabled],fieldset[disabled] html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark fieldset[disabled] .pagination-previous,fieldset[disabled] html.theme--documenter-dark .pagination-next,html.theme--documenter-dark fieldset[disabled] .pagination-next,fieldset[disabled] html.theme--documenter-dark .pagination-link,html.theme--documenter-dark fieldset[disabled] .pagination-link,fieldset[disabled] html.theme--documenter-dark .pagination-ellipsis,html.theme--documenter-dark fieldset[disabled] .pagination-ellipsis,fieldset[disabled] html.theme--documenter-dark .file-cta,html.theme--documenter-dark fieldset[disabled] .file-cta,fieldset[disabled] html.theme--documenter-dark .file-name,html.theme--documenter-dark fieldset[disabled] .file-name,fieldset[disabled] html.theme--documenter-dark .select select,fieldset[disabled] html.theme--documenter-dark .textarea,fieldset[disabled] html.theme--documenter-dark .input,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark fieldset[disabled] .select select,html.theme--documenter-dark .select fieldset[disabled] select,html.theme--documenter-dark fieldset[disabled] .textarea,html.theme--documenter-dark fieldset[disabled] .input,html.theme--documenter-dark fieldset[disabled] #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar fieldset[disabled] form.docs-search>input,fieldset[disabled] html.theme--documenter-dark .button,html.theme--documenter-dark fieldset[disabled] .button{cursor:not-allowed}html.theme--documenter-dark .tabs,html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-ellipsis,html.theme--documenter-dark .breadcrumb,html.theme--documenter-dark .file,html.theme--documenter-dark .button,.is-unselectable{-webkit-touch-callout:none;-webkit-user-select:none;-moz-user-select:none;-ms-user-select:none;user-select:none}html.theme--documenter-dark .navbar-link:not(.is-arrowless)::after,html.theme--documenter-dark .select:not(.is-multiple):not(.is-loading)::after{border:3px solid rgba(0,0,0,0);border-radius:2px;border-right:0;border-top:0;content:" ";display:block;height:0.625em;margin-top:-0.4375em;pointer-events:none;position:absolute;top:50%;transform:rotate(-45deg);transform-origin:center;width:0.625em}html.theme--documenter-dark .admonition:not(:last-child),html.theme--documenter-dark .tabs:not(:last-child),html.theme--documenter-dark .pagination:not(:last-child),html.theme--documenter-dark .message:not(:last-child),html.theme--documenter-dark .level:not(:last-child),html.theme--documenter-dark .breadcrumb:not(:last-child),html.theme--documenter-dark .block:not(:last-child),html.theme--documenter-dark .title:not(:last-child),html.theme--documenter-dark .subtitle:not(:last-child),html.theme--documenter-dark .table-container:not(:last-child),html.theme--documenter-dark .table:not(:last-child),html.theme--documenter-dark .progress:not(:last-child),html.theme--documenter-dark .notification:not(:last-child),html.theme--documenter-dark .content:not(:last-child),html.theme--documenter-dark .box:not(:last-child){margin-bottom:1.5rem}html.theme--documenter-dark .modal-close,html.theme--documenter-dark .delete{-webkit-touch-callout:none;-webkit-user-select:none;-moz-user-select:none;-ms-user-select:none;user-select:none;-moz-appearance:none;-webkit-appearance:none;background-color:rgba(10,10,10,0.2);border:none;border-radius:9999px;cursor:pointer;pointer-events:auto;display:inline-block;flex-grow:0;flex-shrink:0;font-size:0;height:20px;max-height:20px;max-width:20px;min-height:20px;min-width:20px;outline:none;position:relative;vertical-align:top;width:20px}html.theme--documenter-dark .modal-close::before,html.theme--documenter-dark .delete::before,html.theme--documenter-dark .modal-close::after,html.theme--documenter-dark .delete::after{background-color:#fff;content:"";display:block;left:50%;position:absolute;top:50%;transform:translateX(-50%) translateY(-50%) rotate(45deg);transform-origin:center center}html.theme--documenter-dark .modal-close::before,html.theme--documenter-dark .delete::before{height:2px;width:50%}html.theme--documenter-dark .modal-close::after,html.theme--documenter-dark .delete::after{height:50%;width:2px}html.theme--documenter-dark .modal-close:hover,html.theme--documenter-dark .delete:hover,html.theme--documenter-dark .modal-close:focus,html.theme--documenter-dark .delete:focus{background-color:rgba(10,10,10,0.3)}html.theme--documenter-dark .modal-close:active,html.theme--documenter-dark .delete:active{background-color:rgba(10,10,10,0.4)}html.theme--documenter-dark .is-small.modal-close,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.modal-close,html.theme--documenter-dark .is-small.delete,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.delete{height:16px;max-height:16px;max-width:16px;min-height:16px;min-width:16px;width:16px}html.theme--documenter-dark .is-medium.modal-close,html.theme--documenter-dark .is-medium.delete{height:24px;max-height:24px;max-width:24px;min-height:24px;min-width:24px;width:24px}html.theme--documenter-dark .is-large.modal-close,html.theme--documenter-dark .is-large.delete{height:32px;max-height:32px;max-width:32px;min-height:32px;min-width:32px;width:32px}html.theme--documenter-dark .control.is-loading::after,html.theme--documenter-dark .select.is-loading::after,html.theme--documenter-dark .loader,html.theme--documenter-dark .button.is-loading::after{animation:spinAround 500ms infinite linear;border:2px solid #dbdee0;border-radius:9999px;border-right-color:transparent;border-top-color:transparent;content:"";display:block;height:1em;position:relative;width:1em}html.theme--documenter-dark .hero-video,html.theme--documenter-dark .modal-background,html.theme--documenter-dark .modal,html.theme--documenter-dark .image.is-square img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square img,html.theme--documenter-dark .image.is-square .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square .has-ratio,html.theme--documenter-dark .image.is-1by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1 img,html.theme--documenter-dark .image.is-1by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1 .has-ratio,html.theme--documenter-dark .image.is-5by4 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4 img,html.theme--documenter-dark .image.is-5by4 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4 .has-ratio,html.theme--documenter-dark .image.is-4by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3 img,html.theme--documenter-dark .image.is-4by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3 .has-ratio,html.theme--documenter-dark .image.is-3by2 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2 img,html.theme--documenter-dark .image.is-3by2 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2 .has-ratio,html.theme--documenter-dark .image.is-5by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3 img,html.theme--documenter-dark .image.is-5by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3 .has-ratio,html.theme--documenter-dark .image.is-16by9 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9 img,html.theme--documenter-dark .image.is-16by9 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9 .has-ratio,html.theme--documenter-dark .image.is-2by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1 img,html.theme--documenter-dark .image.is-2by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1 .has-ratio,html.theme--documenter-dark .image.is-3by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1 img,html.theme--documenter-dark .image.is-3by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1 .has-ratio,html.theme--documenter-dark .image.is-4by5 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5 img,html.theme--documenter-dark .image.is-4by5 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5 .has-ratio,html.theme--documenter-dark .image.is-3by4 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4 img,html.theme--documenter-dark .image.is-3by4 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4 .has-ratio,html.theme--documenter-dark .image.is-2by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3 img,html.theme--documenter-dark .image.is-2by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3 .has-ratio,html.theme--documenter-dark .image.is-3by5 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5 img,html.theme--documenter-dark .image.is-3by5 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5 .has-ratio,html.theme--documenter-dark .image.is-9by16 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16 img,html.theme--documenter-dark .image.is-9by16 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16 .has-ratio,html.theme--documenter-dark .image.is-1by2 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2 img,html.theme--documenter-dark .image.is-1by2 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2 .has-ratio,html.theme--documenter-dark .image.is-1by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3 img,html.theme--documenter-dark .image.is-1by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3 .has-ratio,.is-overlay{bottom:0;left:0;position:absolute;right:0;top:0}html.theme--documenter-dark .navbar-burger{-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;color:currentColor;font-family:inherit;font-size:1em;margin:0;padding:0}/*! minireset.css v0.0.6 | MIT License | github.com/jgthms/minireset.css */html,body,p,ol,ul,li,dl,dt,dd,blockquote,figure,fieldset,legend,textarea,pre,iframe,hr,h1,h2,h3,h4,h5,h6{margin:0;padding:0}h1,h2,h3,h4,h5,h6{font-size:100%;font-weight:normal}ul{list-style:none}button,input,select,textarea{margin:0}html{box-sizing:border-box}*,*::before,*::after{box-sizing:inherit}img,video{height:auto;max-width:100%}iframe{border:0}table{border-collapse:collapse;border-spacing:0}td,th{padding:0}td:not([align]),th:not([align]){text-align:inherit}.has-text-white{color:#fff !important}a.has-text-white:hover,a.has-text-white:focus{color:#e6e6e6 !important}.has-background-white{background-color:#fff !important}.has-text-black{color:#0a0a0a !important}a.has-text-black:hover,a.has-text-black:focus{color:#000 !important}.has-background-black{background-color:#0a0a0a !important}.has-text-light{color:#ecf0f1 !important}a.has-text-light:hover,a.has-text-light:focus{color:#cfd9db !important}.has-background-light{background-color:#ecf0f1 !important}.has-text-dark{color:#282f2f !important}a.has-text-dark:hover,a.has-text-dark:focus{color:#111414 !important}.has-background-dark{background-color:#282f2f !important}.has-text-primary{color:#375a7f !important}a.has-text-primary:hover,a.has-text-primary:focus{color:#28415b !important}.has-background-primary{background-color:#375a7f !important}.has-text-primary-light{color:#f1f5f9 !important}a.has-text-primary-light:hover,a.has-text-primary-light:focus{color:#cddbe9 !important}.has-background-primary-light{background-color:#f1f5f9 !important}.has-text-primary-dark{color:#4d7eb2 !important}a.has-text-primary-dark:hover,a.has-text-primary-dark:focus{color:#7198c1 !important}.has-background-primary-dark{background-color:#4d7eb2 !important}.has-text-link{color:#1abc9c !important}a.has-text-link:hover,a.has-text-link:focus{color:#148f77 !important}.has-background-link{background-color:#1abc9c !important}.has-text-link-light{color:#edfdf9 !important}a.has-text-link-light:hover,a.has-text-link-light:focus{color:#c0f6ec !important}.has-background-link-light{background-color:#edfdf9 !important}.has-text-link-dark{color:#15987e !important}a.has-text-link-dark:hover,a.has-text-link-dark:focus{color:#1bc5a4 !important}.has-background-link-dark{background-color:#15987e !important}.has-text-info{color:#024c7d !important}a.has-text-info:hover,a.has-text-info:focus{color:#012d4b !important}.has-background-info{background-color:#024c7d !important}.has-text-info-light{color:#ebf7ff !important}a.has-text-info-light:hover,a.has-text-info-light:focus{color:#b9e2fe !important}.has-background-info-light{background-color:#ebf7ff !important}.has-text-info-dark{color:#0e9dfb !important}a.has-text-info-dark:hover,a.has-text-info-dark:focus{color:#40b1fc !important}.has-background-info-dark{background-color:#0e9dfb !important}.has-text-success{color:#008438 !important}a.has-text-success:hover,a.has-text-success:focus{color:#005122 !important}.has-background-success{background-color:#008438 !important}.has-text-success-light{color:#ebfff3 !important}a.has-text-success-light:hover,a.has-text-success-light:focus{color:#b8ffd6 !important}.has-background-success-light{background-color:#ebfff3 !important}.has-text-success-dark{color:#00eb64 !important}a.has-text-success-dark:hover,a.has-text-success-dark:focus{color:#1fff7e !important}.has-background-success-dark{background-color:#00eb64 !important}.has-text-warning{color:#ad8100 !important}a.has-text-warning:hover,a.has-text-warning:focus{color:#7a5b00 !important}.has-background-warning{background-color:#ad8100 !important}.has-text-warning-light{color:#fffaeb !important}a.has-text-warning-light:hover,a.has-text-warning-light:focus{color:#ffedb8 !important}.has-background-warning-light{background-color:#fffaeb !important}.has-text-warning-dark{color:#d19c00 !important}a.has-text-warning-dark:hover,a.has-text-warning-dark:focus{color:#ffbf05 !important}.has-background-warning-dark{background-color:#d19c00 !important}.has-text-danger{color:#9e1b0d !important}a.has-text-danger:hover,a.has-text-danger:focus{color:#6f1309 !important}.has-background-danger{background-color:#9e1b0d !important}.has-text-danger-light{color:#fdeeec !important}a.has-text-danger-light:hover,a.has-text-danger-light:focus{color:#fac3bd !important}.has-background-danger-light{background-color:#fdeeec !important}.has-text-danger-dark{color:#ec311d !important}a.has-text-danger-dark:hover,a.has-text-danger-dark:focus{color:#f05c4c !important}.has-background-danger-dark{background-color:#ec311d !important}.has-text-black-bis{color:#121212 !important}.has-background-black-bis{background-color:#121212 !important}.has-text-black-ter{color:#242424 !important}.has-background-black-ter{background-color:#242424 !important}.has-text-grey-darker{color:#282f2f !important}.has-background-grey-darker{background-color:#282f2f !important}.has-text-grey-dark{color:#343c3d !important}.has-background-grey-dark{background-color:#343c3d !important}.has-text-grey{color:#5e6d6f !important}.has-background-grey{background-color:#5e6d6f !important}.has-text-grey-light{color:#8c9b9d !important}.has-background-grey-light{background-color:#8c9b9d !important}.has-text-grey-lighter{color:#dbdee0 !important}.has-background-grey-lighter{background-color:#dbdee0 !important}.has-text-white-ter{color:#ecf0f1 !important}.has-background-white-ter{background-color:#ecf0f1 !important}.has-text-white-bis{color:#fafafa !important}.has-background-white-bis{background-color:#fafafa !important}.is-flex-direction-row{flex-direction:row !important}.is-flex-direction-row-reverse{flex-direction:row-reverse !important}.is-flex-direction-column{flex-direction:column !important}.is-flex-direction-column-reverse{flex-direction:column-reverse !important}.is-flex-wrap-nowrap{flex-wrap:nowrap !important}.is-flex-wrap-wrap{flex-wrap:wrap !important}.is-flex-wrap-wrap-reverse{flex-wrap:wrap-reverse !important}.is-justify-content-flex-start{justify-content:flex-start !important}.is-justify-content-flex-end{justify-content:flex-end !important}.is-justify-content-center{justify-content:center !important}.is-justify-content-space-between{justify-content:space-between !important}.is-justify-content-space-around{justify-content:space-around !important}.is-justify-content-space-evenly{justify-content:space-evenly !important}.is-justify-content-start{justify-content:start !important}.is-justify-content-end{justify-content:end !important}.is-justify-content-left{justify-content:left !important}.is-justify-content-right{justify-content:right !important}.is-align-content-flex-start{align-content:flex-start !important}.is-align-content-flex-end{align-content:flex-end !important}.is-align-content-center{align-content:center !important}.is-align-content-space-between{align-content:space-between !important}.is-align-content-space-around{align-content:space-around !important}.is-align-content-space-evenly{align-content:space-evenly !important}.is-align-content-stretch{align-content:stretch !important}.is-align-content-start{align-content:start !important}.is-align-content-end{align-content:end !important}.is-align-content-baseline{align-content:baseline !important}.is-align-items-stretch{align-items:stretch !important}.is-align-items-flex-start{align-items:flex-start !important}.is-align-items-flex-end{align-items:flex-end !important}.is-align-items-center{align-items:center !important}.is-align-items-baseline{align-items:baseline !important}.is-align-items-start{align-items:start !important}.is-align-items-end{align-items:end !important}.is-align-items-self-start{align-items:self-start !important}.is-align-items-self-end{align-items:self-end !important}.is-align-self-auto{align-self:auto !important}.is-align-self-flex-start{align-self:flex-start !important}.is-align-self-flex-end{align-self:flex-end !important}.is-align-self-center{align-self:center !important}.is-align-self-baseline{align-self:baseline !important}.is-align-self-stretch{align-self:stretch !important}.is-flex-grow-0{flex-grow:0 !important}.is-flex-grow-1{flex-grow:1 !important}.is-flex-grow-2{flex-grow:2 !important}.is-flex-grow-3{flex-grow:3 !important}.is-flex-grow-4{flex-grow:4 !important}.is-flex-grow-5{flex-grow:5 !important}.is-flex-shrink-0{flex-shrink:0 !important}.is-flex-shrink-1{flex-shrink:1 !important}.is-flex-shrink-2{flex-shrink:2 !important}.is-flex-shrink-3{flex-shrink:3 !important}.is-flex-shrink-4{flex-shrink:4 !important}.is-flex-shrink-5{flex-shrink:5 !important}.is-clearfix::after{clear:both;content:" ";display:table}.is-pulled-left{float:left !important}.is-pulled-right{float:right !important}.is-radiusless{border-radius:0 !important}.is-shadowless{box-shadow:none !important}.is-clickable{cursor:pointer !important;pointer-events:all !important}.is-clipped{overflow:hidden !important}.is-relative{position:relative !important}.is-marginless{margin:0 !important}.is-paddingless{padding:0 !important}.m-0{margin:0 !important}.mt-0{margin-top:0 !important}.mr-0{margin-right:0 !important}.mb-0{margin-bottom:0 !important}.ml-0{margin-left:0 !important}.mx-0{margin-left:0 !important;margin-right:0 !important}.my-0{margin-top:0 !important;margin-bottom:0 !important}.m-1{margin:.25rem !important}.mt-1{margin-top:.25rem !important}.mr-1{margin-right:.25rem !important}.mb-1{margin-bottom:.25rem !important}.ml-1{margin-left:.25rem !important}.mx-1{margin-left:.25rem !important;margin-right:.25rem !important}.my-1{margin-top:.25rem !important;margin-bottom:.25rem !important}.m-2{margin:.5rem !important}.mt-2{margin-top:.5rem !important}.mr-2{margin-right:.5rem !important}.mb-2{margin-bottom:.5rem !important}.ml-2{margin-left:.5rem !important}.mx-2{margin-left:.5rem !important;margin-right:.5rem !important}.my-2{margin-top:.5rem !important;margin-bottom:.5rem !important}.m-3{margin:.75rem !important}.mt-3{margin-top:.75rem !important}.mr-3{margin-right:.75rem !important}.mb-3{margin-bottom:.75rem !important}.ml-3{margin-left:.75rem !important}.mx-3{margin-left:.75rem !important;margin-right:.75rem !important}.my-3{margin-top:.75rem !important;margin-bottom:.75rem !important}.m-4{margin:1rem !important}.mt-4{margin-top:1rem !important}.mr-4{margin-right:1rem !important}.mb-4{margin-bottom:1rem !important}.ml-4{margin-left:1rem !important}.mx-4{margin-left:1rem !important;margin-right:1rem !important}.my-4{margin-top:1rem !important;margin-bottom:1rem !important}.m-5{margin:1.5rem !important}.mt-5{margin-top:1.5rem !important}.mr-5{margin-right:1.5rem !important}.mb-5{margin-bottom:1.5rem !important}.ml-5{margin-left:1.5rem !important}.mx-5{margin-left:1.5rem !important;margin-right:1.5rem !important}.my-5{margin-top:1.5rem !important;margin-bottom:1.5rem !important}.m-6{margin:3rem !important}.mt-6{margin-top:3rem !important}.mr-6{margin-right:3rem !important}.mb-6{margin-bottom:3rem !important}.ml-6{margin-left:3rem !important}.mx-6{margin-left:3rem !important;margin-right:3rem !important}.my-6{margin-top:3rem !important;margin-bottom:3rem !important}.m-auto{margin:auto !important}.mt-auto{margin-top:auto !important}.mr-auto{margin-right:auto !important}.mb-auto{margin-bottom:auto !important}.ml-auto{margin-left:auto !important}.mx-auto{margin-left:auto !important;margin-right:auto !important}.my-auto{margin-top:auto !important;margin-bottom:auto !important}.p-0{padding:0 !important}.pt-0{padding-top:0 !important}.pr-0{padding-right:0 !important}.pb-0{padding-bottom:0 !important}.pl-0{padding-left:0 !important}.px-0{padding-left:0 !important;padding-right:0 !important}.py-0{padding-top:0 !important;padding-bottom:0 !important}.p-1{padding:.25rem !important}.pt-1{padding-top:.25rem !important}.pr-1{padding-right:.25rem !important}.pb-1{padding-bottom:.25rem !important}.pl-1{padding-left:.25rem !important}.px-1{padding-left:.25rem !important;padding-right:.25rem !important}.py-1{padding-top:.25rem !important;padding-bottom:.25rem !important}.p-2{padding:.5rem !important}.pt-2{padding-top:.5rem !important}.pr-2{padding-right:.5rem !important}.pb-2{padding-bottom:.5rem !important}.pl-2{padding-left:.5rem !important}.px-2{padding-left:.5rem !important;padding-right:.5rem !important}.py-2{padding-top:.5rem !important;padding-bottom:.5rem !important}.p-3{padding:.75rem !important}.pt-3{padding-top:.75rem !important}.pr-3{padding-right:.75rem !important}.pb-3{padding-bottom:.75rem !important}.pl-3{padding-left:.75rem !important}.px-3{padding-left:.75rem !important;padding-right:.75rem !important}.py-3{padding-top:.75rem !important;padding-bottom:.75rem !important}.p-4{padding:1rem !important}.pt-4{padding-top:1rem !important}.pr-4{padding-right:1rem !important}.pb-4{padding-bottom:1rem !important}.pl-4{padding-left:1rem !important}.px-4{padding-left:1rem !important;padding-right:1rem !important}.py-4{padding-top:1rem !important;padding-bottom:1rem !important}.p-5{padding:1.5rem !important}.pt-5{padding-top:1.5rem !important}.pr-5{padding-right:1.5rem !important}.pb-5{padding-bottom:1.5rem !important}.pl-5{padding-left:1.5rem !important}.px-5{padding-left:1.5rem !important;padding-right:1.5rem !important}.py-5{padding-top:1.5rem !important;padding-bottom:1.5rem !important}.p-6{padding:3rem !important}.pt-6{padding-top:3rem !important}.pr-6{padding-right:3rem !important}.pb-6{padding-bottom:3rem !important}.pl-6{padding-left:3rem !important}.px-6{padding-left:3rem !important;padding-right:3rem !important}.py-6{padding-top:3rem !important;padding-bottom:3rem !important}.p-auto{padding:auto !important}.pt-auto{padding-top:auto !important}.pr-auto{padding-right:auto !important}.pb-auto{padding-bottom:auto !important}.pl-auto{padding-left:auto !important}.px-auto{padding-left:auto !important;padding-right:auto !important}.py-auto{padding-top:auto !important;padding-bottom:auto !important}.is-size-1{font-size:3rem !important}.is-size-2{font-size:2.5rem !important}.is-size-3{font-size:2rem !important}.is-size-4{font-size:1.5rem !important}.is-size-5{font-size:1.25rem !important}.is-size-6{font-size:1rem !important}.is-size-7,html.theme--documenter-dark .docstring>section>a.docs-sourcelink{font-size:.75rem !important}@media screen and (max-width: 768px){.is-size-1-mobile{font-size:3rem !important}.is-size-2-mobile{font-size:2.5rem !important}.is-size-3-mobile{font-size:2rem !important}.is-size-4-mobile{font-size:1.5rem !important}.is-size-5-mobile{font-size:1.25rem !important}.is-size-6-mobile{font-size:1rem !important}.is-size-7-mobile{font-size:.75rem !important}}@media screen and (min-width: 769px),print{.is-size-1-tablet{font-size:3rem !important}.is-size-2-tablet{font-size:2.5rem !important}.is-size-3-tablet{font-size:2rem !important}.is-size-4-tablet{font-size:1.5rem !important}.is-size-5-tablet{font-size:1.25rem !important}.is-size-6-tablet{font-size:1rem !important}.is-size-7-tablet{font-size:.75rem !important}}@media screen and (max-width: 1055px){.is-size-1-touch{font-size:3rem !important}.is-size-2-touch{font-size:2.5rem !important}.is-size-3-touch{font-size:2rem !important}.is-size-4-touch{font-size:1.5rem !important}.is-size-5-touch{font-size:1.25rem !important}.is-size-6-touch{font-size:1rem !important}.is-size-7-touch{font-size:.75rem !important}}@media screen and (min-width: 1056px){.is-size-1-desktop{font-size:3rem !important}.is-size-2-desktop{font-size:2.5rem !important}.is-size-3-desktop{font-size:2rem !important}.is-size-4-desktop{font-size:1.5rem !important}.is-size-5-desktop{font-size:1.25rem !important}.is-size-6-desktop{font-size:1rem !important}.is-size-7-desktop{font-size:.75rem !important}}@media screen and (min-width: 1216px){.is-size-1-widescreen{font-size:3rem !important}.is-size-2-widescreen{font-size:2.5rem !important}.is-size-3-widescreen{font-size:2rem !important}.is-size-4-widescreen{font-size:1.5rem !important}.is-size-5-widescreen{font-size:1.25rem !important}.is-size-6-widescreen{font-size:1rem !important}.is-size-7-widescreen{font-size:.75rem !important}}@media screen and (min-width: 1408px){.is-size-1-fullhd{font-size:3rem !important}.is-size-2-fullhd{font-size:2.5rem !important}.is-size-3-fullhd{font-size:2rem !important}.is-size-4-fullhd{font-size:1.5rem !important}.is-size-5-fullhd{font-size:1.25rem !important}.is-size-6-fullhd{font-size:1rem !important}.is-size-7-fullhd{font-size:.75rem !important}}.has-text-centered{text-align:center !important}.has-text-justified{text-align:justify !important}.has-text-left{text-align:left !important}.has-text-right{text-align:right !important}@media screen and (max-width: 768px){.has-text-centered-mobile{text-align:center !important}}@media screen and (min-width: 769px),print{.has-text-centered-tablet{text-align:center !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-centered-tablet-only{text-align:center !important}}@media screen and (max-width: 1055px){.has-text-centered-touch{text-align:center !important}}@media screen and (min-width: 1056px){.has-text-centered-desktop{text-align:center !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-centered-desktop-only{text-align:center !important}}@media screen and (min-width: 1216px){.has-text-centered-widescreen{text-align:center !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-centered-widescreen-only{text-align:center !important}}@media screen and (min-width: 1408px){.has-text-centered-fullhd{text-align:center !important}}@media screen and (max-width: 768px){.has-text-justified-mobile{text-align:justify !important}}@media screen and (min-width: 769px),print{.has-text-justified-tablet{text-align:justify !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-justified-tablet-only{text-align:justify !important}}@media screen and (max-width: 1055px){.has-text-justified-touch{text-align:justify !important}}@media screen and (min-width: 1056px){.has-text-justified-desktop{text-align:justify !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-justified-desktop-only{text-align:justify !important}}@media screen and (min-width: 1216px){.has-text-justified-widescreen{text-align:justify !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-justified-widescreen-only{text-align:justify !important}}@media screen and (min-width: 1408px){.has-text-justified-fullhd{text-align:justify !important}}@media screen and (max-width: 768px){.has-text-left-mobile{text-align:left !important}}@media screen and (min-width: 769px),print{.has-text-left-tablet{text-align:left !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-left-tablet-only{text-align:left !important}}@media screen and (max-width: 1055px){.has-text-left-touch{text-align:left !important}}@media screen and (min-width: 1056px){.has-text-left-desktop{text-align:left !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-left-desktop-only{text-align:left !important}}@media screen and (min-width: 1216px){.has-text-left-widescreen{text-align:left !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-left-widescreen-only{text-align:left !important}}@media screen and (min-width: 1408px){.has-text-left-fullhd{text-align:left !important}}@media screen and (max-width: 768px){.has-text-right-mobile{text-align:right !important}}@media screen and (min-width: 769px),print{.has-text-right-tablet{text-align:right !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-right-tablet-only{text-align:right !important}}@media screen and (max-width: 1055px){.has-text-right-touch{text-align:right !important}}@media screen and (min-width: 1056px){.has-text-right-desktop{text-align:right !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-right-desktop-only{text-align:right !important}}@media screen and (min-width: 1216px){.has-text-right-widescreen{text-align:right !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-right-widescreen-only{text-align:right !important}}@media screen and (min-width: 1408px){.has-text-right-fullhd{text-align:right !important}}.is-capitalized{text-transform:capitalize !important}.is-lowercase{text-transform:lowercase !important}.is-uppercase{text-transform:uppercase !important}.is-italic{font-style:italic !important}.is-underlined{text-decoration:underline !important}.has-text-weight-light{font-weight:300 !important}.has-text-weight-normal{font-weight:400 !important}.has-text-weight-medium{font-weight:500 !important}.has-text-weight-semibold{font-weight:600 !important}.has-text-weight-bold{font-weight:700 !important}.is-family-primary{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-secondary{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-sans-serif{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-monospace{font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace !important}.is-family-code{font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace !important}.is-block{display:block !important}@media screen and (max-width: 768px){.is-block-mobile{display:block !important}}@media screen and (min-width: 769px),print{.is-block-tablet{display:block !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-block-tablet-only{display:block !important}}@media screen and (max-width: 1055px){.is-block-touch{display:block !important}}@media screen and (min-width: 1056px){.is-block-desktop{display:block !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-block-desktop-only{display:block !important}}@media screen and (min-width: 1216px){.is-block-widescreen{display:block !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-block-widescreen-only{display:block !important}}@media screen and (min-width: 1408px){.is-block-fullhd{display:block !important}}.is-flex{display:flex !important}@media screen and (max-width: 768px){.is-flex-mobile{display:flex !important}}@media screen and (min-width: 769px),print{.is-flex-tablet{display:flex !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-flex-tablet-only{display:flex !important}}@media screen and (max-width: 1055px){.is-flex-touch{display:flex !important}}@media screen and (min-width: 1056px){.is-flex-desktop{display:flex !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-flex-desktop-only{display:flex !important}}@media screen and (min-width: 1216px){.is-flex-widescreen{display:flex !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-flex-widescreen-only{display:flex !important}}@media screen and (min-width: 1408px){.is-flex-fullhd{display:flex !important}}.is-inline{display:inline !important}@media screen and (max-width: 768px){.is-inline-mobile{display:inline !important}}@media screen and (min-width: 769px),print{.is-inline-tablet{display:inline !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-tablet-only{display:inline !important}}@media screen and (max-width: 1055px){.is-inline-touch{display:inline !important}}@media screen and (min-width: 1056px){.is-inline-desktop{display:inline !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-desktop-only{display:inline !important}}@media screen and (min-width: 1216px){.is-inline-widescreen{display:inline !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-widescreen-only{display:inline !important}}@media screen and (min-width: 1408px){.is-inline-fullhd{display:inline !important}}.is-inline-block{display:inline-block !important}@media screen and (max-width: 768px){.is-inline-block-mobile{display:inline-block !important}}@media screen and (min-width: 769px),print{.is-inline-block-tablet{display:inline-block !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-block-tablet-only{display:inline-block !important}}@media screen and (max-width: 1055px){.is-inline-block-touch{display:inline-block !important}}@media screen and (min-width: 1056px){.is-inline-block-desktop{display:inline-block !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-block-desktop-only{display:inline-block !important}}@media screen and (min-width: 1216px){.is-inline-block-widescreen{display:inline-block !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-block-widescreen-only{display:inline-block !important}}@media screen and (min-width: 1408px){.is-inline-block-fullhd{display:inline-block !important}}.is-inline-flex{display:inline-flex !important}@media screen and (max-width: 768px){.is-inline-flex-mobile{display:inline-flex !important}}@media screen and (min-width: 769px),print{.is-inline-flex-tablet{display:inline-flex !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-flex-tablet-only{display:inline-flex !important}}@media screen and (max-width: 1055px){.is-inline-flex-touch{display:inline-flex !important}}@media screen and (min-width: 1056px){.is-inline-flex-desktop{display:inline-flex !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-flex-desktop-only{display:inline-flex !important}}@media screen and (min-width: 1216px){.is-inline-flex-widescreen{display:inline-flex !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-flex-widescreen-only{display:inline-flex !important}}@media screen and (min-width: 1408px){.is-inline-flex-fullhd{display:inline-flex !important}}.is-hidden{display:none !important}.is-sr-only{border:none !important;clip:rect(0, 0, 0, 0) !important;height:0.01em !important;overflow:hidden !important;padding:0 !important;position:absolute !important;white-space:nowrap !important;width:0.01em !important}@media screen and (max-width: 768px){.is-hidden-mobile{display:none !important}}@media screen and (min-width: 769px),print{.is-hidden-tablet{display:none !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-hidden-tablet-only{display:none !important}}@media screen and (max-width: 1055px){.is-hidden-touch{display:none !important}}@media screen and (min-width: 1056px){.is-hidden-desktop{display:none !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-hidden-desktop-only{display:none !important}}@media screen and (min-width: 1216px){.is-hidden-widescreen{display:none !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-hidden-widescreen-only{display:none !important}}@media screen and (min-width: 1408px){.is-hidden-fullhd{display:none !important}}.is-invisible{visibility:hidden !important}@media screen and (max-width: 768px){.is-invisible-mobile{visibility:hidden !important}}@media screen and (min-width: 769px),print{.is-invisible-tablet{visibility:hidden !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-invisible-tablet-only{visibility:hidden !important}}@media screen and (max-width: 1055px){.is-invisible-touch{visibility:hidden !important}}@media screen and (min-width: 1056px){.is-invisible-desktop{visibility:hidden !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-invisible-desktop-only{visibility:hidden !important}}@media screen and (min-width: 1216px){.is-invisible-widescreen{visibility:hidden !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-invisible-widescreen-only{visibility:hidden !important}}@media screen and (min-width: 1408px){.is-invisible-fullhd{visibility:hidden !important}}html.theme--documenter-dark{/*!
+ Theme: a11y-dark
+ Author: @ericwbailey
+ Maintainer: @ericwbailey
+
+ Based on the Tomorrow Night Eighties theme: https://github.com/isagalaev/highlight.js/blob/master/src/styles/tomorrow-night-eighties.css
+*/}html.theme--documenter-dark html{background-color:#1f2424;font-size:16px;-moz-osx-font-smoothing:grayscale;-webkit-font-smoothing:antialiased;min-width:300px;overflow-x:auto;overflow-y:scroll;text-rendering:optimizeLegibility;text-size-adjust:100%}html.theme--documenter-dark article,html.theme--documenter-dark aside,html.theme--documenter-dark figure,html.theme--documenter-dark footer,html.theme--documenter-dark header,html.theme--documenter-dark hgroup,html.theme--documenter-dark section{display:block}html.theme--documenter-dark body,html.theme--documenter-dark button,html.theme--documenter-dark input,html.theme--documenter-dark optgroup,html.theme--documenter-dark select,html.theme--documenter-dark textarea{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif}html.theme--documenter-dark code,html.theme--documenter-dark pre{-moz-osx-font-smoothing:auto;-webkit-font-smoothing:auto;font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace}html.theme--documenter-dark body{color:#fff;font-size:1em;font-weight:400;line-height:1.5}html.theme--documenter-dark a{color:#1abc9c;cursor:pointer;text-decoration:none}html.theme--documenter-dark a strong{color:currentColor}html.theme--documenter-dark a:hover{color:#1dd2af}html.theme--documenter-dark code{background-color:rgba(255,255,255,0.05);color:#ececec;font-size:.875em;font-weight:normal;padding:.1em}html.theme--documenter-dark hr{background-color:#282f2f;border:none;display:block;height:2px;margin:1.5rem 0}html.theme--documenter-dark img{height:auto;max-width:100%}html.theme--documenter-dark input[type="checkbox"],html.theme--documenter-dark input[type="radio"]{vertical-align:baseline}html.theme--documenter-dark small{font-size:.875em}html.theme--documenter-dark span{font-style:inherit;font-weight:inherit}html.theme--documenter-dark strong{color:#f2f2f2;font-weight:700}html.theme--documenter-dark fieldset{border:none}html.theme--documenter-dark pre{-webkit-overflow-scrolling:touch;background-color:#282f2f;color:#fff;font-size:.875em;overflow-x:auto;padding:1.25rem 1.5rem;white-space:pre;word-wrap:normal}html.theme--documenter-dark pre code{background-color:transparent;color:currentColor;font-size:1em;padding:0}html.theme--documenter-dark table td,html.theme--documenter-dark table th{vertical-align:top}html.theme--documenter-dark table td:not([align]),html.theme--documenter-dark table th:not([align]){text-align:inherit}html.theme--documenter-dark table th{color:#f2f2f2}html.theme--documenter-dark .box{background-color:#343c3d;border-radius:8px;box-shadow:none;color:#fff;display:block;padding:1.25rem}html.theme--documenter-dark a.box:hover,html.theme--documenter-dark a.box:focus{box-shadow:0 0.5em 1em -0.125em rgba(10,10,10,0.1),0 0 0 1px #1abc9c}html.theme--documenter-dark a.box:active{box-shadow:inset 0 1px 2px rgba(10,10,10,0.2),0 0 0 1px #1abc9c}html.theme--documenter-dark .button{background-color:#282f2f;border-color:#4c5759;border-width:1px;color:#375a7f;cursor:pointer;justify-content:center;padding-bottom:calc(0.5em - 1px);padding-left:1em;padding-right:1em;padding-top:calc(0.5em - 1px);text-align:center;white-space:nowrap}html.theme--documenter-dark .button strong{color:inherit}html.theme--documenter-dark .button .icon,html.theme--documenter-dark .button .icon.is-small,html.theme--documenter-dark .button #documenter .docs-sidebar form.docs-search>input.icon,html.theme--documenter-dark #documenter .docs-sidebar .button form.docs-search>input.icon,html.theme--documenter-dark .button .icon.is-medium,html.theme--documenter-dark .button .icon.is-large{height:1.5em;width:1.5em}html.theme--documenter-dark .button .icon:first-child:not(:last-child){margin-left:calc(-0.5em - 1px);margin-right:.25em}html.theme--documenter-dark .button .icon:last-child:not(:first-child){margin-left:.25em;margin-right:calc(-0.5em - 1px)}html.theme--documenter-dark .button .icon:first-child:last-child{margin-left:calc(-0.5em - 1px);margin-right:calc(-0.5em - 1px)}html.theme--documenter-dark .button:hover,html.theme--documenter-dark .button.is-hovered{border-color:#8c9b9d;color:#f2f2f2}html.theme--documenter-dark .button:focus,html.theme--documenter-dark .button.is-focused{border-color:#8c9b9d;color:#17a689}html.theme--documenter-dark .button:focus:not(:active),html.theme--documenter-dark .button.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .button:active,html.theme--documenter-dark .button.is-active{border-color:#343c3d;color:#f2f2f2}html.theme--documenter-dark .button.is-text{background-color:transparent;border-color:transparent;color:#fff;text-decoration:underline}html.theme--documenter-dark .button.is-text:hover,html.theme--documenter-dark .button.is-text.is-hovered,html.theme--documenter-dark .button.is-text:focus,html.theme--documenter-dark .button.is-text.is-focused{background-color:#282f2f;color:#f2f2f2}html.theme--documenter-dark .button.is-text:active,html.theme--documenter-dark .button.is-text.is-active{background-color:#1d2122;color:#f2f2f2}html.theme--documenter-dark .button.is-text[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-text{background-color:transparent;border-color:transparent;box-shadow:none}html.theme--documenter-dark .button.is-ghost{background:none;border-color:rgba(0,0,0,0);color:#1abc9c;text-decoration:none}html.theme--documenter-dark .button.is-ghost:hover,html.theme--documenter-dark .button.is-ghost.is-hovered{color:#1abc9c;text-decoration:underline}html.theme--documenter-dark .button.is-white{background-color:#fff;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .button.is-white:hover,html.theme--documenter-dark .button.is-white.is-hovered{background-color:#f9f9f9;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .button.is-white:focus,html.theme--documenter-dark .button.is-white.is-focused{border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .button.is-white:focus:not(:active),html.theme--documenter-dark .button.is-white.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}html.theme--documenter-dark .button.is-white:active,html.theme--documenter-dark .button.is-white.is-active{background-color:#f2f2f2;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .button.is-white[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-white{background-color:#fff;border-color:#fff;box-shadow:none}html.theme--documenter-dark .button.is-white.is-inverted{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .button.is-white.is-inverted:hover,html.theme--documenter-dark .button.is-white.is-inverted.is-hovered{background-color:#000}html.theme--documenter-dark .button.is-white.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-white.is-inverted{background-color:#0a0a0a;border-color:transparent;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-white.is-loading::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}html.theme--documenter-dark .button.is-white.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-white.is-outlined:hover,html.theme--documenter-dark .button.is-white.is-outlined.is-hovered,html.theme--documenter-dark .button.is-white.is-outlined:focus,html.theme--documenter-dark .button.is-white.is-outlined.is-focused{background-color:#fff;border-color:#fff;color:#0a0a0a}html.theme--documenter-dark .button.is-white.is-outlined.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-white.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-white.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-white.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-white.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}html.theme--documenter-dark .button.is-white.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-white.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-white.is-inverted.is-outlined{background-color:transparent;border-color:#0a0a0a;color:#0a0a0a}html.theme--documenter-dark .button.is-white.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-focused{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-white.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-white.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-white.is-inverted.is-outlined{background-color:transparent;border-color:#0a0a0a;box-shadow:none;color:#0a0a0a}html.theme--documenter-dark .button.is-black{background-color:#0a0a0a;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-black:hover,html.theme--documenter-dark .button.is-black.is-hovered{background-color:#040404;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-black:focus,html.theme--documenter-dark .button.is-black.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-black:focus:not(:active),html.theme--documenter-dark .button.is-black.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}html.theme--documenter-dark .button.is-black:active,html.theme--documenter-dark .button.is-black.is-active{background-color:#000;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-black[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-black{background-color:#0a0a0a;border-color:#0a0a0a;box-shadow:none}html.theme--documenter-dark .button.is-black.is-inverted{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-inverted:hover,html.theme--documenter-dark .button.is-black.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-black.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-black.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-black.is-outlined{background-color:transparent;border-color:#0a0a0a;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-outlined:hover,html.theme--documenter-dark .button.is-black.is-outlined.is-hovered,html.theme--documenter-dark .button.is-black.is-outlined:focus,html.theme--documenter-dark .button.is-black.is-outlined.is-focused{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}html.theme--documenter-dark .button.is-black.is-outlined.is-loading::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}html.theme--documenter-dark .button.is-black.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-black.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-black.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-black.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-black.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-black.is-outlined{background-color:transparent;border-color:#0a0a0a;box-shadow:none;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-black.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-focused{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-black.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}html.theme--documenter-dark .button.is-black.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-black.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-light{background-color:#ecf0f1;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light:hover,html.theme--documenter-dark .button.is-light.is-hovered{background-color:#e5eaec;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light:focus,html.theme--documenter-dark .button.is-light.is-focused{border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light:focus:not(:active),html.theme--documenter-dark .button.is-light.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(236,240,241,0.25)}html.theme--documenter-dark .button.is-light:active,html.theme--documenter-dark .button.is-light.is-active{background-color:#dde4e6;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-light{background-color:#ecf0f1;border-color:#ecf0f1;box-shadow:none}html.theme--documenter-dark .button.is-light.is-inverted{background-color:rgba(0,0,0,0.7);color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-inverted:hover,html.theme--documenter-dark .button.is-light.is-inverted.is-hovered{background-color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-light.is-inverted{background-color:rgba(0,0,0,0.7);border-color:transparent;box-shadow:none;color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-loading::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}html.theme--documenter-dark .button.is-light.is-outlined{background-color:transparent;border-color:#ecf0f1;color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-outlined:hover,html.theme--documenter-dark .button.is-light.is-outlined.is-hovered,html.theme--documenter-dark .button.is-light.is-outlined:focus,html.theme--documenter-dark .button.is-light.is-outlined.is-focused{background-color:#ecf0f1;border-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light.is-outlined.is-loading::after{border-color:transparent transparent #ecf0f1 #ecf0f1 !important}html.theme--documenter-dark .button.is-light.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-light.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-light.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-light.is-outlined.is-loading.is-focused::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}html.theme--documenter-dark .button.is-light.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-light.is-outlined{background-color:transparent;border-color:#ecf0f1;box-shadow:none;color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-light.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-focused{background-color:rgba(0,0,0,0.7);color:#ecf0f1}html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-light.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #ecf0f1 #ecf0f1 !important}html.theme--documenter-dark .button.is-light.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-light.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);box-shadow:none;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .button.is-dark,html.theme--documenter-dark .content kbd.button{background-color:#282f2f;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-dark:hover,html.theme--documenter-dark .content kbd.button:hover,html.theme--documenter-dark .button.is-dark.is-hovered,html.theme--documenter-dark .content kbd.button.is-hovered{background-color:#232829;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-dark:focus,html.theme--documenter-dark .content kbd.button:focus,html.theme--documenter-dark .button.is-dark.is-focused,html.theme--documenter-dark .content kbd.button.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-dark:focus:not(:active),html.theme--documenter-dark .content kbd.button:focus:not(:active),html.theme--documenter-dark .button.is-dark.is-focused:not(:active),html.theme--documenter-dark .content kbd.button.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(40,47,47,0.25)}html.theme--documenter-dark .button.is-dark:active,html.theme--documenter-dark .content kbd.button:active,html.theme--documenter-dark .button.is-dark.is-active,html.theme--documenter-dark .content kbd.button.is-active{background-color:#1d2122;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-dark[disabled],html.theme--documenter-dark .content kbd.button[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-dark,fieldset[disabled] html.theme--documenter-dark .content kbd.button{background-color:#282f2f;border-color:#282f2f;box-shadow:none}html.theme--documenter-dark .button.is-dark.is-inverted,html.theme--documenter-dark .content kbd.button.is-inverted{background-color:#fff;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-inverted:hover,html.theme--documenter-dark .content kbd.button.is-inverted:hover,html.theme--documenter-dark .button.is-dark.is-inverted.is-hovered,html.theme--documenter-dark .content kbd.button.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-dark.is-inverted[disabled],html.theme--documenter-dark .content kbd.button.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-dark.is-inverted,fieldset[disabled] html.theme--documenter-dark .content kbd.button.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-loading::after,html.theme--documenter-dark .content kbd.button.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-dark.is-outlined,html.theme--documenter-dark .content kbd.button.is-outlined{background-color:transparent;border-color:#282f2f;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-outlined:hover,html.theme--documenter-dark .content kbd.button.is-outlined:hover,html.theme--documenter-dark .button.is-dark.is-outlined.is-hovered,html.theme--documenter-dark .content kbd.button.is-outlined.is-hovered,html.theme--documenter-dark .button.is-dark.is-outlined:focus,html.theme--documenter-dark .content kbd.button.is-outlined:focus,html.theme--documenter-dark .button.is-dark.is-outlined.is-focused,html.theme--documenter-dark .content kbd.button.is-outlined.is-focused{background-color:#282f2f;border-color:#282f2f;color:#fff}html.theme--documenter-dark .button.is-dark.is-outlined.is-loading::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading::after{border-color:transparent transparent #282f2f #282f2f !important}html.theme--documenter-dark .button.is-dark.is-outlined.is-loading:hover::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-dark.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-dark.is-outlined.is-loading:focus::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-dark.is-outlined.is-loading.is-focused::after,html.theme--documenter-dark .content kbd.button.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-dark.is-outlined[disabled],html.theme--documenter-dark .content kbd.button.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-dark.is-outlined,fieldset[disabled] html.theme--documenter-dark .content kbd.button.is-outlined{background-color:transparent;border-color:#282f2f;box-shadow:none;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined:hover,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined:focus,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-focused,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-focused{background-color:#fff;color:#282f2f}html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined.is-loading.is-focused::after,html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #282f2f #282f2f !important}html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined[disabled],html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-dark.is-inverted.is-outlined,fieldset[disabled] html.theme--documenter-dark .content kbd.button.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-primary,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink{background-color:#375a7f;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-primary:hover,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-hovered.docs-sourcelink{background-color:#335476;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-primary:focus,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:focus,html.theme--documenter-dark .button.is-primary.is-focused,html.theme--documenter-dark .docstring>section>a.button.is-focused.docs-sourcelink{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-primary:focus:not(:active),html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:focus:not(:active),html.theme--documenter-dark .button.is-primary.is-focused:not(:active),html.theme--documenter-dark .docstring>section>a.button.is-focused.docs-sourcelink:not(:active){box-shadow:0 0 0 0.125em rgba(55,90,127,0.25)}html.theme--documenter-dark .button.is-primary:active,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:active,html.theme--documenter-dark .button.is-primary.is-active,html.theme--documenter-dark .docstring>section>a.button.is-active.docs-sourcelink{background-color:#2f4d6d;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-primary[disabled],html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-primary,fieldset[disabled] html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink{background-color:#375a7f;border-color:#375a7f;box-shadow:none}html.theme--documenter-dark .button.is-primary.is-inverted,html.theme--documenter-dark .docstring>section>a.button.is-inverted.docs-sourcelink{background-color:#fff;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-inverted:hover,html.theme--documenter-dark .docstring>section>a.button.is-inverted.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-inverted.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-hovered.docs-sourcelink{background-color:#f2f2f2}html.theme--documenter-dark .button.is-primary.is-inverted[disabled],html.theme--documenter-dark .docstring>section>a.button.is-inverted.docs-sourcelink[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-primary.is-inverted,fieldset[disabled] html.theme--documenter-dark .docstring>section>a.button.is-inverted.docs-sourcelink{background-color:#fff;border-color:transparent;box-shadow:none;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-loading::after,html.theme--documenter-dark .docstring>section>a.button.is-loading.docs-sourcelink::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-primary.is-outlined,html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink{background-color:transparent;border-color:#375a7f;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-outlined:hover,html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-outlined.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-hovered.docs-sourcelink,html.theme--documenter-dark .button.is-primary.is-outlined:focus,html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink:focus,html.theme--documenter-dark .button.is-primary.is-outlined.is-focused,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-focused.docs-sourcelink{background-color:#375a7f;border-color:#375a7f;color:#fff}html.theme--documenter-dark .button.is-primary.is-outlined.is-loading::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.docs-sourcelink::after{border-color:transparent transparent #375a7f #375a7f !important}html.theme--documenter-dark .button.is-primary.is-outlined.is-loading:hover::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.docs-sourcelink:hover::after,html.theme--documenter-dark .button.is-primary.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.is-hovered.docs-sourcelink::after,html.theme--documenter-dark .button.is-primary.is-outlined.is-loading:focus::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.docs-sourcelink:focus::after,html.theme--documenter-dark .button.is-primary.is-outlined.is-loading.is-focused::after,html.theme--documenter-dark .docstring>section>a.button.is-outlined.is-loading.is-focused.docs-sourcelink::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-primary.is-outlined[disabled],html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-primary.is-outlined,fieldset[disabled] html.theme--documenter-dark .docstring>section>a.button.is-outlined.docs-sourcelink{background-color:transparent;border-color:#375a7f;box-shadow:none;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined:hover,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-hovered.docs-sourcelink,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined:focus,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink:focus,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-focused,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-focused.docs-sourcelink{background-color:#fff;color:#375a7f}html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-loading.docs-sourcelink:hover::after,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-loading.is-hovered.docs-sourcelink::after,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-loading.docs-sourcelink:focus::after,html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined.is-loading.is-focused::after,html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.is-loading.is-focused.docs-sourcelink::after{border-color:transparent transparent #375a7f #375a7f !important}html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined[disabled],html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-primary.is-inverted.is-outlined,fieldset[disabled] html.theme--documenter-dark .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-primary.is-light,html.theme--documenter-dark .docstring>section>a.button.is-light.docs-sourcelink{background-color:#f1f5f9;color:#4d7eb2}html.theme--documenter-dark .button.is-primary.is-light:hover,html.theme--documenter-dark .docstring>section>a.button.is-light.docs-sourcelink:hover,html.theme--documenter-dark .button.is-primary.is-light.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-light.is-hovered.docs-sourcelink{background-color:#e8eef5;border-color:transparent;color:#4d7eb2}html.theme--documenter-dark .button.is-primary.is-light:active,html.theme--documenter-dark .docstring>section>a.button.is-light.docs-sourcelink:active,html.theme--documenter-dark .button.is-primary.is-light.is-active,html.theme--documenter-dark .docstring>section>a.button.is-light.is-active.docs-sourcelink{background-color:#dfe8f1;border-color:transparent;color:#4d7eb2}html.theme--documenter-dark .button.is-link{background-color:#1abc9c;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-link:hover,html.theme--documenter-dark .button.is-link.is-hovered{background-color:#18b193;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-link:focus,html.theme--documenter-dark .button.is-link.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-link:focus:not(:active),html.theme--documenter-dark .button.is-link.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .button.is-link:active,html.theme--documenter-dark .button.is-link.is-active{background-color:#17a689;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-link[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-link{background-color:#1abc9c;border-color:#1abc9c;box-shadow:none}html.theme--documenter-dark .button.is-link.is-inverted{background-color:#fff;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-inverted:hover,html.theme--documenter-dark .button.is-link.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-link.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-link.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-link.is-outlined{background-color:transparent;border-color:#1abc9c;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-outlined:hover,html.theme--documenter-dark .button.is-link.is-outlined.is-hovered,html.theme--documenter-dark .button.is-link.is-outlined:focus,html.theme--documenter-dark .button.is-link.is-outlined.is-focused{background-color:#1abc9c;border-color:#1abc9c;color:#fff}html.theme--documenter-dark .button.is-link.is-outlined.is-loading::after{border-color:transparent transparent #1abc9c #1abc9c !important}html.theme--documenter-dark .button.is-link.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-link.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-link.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-link.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-link.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-link.is-outlined{background-color:transparent;border-color:#1abc9c;box-shadow:none;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-link.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-focused{background-color:#fff;color:#1abc9c}html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-link.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #1abc9c #1abc9c !important}html.theme--documenter-dark .button.is-link.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-link.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-link.is-light{background-color:#edfdf9;color:#15987e}html.theme--documenter-dark .button.is-link.is-light:hover,html.theme--documenter-dark .button.is-link.is-light.is-hovered{background-color:#e2fbf6;border-color:transparent;color:#15987e}html.theme--documenter-dark .button.is-link.is-light:active,html.theme--documenter-dark .button.is-link.is-light.is-active{background-color:#d7f9f3;border-color:transparent;color:#15987e}html.theme--documenter-dark .button.is-info{background-color:#024c7d;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-info:hover,html.theme--documenter-dark .button.is-info.is-hovered{background-color:#024470;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-info:focus,html.theme--documenter-dark .button.is-info.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-info:focus:not(:active),html.theme--documenter-dark .button.is-info.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(2,76,125,0.25)}html.theme--documenter-dark .button.is-info:active,html.theme--documenter-dark .button.is-info.is-active{background-color:#023d64;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-info[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-info{background-color:#024c7d;border-color:#024c7d;box-shadow:none}html.theme--documenter-dark .button.is-info.is-inverted{background-color:#fff;color:#024c7d}html.theme--documenter-dark .button.is-info.is-inverted:hover,html.theme--documenter-dark .button.is-info.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-info.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-info.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#024c7d}html.theme--documenter-dark .button.is-info.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-info.is-outlined{background-color:transparent;border-color:#024c7d;color:#024c7d}html.theme--documenter-dark .button.is-info.is-outlined:hover,html.theme--documenter-dark .button.is-info.is-outlined.is-hovered,html.theme--documenter-dark .button.is-info.is-outlined:focus,html.theme--documenter-dark .button.is-info.is-outlined.is-focused{background-color:#024c7d;border-color:#024c7d;color:#fff}html.theme--documenter-dark .button.is-info.is-outlined.is-loading::after{border-color:transparent transparent #024c7d #024c7d !important}html.theme--documenter-dark .button.is-info.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-info.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-info.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-info.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-info.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-info.is-outlined{background-color:transparent;border-color:#024c7d;box-shadow:none;color:#024c7d}html.theme--documenter-dark .button.is-info.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-info.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-focused{background-color:#fff;color:#024c7d}html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-info.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #024c7d #024c7d !important}html.theme--documenter-dark .button.is-info.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-info.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-info.is-light{background-color:#ebf7ff;color:#0e9dfb}html.theme--documenter-dark .button.is-info.is-light:hover,html.theme--documenter-dark .button.is-info.is-light.is-hovered{background-color:#def2fe;border-color:transparent;color:#0e9dfb}html.theme--documenter-dark .button.is-info.is-light:active,html.theme--documenter-dark .button.is-info.is-light.is-active{background-color:#d2edfe;border-color:transparent;color:#0e9dfb}html.theme--documenter-dark .button.is-success{background-color:#008438;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-success:hover,html.theme--documenter-dark .button.is-success.is-hovered{background-color:#073;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-success:focus,html.theme--documenter-dark .button.is-success.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-success:focus:not(:active),html.theme--documenter-dark .button.is-success.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(0,132,56,0.25)}html.theme--documenter-dark .button.is-success:active,html.theme--documenter-dark .button.is-success.is-active{background-color:#006b2d;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-success[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-success{background-color:#008438;border-color:#008438;box-shadow:none}html.theme--documenter-dark .button.is-success.is-inverted{background-color:#fff;color:#008438}html.theme--documenter-dark .button.is-success.is-inverted:hover,html.theme--documenter-dark .button.is-success.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-success.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-success.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#008438}html.theme--documenter-dark .button.is-success.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-success.is-outlined{background-color:transparent;border-color:#008438;color:#008438}html.theme--documenter-dark .button.is-success.is-outlined:hover,html.theme--documenter-dark .button.is-success.is-outlined.is-hovered,html.theme--documenter-dark .button.is-success.is-outlined:focus,html.theme--documenter-dark .button.is-success.is-outlined.is-focused{background-color:#008438;border-color:#008438;color:#fff}html.theme--documenter-dark .button.is-success.is-outlined.is-loading::after{border-color:transparent transparent #008438 #008438 !important}html.theme--documenter-dark .button.is-success.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-success.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-success.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-success.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-success.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-success.is-outlined{background-color:transparent;border-color:#008438;box-shadow:none;color:#008438}html.theme--documenter-dark .button.is-success.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-success.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-focused{background-color:#fff;color:#008438}html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-success.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #008438 #008438 !important}html.theme--documenter-dark .button.is-success.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-success.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-success.is-light{background-color:#ebfff3;color:#00eb64}html.theme--documenter-dark .button.is-success.is-light:hover,html.theme--documenter-dark .button.is-success.is-light.is-hovered{background-color:#deffec;border-color:transparent;color:#00eb64}html.theme--documenter-dark .button.is-success.is-light:active,html.theme--documenter-dark .button.is-success.is-light.is-active{background-color:#d1ffe5;border-color:transparent;color:#00eb64}html.theme--documenter-dark .button.is-warning{background-color:#ad8100;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-warning:hover,html.theme--documenter-dark .button.is-warning.is-hovered{background-color:#a07700;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-warning:focus,html.theme--documenter-dark .button.is-warning.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-warning:focus:not(:active),html.theme--documenter-dark .button.is-warning.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(173,129,0,0.25)}html.theme--documenter-dark .button.is-warning:active,html.theme--documenter-dark .button.is-warning.is-active{background-color:#946e00;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-warning[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-warning{background-color:#ad8100;border-color:#ad8100;box-shadow:none}html.theme--documenter-dark .button.is-warning.is-inverted{background-color:#fff;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-inverted:hover,html.theme--documenter-dark .button.is-warning.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-warning.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-warning.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-warning.is-outlined{background-color:transparent;border-color:#ad8100;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-outlined:hover,html.theme--documenter-dark .button.is-warning.is-outlined.is-hovered,html.theme--documenter-dark .button.is-warning.is-outlined:focus,html.theme--documenter-dark .button.is-warning.is-outlined.is-focused{background-color:#ad8100;border-color:#ad8100;color:#fff}html.theme--documenter-dark .button.is-warning.is-outlined.is-loading::after{border-color:transparent transparent #ad8100 #ad8100 !important}html.theme--documenter-dark .button.is-warning.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-warning.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-warning.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-warning.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-warning.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-warning.is-outlined{background-color:transparent;border-color:#ad8100;box-shadow:none;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-focused{background-color:#fff;color:#ad8100}html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #ad8100 #ad8100 !important}html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-warning.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-warning.is-light{background-color:#fffaeb;color:#d19c00}html.theme--documenter-dark .button.is-warning.is-light:hover,html.theme--documenter-dark .button.is-warning.is-light.is-hovered{background-color:#fff7de;border-color:transparent;color:#d19c00}html.theme--documenter-dark .button.is-warning.is-light:active,html.theme--documenter-dark .button.is-warning.is-light.is-active{background-color:#fff3d1;border-color:transparent;color:#d19c00}html.theme--documenter-dark .button.is-danger{background-color:#9e1b0d;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-danger:hover,html.theme--documenter-dark .button.is-danger.is-hovered{background-color:#92190c;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-danger:focus,html.theme--documenter-dark .button.is-danger.is-focused{border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-danger:focus:not(:active),html.theme--documenter-dark .button.is-danger.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(158,27,13,0.25)}html.theme--documenter-dark .button.is-danger:active,html.theme--documenter-dark .button.is-danger.is-active{background-color:#86170b;border-color:transparent;color:#fff}html.theme--documenter-dark .button.is-danger[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-danger{background-color:#9e1b0d;border-color:#9e1b0d;box-shadow:none}html.theme--documenter-dark .button.is-danger.is-inverted{background-color:#fff;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-inverted:hover,html.theme--documenter-dark .button.is-danger.is-inverted.is-hovered{background-color:#f2f2f2}html.theme--documenter-dark .button.is-danger.is-inverted[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-danger.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-loading::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-danger.is-outlined{background-color:transparent;border-color:#9e1b0d;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-outlined:hover,html.theme--documenter-dark .button.is-danger.is-outlined.is-hovered,html.theme--documenter-dark .button.is-danger.is-outlined:focus,html.theme--documenter-dark .button.is-danger.is-outlined.is-focused{background-color:#9e1b0d;border-color:#9e1b0d;color:#fff}html.theme--documenter-dark .button.is-danger.is-outlined.is-loading::after{border-color:transparent transparent #9e1b0d #9e1b0d !important}html.theme--documenter-dark .button.is-danger.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-danger.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-danger.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-danger.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}html.theme--documenter-dark .button.is-danger.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-danger.is-outlined{background-color:transparent;border-color:#9e1b0d;box-shadow:none;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined:hover,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-hovered,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined:focus,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-focused{background-color:#fff;color:#9e1b0d}html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-loading:hover::after,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-loading.is-hovered::after,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-loading:focus::after,html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #9e1b0d #9e1b0d !important}html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined[disabled],fieldset[disabled] html.theme--documenter-dark .button.is-danger.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}html.theme--documenter-dark .button.is-danger.is-light{background-color:#fdeeec;color:#ec311d}html.theme--documenter-dark .button.is-danger.is-light:hover,html.theme--documenter-dark .button.is-danger.is-light.is-hovered{background-color:#fce3e0;border-color:transparent;color:#ec311d}html.theme--documenter-dark .button.is-danger.is-light:active,html.theme--documenter-dark .button.is-danger.is-light.is-active{background-color:#fcd8d5;border-color:transparent;color:#ec311d}html.theme--documenter-dark .button.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.button{font-size:.75rem}html.theme--documenter-dark .button.is-small:not(.is-rounded),html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.button:not(.is-rounded){border-radius:3px}html.theme--documenter-dark .button.is-normal{font-size:1rem}html.theme--documenter-dark .button.is-medium{font-size:1.25rem}html.theme--documenter-dark .button.is-large{font-size:1.5rem}html.theme--documenter-dark .button[disabled],fieldset[disabled] html.theme--documenter-dark .button{background-color:#8c9b9d;border-color:#5e6d6f;box-shadow:none;opacity:.5}html.theme--documenter-dark .button.is-fullwidth{display:flex;width:100%}html.theme--documenter-dark .button.is-loading{color:transparent !important;pointer-events:none}html.theme--documenter-dark .button.is-loading::after{position:absolute;left:calc(50% - (1em * 0.5));top:calc(50% - (1em * 0.5));position:absolute !important}html.theme--documenter-dark .button.is-static{background-color:#282f2f;border-color:#5e6d6f;color:#dbdee0;box-shadow:none;pointer-events:none}html.theme--documenter-dark .button.is-rounded,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.button{border-radius:9999px;padding-left:calc(1em + 0.25em);padding-right:calc(1em + 0.25em)}html.theme--documenter-dark .buttons{align-items:center;display:flex;flex-wrap:wrap;justify-content:flex-start}html.theme--documenter-dark .buttons .button{margin-bottom:0.5rem}html.theme--documenter-dark .buttons .button:not(:last-child):not(.is-fullwidth){margin-right:.5rem}html.theme--documenter-dark .buttons:last-child{margin-bottom:-0.5rem}html.theme--documenter-dark .buttons:not(:last-child){margin-bottom:1rem}html.theme--documenter-dark .buttons.are-small .button:not(.is-normal):not(.is-medium):not(.is-large){font-size:.75rem}html.theme--documenter-dark .buttons.are-small .button:not(.is-normal):not(.is-medium):not(.is-large):not(.is-rounded){border-radius:3px}html.theme--documenter-dark .buttons.are-medium .button:not(.is-small):not(.is-normal):not(.is-large){font-size:1.25rem}html.theme--documenter-dark .buttons.are-large .button:not(.is-small):not(.is-normal):not(.is-medium){font-size:1.5rem}html.theme--documenter-dark .buttons.has-addons .button:not(:first-child){border-bottom-left-radius:0;border-top-left-radius:0}html.theme--documenter-dark .buttons.has-addons .button:not(:last-child){border-bottom-right-radius:0;border-top-right-radius:0;margin-right:-1px}html.theme--documenter-dark .buttons.has-addons .button:last-child{margin-right:0}html.theme--documenter-dark .buttons.has-addons .button:hover,html.theme--documenter-dark .buttons.has-addons .button.is-hovered{z-index:2}html.theme--documenter-dark .buttons.has-addons .button:focus,html.theme--documenter-dark .buttons.has-addons .button.is-focused,html.theme--documenter-dark .buttons.has-addons .button:active,html.theme--documenter-dark .buttons.has-addons .button.is-active,html.theme--documenter-dark .buttons.has-addons .button.is-selected{z-index:3}html.theme--documenter-dark .buttons.has-addons .button:focus:hover,html.theme--documenter-dark .buttons.has-addons .button.is-focused:hover,html.theme--documenter-dark .buttons.has-addons .button:active:hover,html.theme--documenter-dark .buttons.has-addons .button.is-active:hover,html.theme--documenter-dark .buttons.has-addons .button.is-selected:hover{z-index:4}html.theme--documenter-dark .buttons.has-addons .button.is-expanded{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .buttons.is-centered{justify-content:center}html.theme--documenter-dark .buttons.is-centered:not(.has-addons) .button:not(.is-fullwidth){margin-left:0.25rem;margin-right:0.25rem}html.theme--documenter-dark .buttons.is-right{justify-content:flex-end}html.theme--documenter-dark .buttons.is-right:not(.has-addons) .button:not(.is-fullwidth){margin-left:0.25rem;margin-right:0.25rem}@media screen and (max-width: 768px){html.theme--documenter-dark .button.is-responsive.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-responsive{font-size:.5625rem}html.theme--documenter-dark .button.is-responsive,html.theme--documenter-dark .button.is-responsive.is-normal{font-size:.65625rem}html.theme--documenter-dark .button.is-responsive.is-medium{font-size:.75rem}html.theme--documenter-dark .button.is-responsive.is-large{font-size:1rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .button.is-responsive.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-responsive{font-size:.65625rem}html.theme--documenter-dark .button.is-responsive,html.theme--documenter-dark .button.is-responsive.is-normal{font-size:.75rem}html.theme--documenter-dark .button.is-responsive.is-medium{font-size:1rem}html.theme--documenter-dark .button.is-responsive.is-large{font-size:1.25rem}}html.theme--documenter-dark .container{flex-grow:1;margin:0 auto;position:relative;width:auto}html.theme--documenter-dark .container.is-fluid{max-width:none !important;padding-left:32px;padding-right:32px;width:100%}@media screen and (min-width: 1056px){html.theme--documenter-dark .container{max-width:992px}}@media screen and (max-width: 1215px){html.theme--documenter-dark .container.is-widescreen:not(.is-max-desktop){max-width:1152px}}@media screen and (max-width: 1407px){html.theme--documenter-dark .container.is-fullhd:not(.is-max-desktop):not(.is-max-widescreen){max-width:1344px}}@media screen and (min-width: 1216px){html.theme--documenter-dark .container:not(.is-max-desktop){max-width:1152px}}@media screen and (min-width: 1408px){html.theme--documenter-dark .container:not(.is-max-desktop):not(.is-max-widescreen){max-width:1344px}}html.theme--documenter-dark .content li+li{margin-top:0.25em}html.theme--documenter-dark .content p:not(:last-child),html.theme--documenter-dark .content dl:not(:last-child),html.theme--documenter-dark .content ol:not(:last-child),html.theme--documenter-dark .content ul:not(:last-child),html.theme--documenter-dark .content blockquote:not(:last-child),html.theme--documenter-dark .content pre:not(:last-child),html.theme--documenter-dark .content table:not(:last-child){margin-bottom:1em}html.theme--documenter-dark .content h1,html.theme--documenter-dark .content h2,html.theme--documenter-dark .content h3,html.theme--documenter-dark .content h4,html.theme--documenter-dark .content h5,html.theme--documenter-dark .content h6{color:#f2f2f2;font-weight:600;line-height:1.125}html.theme--documenter-dark .content h1{font-size:2em;margin-bottom:0.5em}html.theme--documenter-dark .content h1:not(:first-child){margin-top:1em}html.theme--documenter-dark .content h2{font-size:1.75em;margin-bottom:0.5714em}html.theme--documenter-dark .content h2:not(:first-child){margin-top:1.1428em}html.theme--documenter-dark .content h3{font-size:1.5em;margin-bottom:0.6666em}html.theme--documenter-dark .content h3:not(:first-child){margin-top:1.3333em}html.theme--documenter-dark .content h4{font-size:1.25em;margin-bottom:0.8em}html.theme--documenter-dark .content h5{font-size:1.125em;margin-bottom:0.8888em}html.theme--documenter-dark .content h6{font-size:1em;margin-bottom:1em}html.theme--documenter-dark .content blockquote{background-color:#282f2f;border-left:5px solid #5e6d6f;padding:1.25em 1.5em}html.theme--documenter-dark .content ol{list-style-position:outside;margin-left:2em;margin-top:1em}html.theme--documenter-dark .content ol:not([type]){list-style-type:decimal}html.theme--documenter-dark .content ol.is-lower-alpha:not([type]){list-style-type:lower-alpha}html.theme--documenter-dark .content ol.is-lower-roman:not([type]){list-style-type:lower-roman}html.theme--documenter-dark .content ol.is-upper-alpha:not([type]){list-style-type:upper-alpha}html.theme--documenter-dark .content ol.is-upper-roman:not([type]){list-style-type:upper-roman}html.theme--documenter-dark .content ul{list-style:disc outside;margin-left:2em;margin-top:1em}html.theme--documenter-dark .content ul ul{list-style-type:circle;margin-top:0.5em}html.theme--documenter-dark .content ul ul ul{list-style-type:square}html.theme--documenter-dark .content dd{margin-left:2em}html.theme--documenter-dark .content figure{margin-left:2em;margin-right:2em;text-align:center}html.theme--documenter-dark .content figure:not(:first-child){margin-top:2em}html.theme--documenter-dark .content figure:not(:last-child){margin-bottom:2em}html.theme--documenter-dark .content figure img{display:inline-block}html.theme--documenter-dark .content figure figcaption{font-style:italic}html.theme--documenter-dark .content pre{-webkit-overflow-scrolling:touch;overflow-x:auto;padding:0;white-space:pre;word-wrap:normal}html.theme--documenter-dark .content sup,html.theme--documenter-dark .content sub{font-size:75%}html.theme--documenter-dark .content table{width:100%}html.theme--documenter-dark .content table td,html.theme--documenter-dark .content table th{border:1px solid #5e6d6f;border-width:0 0 1px;padding:0.5em 0.75em;vertical-align:top}html.theme--documenter-dark .content table th{color:#f2f2f2}html.theme--documenter-dark .content table th:not([align]){text-align:inherit}html.theme--documenter-dark .content table thead td,html.theme--documenter-dark .content table thead th{border-width:0 0 2px;color:#f2f2f2}html.theme--documenter-dark .content table tfoot td,html.theme--documenter-dark .content table tfoot th{border-width:2px 0 0;color:#f2f2f2}html.theme--documenter-dark .content table tbody tr:last-child td,html.theme--documenter-dark .content table tbody tr:last-child th{border-bottom-width:0}html.theme--documenter-dark .content .tabs li+li{margin-top:0}html.theme--documenter-dark .content.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.content{font-size:.75rem}html.theme--documenter-dark .content.is-normal{font-size:1rem}html.theme--documenter-dark .content.is-medium{font-size:1.25rem}html.theme--documenter-dark .content.is-large{font-size:1.5rem}html.theme--documenter-dark .icon{align-items:center;display:inline-flex;justify-content:center;height:1.5rem;width:1.5rem}html.theme--documenter-dark .icon.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.icon{height:1rem;width:1rem}html.theme--documenter-dark .icon.is-medium{height:2rem;width:2rem}html.theme--documenter-dark .icon.is-large{height:3rem;width:3rem}html.theme--documenter-dark .icon-text{align-items:flex-start;color:inherit;display:inline-flex;flex-wrap:wrap;line-height:1.5rem;vertical-align:top}html.theme--documenter-dark .icon-text .icon{flex-grow:0;flex-shrink:0}html.theme--documenter-dark .icon-text .icon:not(:last-child){margin-right:.25em}html.theme--documenter-dark .icon-text .icon:not(:first-child){margin-left:.25em}html.theme--documenter-dark div.icon-text{display:flex}html.theme--documenter-dark .image,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img{display:block;position:relative}html.theme--documenter-dark .image img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img img{display:block;height:auto;width:100%}html.theme--documenter-dark .image img.is-rounded,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img img.is-rounded{border-radius:9999px}html.theme--documenter-dark .image.is-fullwidth,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-fullwidth{width:100%}html.theme--documenter-dark .image.is-square img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square img,html.theme--documenter-dark .image.is-square .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square .has-ratio,html.theme--documenter-dark .image.is-1by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1 img,html.theme--documenter-dark .image.is-1by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1 .has-ratio,html.theme--documenter-dark .image.is-5by4 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4 img,html.theme--documenter-dark .image.is-5by4 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4 .has-ratio,html.theme--documenter-dark .image.is-4by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3 img,html.theme--documenter-dark .image.is-4by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3 .has-ratio,html.theme--documenter-dark .image.is-3by2 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2 img,html.theme--documenter-dark .image.is-3by2 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2 .has-ratio,html.theme--documenter-dark .image.is-5by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3 img,html.theme--documenter-dark .image.is-5by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3 .has-ratio,html.theme--documenter-dark .image.is-16by9 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9 img,html.theme--documenter-dark .image.is-16by9 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9 .has-ratio,html.theme--documenter-dark .image.is-2by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1 img,html.theme--documenter-dark .image.is-2by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1 .has-ratio,html.theme--documenter-dark .image.is-3by1 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1 img,html.theme--documenter-dark .image.is-3by1 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1 .has-ratio,html.theme--documenter-dark .image.is-4by5 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5 img,html.theme--documenter-dark .image.is-4by5 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5 .has-ratio,html.theme--documenter-dark .image.is-3by4 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4 img,html.theme--documenter-dark .image.is-3by4 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4 .has-ratio,html.theme--documenter-dark .image.is-2by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3 img,html.theme--documenter-dark .image.is-2by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3 .has-ratio,html.theme--documenter-dark .image.is-3by5 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5 img,html.theme--documenter-dark .image.is-3by5 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5 .has-ratio,html.theme--documenter-dark .image.is-9by16 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16 img,html.theme--documenter-dark .image.is-9by16 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16 .has-ratio,html.theme--documenter-dark .image.is-1by2 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2 img,html.theme--documenter-dark .image.is-1by2 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2 .has-ratio,html.theme--documenter-dark .image.is-1by3 img,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3 img,html.theme--documenter-dark .image.is-1by3 .has-ratio,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3 .has-ratio{height:100%;width:100%}html.theme--documenter-dark .image.is-square,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-square,html.theme--documenter-dark .image.is-1by1,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by1{padding-top:100%}html.theme--documenter-dark .image.is-5by4,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by4{padding-top:80%}html.theme--documenter-dark .image.is-4by3,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by3{padding-top:75%}html.theme--documenter-dark .image.is-3by2,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by2{padding-top:66.6666%}html.theme--documenter-dark .image.is-5by3,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-5by3{padding-top:60%}html.theme--documenter-dark .image.is-16by9,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16by9{padding-top:56.25%}html.theme--documenter-dark .image.is-2by1,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by1{padding-top:50%}html.theme--documenter-dark .image.is-3by1,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by1{padding-top:33.3333%}html.theme--documenter-dark .image.is-4by5,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-4by5{padding-top:125%}html.theme--documenter-dark .image.is-3by4,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by4{padding-top:133.3333%}html.theme--documenter-dark .image.is-2by3,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-2by3{padding-top:150%}html.theme--documenter-dark .image.is-3by5,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-3by5{padding-top:166.6666%}html.theme--documenter-dark .image.is-9by16,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-9by16{padding-top:177.7777%}html.theme--documenter-dark .image.is-1by2,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by2{padding-top:200%}html.theme--documenter-dark .image.is-1by3,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-1by3{padding-top:300%}html.theme--documenter-dark .image.is-16x16,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-16x16{height:16px;width:16px}html.theme--documenter-dark .image.is-24x24,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-24x24{height:24px;width:24px}html.theme--documenter-dark .image.is-32x32,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-32x32{height:32px;width:32px}html.theme--documenter-dark .image.is-48x48,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-48x48{height:48px;width:48px}html.theme--documenter-dark .image.is-64x64,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-64x64{height:64px;width:64px}html.theme--documenter-dark .image.is-96x96,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-96x96{height:96px;width:96px}html.theme--documenter-dark .image.is-128x128,html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img.is-128x128{height:128px;width:128px}html.theme--documenter-dark .notification{background-color:#282f2f;border-radius:.4em;position:relative;padding:1.25rem 2.5rem 1.25rem 1.5rem}html.theme--documenter-dark .notification a:not(.button):not(.dropdown-item){color:currentColor;text-decoration:underline}html.theme--documenter-dark .notification strong{color:currentColor}html.theme--documenter-dark .notification code,html.theme--documenter-dark .notification pre{background:#fff}html.theme--documenter-dark .notification pre code{background:transparent}html.theme--documenter-dark .notification>.delete{right:.5rem;position:absolute;top:0.5rem}html.theme--documenter-dark .notification .title,html.theme--documenter-dark .notification .subtitle,html.theme--documenter-dark .notification .content{color:currentColor}html.theme--documenter-dark .notification.is-white{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .notification.is-black{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .notification.is-light{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .notification.is-dark,html.theme--documenter-dark .content kbd.notification{background-color:#282f2f;color:#fff}html.theme--documenter-dark .notification.is-primary,html.theme--documenter-dark .docstring>section>a.notification.docs-sourcelink{background-color:#375a7f;color:#fff}html.theme--documenter-dark .notification.is-primary.is-light,html.theme--documenter-dark .docstring>section>a.notification.is-light.docs-sourcelink{background-color:#f1f5f9;color:#4d7eb2}html.theme--documenter-dark .notification.is-link{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .notification.is-link.is-light{background-color:#edfdf9;color:#15987e}html.theme--documenter-dark .notification.is-info{background-color:#024c7d;color:#fff}html.theme--documenter-dark .notification.is-info.is-light{background-color:#ebf7ff;color:#0e9dfb}html.theme--documenter-dark .notification.is-success{background-color:#008438;color:#fff}html.theme--documenter-dark .notification.is-success.is-light{background-color:#ebfff3;color:#00eb64}html.theme--documenter-dark .notification.is-warning{background-color:#ad8100;color:#fff}html.theme--documenter-dark .notification.is-warning.is-light{background-color:#fffaeb;color:#d19c00}html.theme--documenter-dark .notification.is-danger{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .notification.is-danger.is-light{background-color:#fdeeec;color:#ec311d}html.theme--documenter-dark .progress{-moz-appearance:none;-webkit-appearance:none;border:none;border-radius:9999px;display:block;height:1rem;overflow:hidden;padding:0;width:100%}html.theme--documenter-dark .progress::-webkit-progress-bar{background-color:#343c3d}html.theme--documenter-dark .progress::-webkit-progress-value{background-color:#dbdee0}html.theme--documenter-dark .progress::-moz-progress-bar{background-color:#dbdee0}html.theme--documenter-dark .progress::-ms-fill{background-color:#dbdee0;border:none}html.theme--documenter-dark .progress.is-white::-webkit-progress-value{background-color:#fff}html.theme--documenter-dark .progress.is-white::-moz-progress-bar{background-color:#fff}html.theme--documenter-dark .progress.is-white::-ms-fill{background-color:#fff}html.theme--documenter-dark .progress.is-white:indeterminate{background-image:linear-gradient(to right, #fff 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-black::-webkit-progress-value{background-color:#0a0a0a}html.theme--documenter-dark .progress.is-black::-moz-progress-bar{background-color:#0a0a0a}html.theme--documenter-dark .progress.is-black::-ms-fill{background-color:#0a0a0a}html.theme--documenter-dark .progress.is-black:indeterminate{background-image:linear-gradient(to right, #0a0a0a 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-light::-webkit-progress-value{background-color:#ecf0f1}html.theme--documenter-dark .progress.is-light::-moz-progress-bar{background-color:#ecf0f1}html.theme--documenter-dark .progress.is-light::-ms-fill{background-color:#ecf0f1}html.theme--documenter-dark .progress.is-light:indeterminate{background-image:linear-gradient(to right, #ecf0f1 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-dark::-webkit-progress-value,html.theme--documenter-dark .content kbd.progress::-webkit-progress-value{background-color:#282f2f}html.theme--documenter-dark .progress.is-dark::-moz-progress-bar,html.theme--documenter-dark .content kbd.progress::-moz-progress-bar{background-color:#282f2f}html.theme--documenter-dark .progress.is-dark::-ms-fill,html.theme--documenter-dark .content kbd.progress::-ms-fill{background-color:#282f2f}html.theme--documenter-dark .progress.is-dark:indeterminate,html.theme--documenter-dark .content kbd.progress:indeterminate{background-image:linear-gradient(to right, #282f2f 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-primary::-webkit-progress-value,html.theme--documenter-dark .docstring>section>a.progress.docs-sourcelink::-webkit-progress-value{background-color:#375a7f}html.theme--documenter-dark .progress.is-primary::-moz-progress-bar,html.theme--documenter-dark .docstring>section>a.progress.docs-sourcelink::-moz-progress-bar{background-color:#375a7f}html.theme--documenter-dark .progress.is-primary::-ms-fill,html.theme--documenter-dark .docstring>section>a.progress.docs-sourcelink::-ms-fill{background-color:#375a7f}html.theme--documenter-dark .progress.is-primary:indeterminate,html.theme--documenter-dark .docstring>section>a.progress.docs-sourcelink:indeterminate{background-image:linear-gradient(to right, #375a7f 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-link::-webkit-progress-value{background-color:#1abc9c}html.theme--documenter-dark .progress.is-link::-moz-progress-bar{background-color:#1abc9c}html.theme--documenter-dark .progress.is-link::-ms-fill{background-color:#1abc9c}html.theme--documenter-dark .progress.is-link:indeterminate{background-image:linear-gradient(to right, #1abc9c 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-info::-webkit-progress-value{background-color:#024c7d}html.theme--documenter-dark .progress.is-info::-moz-progress-bar{background-color:#024c7d}html.theme--documenter-dark .progress.is-info::-ms-fill{background-color:#024c7d}html.theme--documenter-dark .progress.is-info:indeterminate{background-image:linear-gradient(to right, #024c7d 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-success::-webkit-progress-value{background-color:#008438}html.theme--documenter-dark .progress.is-success::-moz-progress-bar{background-color:#008438}html.theme--documenter-dark .progress.is-success::-ms-fill{background-color:#008438}html.theme--documenter-dark .progress.is-success:indeterminate{background-image:linear-gradient(to right, #008438 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-warning::-webkit-progress-value{background-color:#ad8100}html.theme--documenter-dark .progress.is-warning::-moz-progress-bar{background-color:#ad8100}html.theme--documenter-dark .progress.is-warning::-ms-fill{background-color:#ad8100}html.theme--documenter-dark .progress.is-warning:indeterminate{background-image:linear-gradient(to right, #ad8100 30%, #343c3d 30%)}html.theme--documenter-dark .progress.is-danger::-webkit-progress-value{background-color:#9e1b0d}html.theme--documenter-dark .progress.is-danger::-moz-progress-bar{background-color:#9e1b0d}html.theme--documenter-dark .progress.is-danger::-ms-fill{background-color:#9e1b0d}html.theme--documenter-dark .progress.is-danger:indeterminate{background-image:linear-gradient(to right, #9e1b0d 30%, #343c3d 30%)}html.theme--documenter-dark .progress:indeterminate{animation-duration:1.5s;animation-iteration-count:infinite;animation-name:moveIndeterminate;animation-timing-function:linear;background-color:#343c3d;background-image:linear-gradient(to right, #fff 30%, #343c3d 30%);background-position:top left;background-repeat:no-repeat;background-size:150% 150%}html.theme--documenter-dark .progress:indeterminate::-webkit-progress-bar{background-color:transparent}html.theme--documenter-dark .progress:indeterminate::-moz-progress-bar{background-color:transparent}html.theme--documenter-dark .progress:indeterminate::-ms-fill{animation-name:none}html.theme--documenter-dark .progress.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.progress{height:.75rem}html.theme--documenter-dark .progress.is-medium{height:1.25rem}html.theme--documenter-dark .progress.is-large{height:1.5rem}@keyframes moveIndeterminate{from{background-position:200% 0}to{background-position:-200% 0}}html.theme--documenter-dark .table{background-color:#343c3d;color:#fff}html.theme--documenter-dark .table td,html.theme--documenter-dark .table th{border:1px solid #5e6d6f;border-width:0 0 1px;padding:0.5em 0.75em;vertical-align:top}html.theme--documenter-dark .table td.is-white,html.theme--documenter-dark .table th.is-white{background-color:#fff;border-color:#fff;color:#0a0a0a}html.theme--documenter-dark .table td.is-black,html.theme--documenter-dark .table th.is-black{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}html.theme--documenter-dark .table td.is-light,html.theme--documenter-dark .table th.is-light{background-color:#ecf0f1;border-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .table td.is-dark,html.theme--documenter-dark .table th.is-dark{background-color:#282f2f;border-color:#282f2f;color:#fff}html.theme--documenter-dark .table td.is-primary,html.theme--documenter-dark .table th.is-primary{background-color:#375a7f;border-color:#375a7f;color:#fff}html.theme--documenter-dark .table td.is-link,html.theme--documenter-dark .table th.is-link{background-color:#1abc9c;border-color:#1abc9c;color:#fff}html.theme--documenter-dark .table td.is-info,html.theme--documenter-dark .table th.is-info{background-color:#024c7d;border-color:#024c7d;color:#fff}html.theme--documenter-dark .table td.is-success,html.theme--documenter-dark .table th.is-success{background-color:#008438;border-color:#008438;color:#fff}html.theme--documenter-dark .table td.is-warning,html.theme--documenter-dark .table th.is-warning{background-color:#ad8100;border-color:#ad8100;color:#fff}html.theme--documenter-dark .table td.is-danger,html.theme--documenter-dark .table th.is-danger{background-color:#9e1b0d;border-color:#9e1b0d;color:#fff}html.theme--documenter-dark .table td.is-narrow,html.theme--documenter-dark .table th.is-narrow{white-space:nowrap;width:1%}html.theme--documenter-dark .table td.is-selected,html.theme--documenter-dark .table th.is-selected{background-color:#375a7f;color:#fff}html.theme--documenter-dark .table td.is-selected a,html.theme--documenter-dark .table td.is-selected strong,html.theme--documenter-dark .table th.is-selected a,html.theme--documenter-dark .table th.is-selected strong{color:currentColor}html.theme--documenter-dark .table td.is-vcentered,html.theme--documenter-dark .table th.is-vcentered{vertical-align:middle}html.theme--documenter-dark .table th{color:#f2f2f2}html.theme--documenter-dark .table th:not([align]){text-align:left}html.theme--documenter-dark .table tr.is-selected{background-color:#375a7f;color:#fff}html.theme--documenter-dark .table tr.is-selected a,html.theme--documenter-dark .table tr.is-selected strong{color:currentColor}html.theme--documenter-dark .table tr.is-selected td,html.theme--documenter-dark .table tr.is-selected th{border-color:#fff;color:currentColor}html.theme--documenter-dark .table thead{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .table thead td,html.theme--documenter-dark .table thead th{border-width:0 0 2px;color:#f2f2f2}html.theme--documenter-dark .table tfoot{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .table tfoot td,html.theme--documenter-dark .table tfoot th{border-width:2px 0 0;color:#f2f2f2}html.theme--documenter-dark .table tbody{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .table tbody tr:last-child td,html.theme--documenter-dark .table tbody tr:last-child th{border-bottom-width:0}html.theme--documenter-dark .table.is-bordered td,html.theme--documenter-dark .table.is-bordered th{border-width:1px}html.theme--documenter-dark .table.is-bordered tr:last-child td,html.theme--documenter-dark .table.is-bordered tr:last-child th{border-bottom-width:1px}html.theme--documenter-dark .table.is-fullwidth{width:100%}html.theme--documenter-dark .table.is-hoverable tbody tr:not(.is-selected):hover{background-color:#282f2f}html.theme--documenter-dark .table.is-hoverable.is-striped tbody tr:not(.is-selected):hover{background-color:#282f2f}html.theme--documenter-dark .table.is-hoverable.is-striped tbody tr:not(.is-selected):hover:nth-child(even){background-color:#2d3435}html.theme--documenter-dark .table.is-narrow td,html.theme--documenter-dark .table.is-narrow th{padding:0.25em 0.5em}html.theme--documenter-dark .table.is-striped tbody tr:not(.is-selected):nth-child(even){background-color:#282f2f}html.theme--documenter-dark .table-container{-webkit-overflow-scrolling:touch;overflow:auto;overflow-y:hidden;max-width:100%}html.theme--documenter-dark .tags{align-items:center;display:flex;flex-wrap:wrap;justify-content:flex-start}html.theme--documenter-dark .tags .tag,html.theme--documenter-dark .tags .content kbd,html.theme--documenter-dark .content .tags kbd,html.theme--documenter-dark .tags .docstring>section>a.docs-sourcelink{margin-bottom:0.5rem}html.theme--documenter-dark .tags .tag:not(:last-child),html.theme--documenter-dark .tags .content kbd:not(:last-child),html.theme--documenter-dark .content .tags kbd:not(:last-child),html.theme--documenter-dark .tags .docstring>section>a.docs-sourcelink:not(:last-child){margin-right:.5rem}html.theme--documenter-dark .tags:last-child{margin-bottom:-0.5rem}html.theme--documenter-dark .tags:not(:last-child){margin-bottom:1rem}html.theme--documenter-dark .tags.are-medium .tag:not(.is-normal):not(.is-large),html.theme--documenter-dark .tags.are-medium .content kbd:not(.is-normal):not(.is-large),html.theme--documenter-dark .content .tags.are-medium kbd:not(.is-normal):not(.is-large),html.theme--documenter-dark .tags.are-medium .docstring>section>a.docs-sourcelink:not(.is-normal):not(.is-large){font-size:1rem}html.theme--documenter-dark .tags.are-large .tag:not(.is-normal):not(.is-medium),html.theme--documenter-dark .tags.are-large .content kbd:not(.is-normal):not(.is-medium),html.theme--documenter-dark .content .tags.are-large kbd:not(.is-normal):not(.is-medium),html.theme--documenter-dark .tags.are-large .docstring>section>a.docs-sourcelink:not(.is-normal):not(.is-medium){font-size:1.25rem}html.theme--documenter-dark .tags.is-centered{justify-content:center}html.theme--documenter-dark .tags.is-centered .tag,html.theme--documenter-dark .tags.is-centered .content kbd,html.theme--documenter-dark .content .tags.is-centered kbd,html.theme--documenter-dark .tags.is-centered .docstring>section>a.docs-sourcelink{margin-right:0.25rem;margin-left:0.25rem}html.theme--documenter-dark .tags.is-right{justify-content:flex-end}html.theme--documenter-dark .tags.is-right .tag:not(:first-child),html.theme--documenter-dark .tags.is-right .content kbd:not(:first-child),html.theme--documenter-dark .content .tags.is-right kbd:not(:first-child),html.theme--documenter-dark .tags.is-right .docstring>section>a.docs-sourcelink:not(:first-child){margin-left:0.5rem}html.theme--documenter-dark .tags.is-right .tag:not(:last-child),html.theme--documenter-dark .tags.is-right .content kbd:not(:last-child),html.theme--documenter-dark .content .tags.is-right kbd:not(:last-child),html.theme--documenter-dark .tags.is-right .docstring>section>a.docs-sourcelink:not(:last-child){margin-right:0}html.theme--documenter-dark .tags.has-addons .tag,html.theme--documenter-dark .tags.has-addons .content kbd,html.theme--documenter-dark .content .tags.has-addons kbd,html.theme--documenter-dark .tags.has-addons .docstring>section>a.docs-sourcelink{margin-right:0}html.theme--documenter-dark .tags.has-addons .tag:not(:first-child),html.theme--documenter-dark .tags.has-addons .content kbd:not(:first-child),html.theme--documenter-dark .content .tags.has-addons kbd:not(:first-child),html.theme--documenter-dark .tags.has-addons .docstring>section>a.docs-sourcelink:not(:first-child){margin-left:0;border-top-left-radius:0;border-bottom-left-radius:0}html.theme--documenter-dark .tags.has-addons .tag:not(:last-child),html.theme--documenter-dark .tags.has-addons .content kbd:not(:last-child),html.theme--documenter-dark .content .tags.has-addons kbd:not(:last-child),html.theme--documenter-dark .tags.has-addons .docstring>section>a.docs-sourcelink:not(:last-child){border-top-right-radius:0;border-bottom-right-radius:0}html.theme--documenter-dark .tag:not(body),html.theme--documenter-dark .content kbd:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body){align-items:center;background-color:#282f2f;border-radius:.4em;color:#fff;display:inline-flex;font-size:.75rem;height:2em;justify-content:center;line-height:1.5;padding-left:0.75em;padding-right:0.75em;white-space:nowrap}html.theme--documenter-dark .tag:not(body) .delete,html.theme--documenter-dark .content kbd:not(body) .delete,html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body) .delete{margin-left:.25rem;margin-right:-.375rem}html.theme--documenter-dark .tag.is-white:not(body),html.theme--documenter-dark .content kbd.is-white:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-white:not(body){background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .tag.is-black:not(body),html.theme--documenter-dark .content kbd.is-black:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-black:not(body){background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .tag.is-light:not(body),html.theme--documenter-dark .content kbd.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-light:not(body){background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .tag.is-dark:not(body),html.theme--documenter-dark .content kbd:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-dark:not(body),html.theme--documenter-dark .content .docstring>section>kbd:not(body){background-color:#282f2f;color:#fff}html.theme--documenter-dark .tag.is-primary:not(body),html.theme--documenter-dark .content kbd.is-primary:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body){background-color:#375a7f;color:#fff}html.theme--documenter-dark .tag.is-primary.is-light:not(body),html.theme--documenter-dark .content kbd.is-primary.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-light:not(body){background-color:#f1f5f9;color:#4d7eb2}html.theme--documenter-dark .tag.is-link:not(body),html.theme--documenter-dark .content kbd.is-link:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-link:not(body){background-color:#1abc9c;color:#fff}html.theme--documenter-dark .tag.is-link.is-light:not(body),html.theme--documenter-dark .content kbd.is-link.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-link.is-light:not(body){background-color:#edfdf9;color:#15987e}html.theme--documenter-dark .tag.is-info:not(body),html.theme--documenter-dark .content kbd.is-info:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-info:not(body){background-color:#024c7d;color:#fff}html.theme--documenter-dark .tag.is-info.is-light:not(body),html.theme--documenter-dark .content kbd.is-info.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-info.is-light:not(body){background-color:#ebf7ff;color:#0e9dfb}html.theme--documenter-dark .tag.is-success:not(body),html.theme--documenter-dark .content kbd.is-success:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-success:not(body){background-color:#008438;color:#fff}html.theme--documenter-dark .tag.is-success.is-light:not(body),html.theme--documenter-dark .content kbd.is-success.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-success.is-light:not(body){background-color:#ebfff3;color:#00eb64}html.theme--documenter-dark .tag.is-warning:not(body),html.theme--documenter-dark .content kbd.is-warning:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-warning:not(body){background-color:#ad8100;color:#fff}html.theme--documenter-dark .tag.is-warning.is-light:not(body),html.theme--documenter-dark .content kbd.is-warning.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-warning.is-light:not(body){background-color:#fffaeb;color:#d19c00}html.theme--documenter-dark .tag.is-danger:not(body),html.theme--documenter-dark .content kbd.is-danger:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-danger:not(body){background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .tag.is-danger.is-light:not(body),html.theme--documenter-dark .content kbd.is-danger.is-light:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-danger.is-light:not(body){background-color:#fdeeec;color:#ec311d}html.theme--documenter-dark .tag.is-normal:not(body),html.theme--documenter-dark .content kbd.is-normal:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-normal:not(body){font-size:.75rem}html.theme--documenter-dark .tag.is-medium:not(body),html.theme--documenter-dark .content kbd.is-medium:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-medium:not(body){font-size:1rem}html.theme--documenter-dark .tag.is-large:not(body),html.theme--documenter-dark .content kbd.is-large:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-large:not(body){font-size:1.25rem}html.theme--documenter-dark .tag:not(body) .icon:first-child:not(:last-child),html.theme--documenter-dark .content kbd:not(body) .icon:first-child:not(:last-child),html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body) .icon:first-child:not(:last-child){margin-left:-.375em;margin-right:.1875em}html.theme--documenter-dark .tag:not(body) .icon:last-child:not(:first-child),html.theme--documenter-dark .content kbd:not(body) .icon:last-child:not(:first-child),html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body) .icon:last-child:not(:first-child){margin-left:.1875em;margin-right:-.375em}html.theme--documenter-dark .tag:not(body) .icon:first-child:last-child,html.theme--documenter-dark .content kbd:not(body) .icon:first-child:last-child,html.theme--documenter-dark .docstring>section>a.docs-sourcelink:not(body) .icon:first-child:last-child{margin-left:-.375em;margin-right:-.375em}html.theme--documenter-dark .tag.is-delete:not(body),html.theme--documenter-dark .content kbd.is-delete:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body){margin-left:1px;padding:0;position:relative;width:2em}html.theme--documenter-dark .tag.is-delete:not(body)::before,html.theme--documenter-dark .content kbd.is-delete:not(body)::before,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body)::before,html.theme--documenter-dark .tag.is-delete:not(body)::after,html.theme--documenter-dark .content kbd.is-delete:not(body)::after,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body)::after{background-color:currentColor;content:"";display:block;left:50%;position:absolute;top:50%;transform:translateX(-50%) translateY(-50%) rotate(45deg);transform-origin:center center}html.theme--documenter-dark .tag.is-delete:not(body)::before,html.theme--documenter-dark .content kbd.is-delete:not(body)::before,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body)::before{height:1px;width:50%}html.theme--documenter-dark .tag.is-delete:not(body)::after,html.theme--documenter-dark .content kbd.is-delete:not(body)::after,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body)::after{height:50%;width:1px}html.theme--documenter-dark .tag.is-delete:not(body):hover,html.theme--documenter-dark .content kbd.is-delete:not(body):hover,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body):hover,html.theme--documenter-dark .tag.is-delete:not(body):focus,html.theme--documenter-dark .content kbd.is-delete:not(body):focus,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body):focus{background-color:#1d2122}html.theme--documenter-dark .tag.is-delete:not(body):active,html.theme--documenter-dark .content kbd.is-delete:not(body):active,html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-delete:not(body):active{background-color:#111414}html.theme--documenter-dark .tag.is-rounded:not(body),html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:not(body),html.theme--documenter-dark .content kbd.is-rounded:not(body),html.theme--documenter-dark #documenter .docs-sidebar .content form.docs-search>input:not(body),html.theme--documenter-dark .docstring>section>a.docs-sourcelink.is-rounded:not(body){border-radius:9999px}html.theme--documenter-dark a.tag:hover,html.theme--documenter-dark .docstring>section>a.docs-sourcelink:hover{text-decoration:underline}html.theme--documenter-dark .title,html.theme--documenter-dark .subtitle{word-break:break-word}html.theme--documenter-dark .title em,html.theme--documenter-dark .title span,html.theme--documenter-dark .subtitle em,html.theme--documenter-dark .subtitle span{font-weight:inherit}html.theme--documenter-dark .title sub,html.theme--documenter-dark .subtitle sub{font-size:.75em}html.theme--documenter-dark .title sup,html.theme--documenter-dark .subtitle sup{font-size:.75em}html.theme--documenter-dark .title .tag,html.theme--documenter-dark .title .content kbd,html.theme--documenter-dark .content .title kbd,html.theme--documenter-dark .title .docstring>section>a.docs-sourcelink,html.theme--documenter-dark .subtitle .tag,html.theme--documenter-dark .subtitle .content kbd,html.theme--documenter-dark .content .subtitle kbd,html.theme--documenter-dark .subtitle .docstring>section>a.docs-sourcelink{vertical-align:middle}html.theme--documenter-dark .title{color:#fff;font-size:2rem;font-weight:500;line-height:1.125}html.theme--documenter-dark .title strong{color:inherit;font-weight:inherit}html.theme--documenter-dark .title:not(.is-spaced)+.subtitle{margin-top:-1.25rem}html.theme--documenter-dark .title.is-1{font-size:3rem}html.theme--documenter-dark .title.is-2{font-size:2.5rem}html.theme--documenter-dark .title.is-3{font-size:2rem}html.theme--documenter-dark .title.is-4{font-size:1.5rem}html.theme--documenter-dark .title.is-5{font-size:1.25rem}html.theme--documenter-dark .title.is-6{font-size:1rem}html.theme--documenter-dark .title.is-7{font-size:.75rem}html.theme--documenter-dark .subtitle{color:#8c9b9d;font-size:1.25rem;font-weight:400;line-height:1.25}html.theme--documenter-dark .subtitle strong{color:#8c9b9d;font-weight:600}html.theme--documenter-dark .subtitle:not(.is-spaced)+.title{margin-top:-1.25rem}html.theme--documenter-dark .subtitle.is-1{font-size:3rem}html.theme--documenter-dark .subtitle.is-2{font-size:2.5rem}html.theme--documenter-dark .subtitle.is-3{font-size:2rem}html.theme--documenter-dark .subtitle.is-4{font-size:1.5rem}html.theme--documenter-dark .subtitle.is-5{font-size:1.25rem}html.theme--documenter-dark .subtitle.is-6{font-size:1rem}html.theme--documenter-dark .subtitle.is-7{font-size:.75rem}html.theme--documenter-dark .heading{display:block;font-size:11px;letter-spacing:1px;margin-bottom:5px;text-transform:uppercase}html.theme--documenter-dark .number{align-items:center;background-color:#282f2f;border-radius:9999px;display:inline-flex;font-size:1.25rem;height:2em;justify-content:center;margin-right:1.5rem;min-width:2.5em;padding:0.25rem 0.5rem;text-align:center;vertical-align:top}html.theme--documenter-dark .select select,html.theme--documenter-dark .textarea,html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{background-color:#1f2424;border-color:#5e6d6f;border-radius:.4em;color:#dbdee0}html.theme--documenter-dark .select select::-moz-placeholder,html.theme--documenter-dark .textarea::-moz-placeholder,html.theme--documenter-dark .input::-moz-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input::-moz-placeholder{color:#868c98}html.theme--documenter-dark .select select::-webkit-input-placeholder,html.theme--documenter-dark .textarea::-webkit-input-placeholder,html.theme--documenter-dark .input::-webkit-input-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input::-webkit-input-placeholder{color:#868c98}html.theme--documenter-dark .select select:-moz-placeholder,html.theme--documenter-dark .textarea:-moz-placeholder,html.theme--documenter-dark .input:-moz-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:-moz-placeholder{color:#868c98}html.theme--documenter-dark .select select:-ms-input-placeholder,html.theme--documenter-dark .textarea:-ms-input-placeholder,html.theme--documenter-dark .input:-ms-input-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:-ms-input-placeholder{color:#868c98}html.theme--documenter-dark .select select:hover,html.theme--documenter-dark .textarea:hover,html.theme--documenter-dark .input:hover,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:hover,html.theme--documenter-dark .select select.is-hovered,html.theme--documenter-dark .is-hovered.textarea,html.theme--documenter-dark .is-hovered.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-hovered{border-color:#8c9b9d}html.theme--documenter-dark .select select:focus,html.theme--documenter-dark .textarea:focus,html.theme--documenter-dark .input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:focus,html.theme--documenter-dark .select select.is-focused,html.theme--documenter-dark .is-focused.textarea,html.theme--documenter-dark .is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .select select:active,html.theme--documenter-dark .textarea:active,html.theme--documenter-dark .input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:active,html.theme--documenter-dark .select select.is-active,html.theme--documenter-dark .is-active.textarea,html.theme--documenter-dark .is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{border-color:#1abc9c;box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .select select[disabled],html.theme--documenter-dark .textarea[disabled],html.theme--documenter-dark .input[disabled],html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled],fieldset[disabled] html.theme--documenter-dark .select select,fieldset[disabled] html.theme--documenter-dark .textarea,fieldset[disabled] html.theme--documenter-dark .input,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{background-color:#8c9b9d;border-color:#282f2f;box-shadow:none;color:#fff}html.theme--documenter-dark .select select[disabled]::-moz-placeholder,html.theme--documenter-dark .textarea[disabled]::-moz-placeholder,html.theme--documenter-dark .input[disabled]::-moz-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled]::-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .select select::-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .textarea::-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .input::-moz-placeholder,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input::-moz-placeholder{color:rgba(255,255,255,0.3)}html.theme--documenter-dark .select select[disabled]::-webkit-input-placeholder,html.theme--documenter-dark .textarea[disabled]::-webkit-input-placeholder,html.theme--documenter-dark .input[disabled]::-webkit-input-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled]::-webkit-input-placeholder,fieldset[disabled] html.theme--documenter-dark .select select::-webkit-input-placeholder,fieldset[disabled] html.theme--documenter-dark .textarea::-webkit-input-placeholder,fieldset[disabled] html.theme--documenter-dark .input::-webkit-input-placeholder,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input::-webkit-input-placeholder{color:rgba(255,255,255,0.3)}html.theme--documenter-dark .select select[disabled]:-moz-placeholder,html.theme--documenter-dark .textarea[disabled]:-moz-placeholder,html.theme--documenter-dark .input[disabled]:-moz-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled]:-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .select select:-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .textarea:-moz-placeholder,fieldset[disabled] html.theme--documenter-dark .input:-moz-placeholder,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:-moz-placeholder{color:rgba(255,255,255,0.3)}html.theme--documenter-dark .select select[disabled]:-ms-input-placeholder,html.theme--documenter-dark .textarea[disabled]:-ms-input-placeholder,html.theme--documenter-dark .input[disabled]:-ms-input-placeholder,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[disabled]:-ms-input-placeholder,fieldset[disabled] html.theme--documenter-dark .select select:-ms-input-placeholder,fieldset[disabled] html.theme--documenter-dark .textarea:-ms-input-placeholder,fieldset[disabled] html.theme--documenter-dark .input:-ms-input-placeholder,fieldset[disabled] html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input:-ms-input-placeholder{color:rgba(255,255,255,0.3)}html.theme--documenter-dark .textarea,html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{box-shadow:inset 0 0.0625em 0.125em rgba(10,10,10,0.05);max-width:100%;width:100%}html.theme--documenter-dark .textarea[readonly],html.theme--documenter-dark .input[readonly],html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input[readonly]{box-shadow:none}html.theme--documenter-dark .is-white.textarea,html.theme--documenter-dark .is-white.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-white{border-color:#fff}html.theme--documenter-dark .is-white.textarea:focus,html.theme--documenter-dark .is-white.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-white:focus,html.theme--documenter-dark .is-white.is-focused.textarea,html.theme--documenter-dark .is-white.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-white.textarea:active,html.theme--documenter-dark .is-white.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-white:active,html.theme--documenter-dark .is-white.is-active.textarea,html.theme--documenter-dark .is-white.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}html.theme--documenter-dark .is-black.textarea,html.theme--documenter-dark .is-black.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-black{border-color:#0a0a0a}html.theme--documenter-dark .is-black.textarea:focus,html.theme--documenter-dark .is-black.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-black:focus,html.theme--documenter-dark .is-black.is-focused.textarea,html.theme--documenter-dark .is-black.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-black.textarea:active,html.theme--documenter-dark .is-black.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-black:active,html.theme--documenter-dark .is-black.is-active.textarea,html.theme--documenter-dark .is-black.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}html.theme--documenter-dark .is-light.textarea,html.theme--documenter-dark .is-light.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-light{border-color:#ecf0f1}html.theme--documenter-dark .is-light.textarea:focus,html.theme--documenter-dark .is-light.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-light:focus,html.theme--documenter-dark .is-light.is-focused.textarea,html.theme--documenter-dark .is-light.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-light.textarea:active,html.theme--documenter-dark .is-light.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-light:active,html.theme--documenter-dark .is-light.is-active.textarea,html.theme--documenter-dark .is-light.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(236,240,241,0.25)}html.theme--documenter-dark .is-dark.textarea,html.theme--documenter-dark .content kbd.textarea,html.theme--documenter-dark .is-dark.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-dark,html.theme--documenter-dark .content kbd.input{border-color:#282f2f}html.theme--documenter-dark .is-dark.textarea:focus,html.theme--documenter-dark .content kbd.textarea:focus,html.theme--documenter-dark .is-dark.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-dark:focus,html.theme--documenter-dark .content kbd.input:focus,html.theme--documenter-dark .is-dark.is-focused.textarea,html.theme--documenter-dark .content kbd.is-focused.textarea,html.theme--documenter-dark .is-dark.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .content kbd.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar .content form.docs-search>input.is-focused,html.theme--documenter-dark .is-dark.textarea:active,html.theme--documenter-dark .content kbd.textarea:active,html.theme--documenter-dark .is-dark.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-dark:active,html.theme--documenter-dark .content kbd.input:active,html.theme--documenter-dark .is-dark.is-active.textarea,html.theme--documenter-dark .content kbd.is-active.textarea,html.theme--documenter-dark .is-dark.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active,html.theme--documenter-dark .content kbd.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar .content form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(40,47,47,0.25)}html.theme--documenter-dark .is-primary.textarea,html.theme--documenter-dark .docstring>section>a.textarea.docs-sourcelink,html.theme--documenter-dark .is-primary.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-primary,html.theme--documenter-dark .docstring>section>a.input.docs-sourcelink{border-color:#375a7f}html.theme--documenter-dark .is-primary.textarea:focus,html.theme--documenter-dark .docstring>section>a.textarea.docs-sourcelink:focus,html.theme--documenter-dark .is-primary.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-primary:focus,html.theme--documenter-dark .docstring>section>a.input.docs-sourcelink:focus,html.theme--documenter-dark .is-primary.is-focused.textarea,html.theme--documenter-dark .docstring>section>a.is-focused.textarea.docs-sourcelink,html.theme--documenter-dark .is-primary.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .docstring>section>a.is-focused.input.docs-sourcelink,html.theme--documenter-dark .is-primary.textarea:active,html.theme--documenter-dark .docstring>section>a.textarea.docs-sourcelink:active,html.theme--documenter-dark .is-primary.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-primary:active,html.theme--documenter-dark .docstring>section>a.input.docs-sourcelink:active,html.theme--documenter-dark .is-primary.is-active.textarea,html.theme--documenter-dark .docstring>section>a.is-active.textarea.docs-sourcelink,html.theme--documenter-dark .is-primary.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active,html.theme--documenter-dark .docstring>section>a.is-active.input.docs-sourcelink{box-shadow:0 0 0 0.125em rgba(55,90,127,0.25)}html.theme--documenter-dark .is-link.textarea,html.theme--documenter-dark .is-link.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-link{border-color:#1abc9c}html.theme--documenter-dark .is-link.textarea:focus,html.theme--documenter-dark .is-link.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-link:focus,html.theme--documenter-dark .is-link.is-focused.textarea,html.theme--documenter-dark .is-link.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-link.textarea:active,html.theme--documenter-dark .is-link.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-link:active,html.theme--documenter-dark .is-link.is-active.textarea,html.theme--documenter-dark .is-link.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .is-info.textarea,html.theme--documenter-dark .is-info.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-info{border-color:#024c7d}html.theme--documenter-dark .is-info.textarea:focus,html.theme--documenter-dark .is-info.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-info:focus,html.theme--documenter-dark .is-info.is-focused.textarea,html.theme--documenter-dark .is-info.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-info.textarea:active,html.theme--documenter-dark .is-info.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-info:active,html.theme--documenter-dark .is-info.is-active.textarea,html.theme--documenter-dark .is-info.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(2,76,125,0.25)}html.theme--documenter-dark .is-success.textarea,html.theme--documenter-dark .is-success.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-success{border-color:#008438}html.theme--documenter-dark .is-success.textarea:focus,html.theme--documenter-dark .is-success.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-success:focus,html.theme--documenter-dark .is-success.is-focused.textarea,html.theme--documenter-dark .is-success.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-success.textarea:active,html.theme--documenter-dark .is-success.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-success:active,html.theme--documenter-dark .is-success.is-active.textarea,html.theme--documenter-dark .is-success.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(0,132,56,0.25)}html.theme--documenter-dark .is-warning.textarea,html.theme--documenter-dark .is-warning.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-warning{border-color:#ad8100}html.theme--documenter-dark .is-warning.textarea:focus,html.theme--documenter-dark .is-warning.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-warning:focus,html.theme--documenter-dark .is-warning.is-focused.textarea,html.theme--documenter-dark .is-warning.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-warning.textarea:active,html.theme--documenter-dark .is-warning.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-warning:active,html.theme--documenter-dark .is-warning.is-active.textarea,html.theme--documenter-dark .is-warning.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(173,129,0,0.25)}html.theme--documenter-dark .is-danger.textarea,html.theme--documenter-dark .is-danger.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-danger{border-color:#9e1b0d}html.theme--documenter-dark .is-danger.textarea:focus,html.theme--documenter-dark .is-danger.input:focus,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-danger:focus,html.theme--documenter-dark .is-danger.is-focused.textarea,html.theme--documenter-dark .is-danger.is-focused.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-focused,html.theme--documenter-dark .is-danger.textarea:active,html.theme--documenter-dark .is-danger.input:active,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-danger:active,html.theme--documenter-dark .is-danger.is-active.textarea,html.theme--documenter-dark .is-danger.is-active.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(158,27,13,0.25)}html.theme--documenter-dark .is-small.textarea,html.theme--documenter-dark .is-small.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{border-radius:3px;font-size:.75rem}html.theme--documenter-dark .is-medium.textarea,html.theme--documenter-dark .is-medium.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-medium{font-size:1.25rem}html.theme--documenter-dark .is-large.textarea,html.theme--documenter-dark .is-large.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-large{font-size:1.5rem}html.theme--documenter-dark .is-fullwidth.textarea,html.theme--documenter-dark .is-fullwidth.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-fullwidth{display:block;width:100%}html.theme--documenter-dark .is-inline.textarea,html.theme--documenter-dark .is-inline.input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-inline{display:inline;width:auto}html.theme--documenter-dark .input.is-rounded,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{border-radius:9999px;padding-left:calc(calc(0.75em - 1px) + 0.375em);padding-right:calc(calc(0.75em - 1px) + 0.375em)}html.theme--documenter-dark .input.is-static,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-static{background-color:transparent;border-color:transparent;box-shadow:none;padding-left:0;padding-right:0}html.theme--documenter-dark .textarea{display:block;max-width:100%;min-width:100%;padding:calc(0.75em - 1px);resize:vertical}html.theme--documenter-dark .textarea:not([rows]){max-height:40em;min-height:8em}html.theme--documenter-dark .textarea[rows]{height:initial}html.theme--documenter-dark .textarea.has-fixed-size{resize:none}html.theme--documenter-dark .radio,html.theme--documenter-dark .checkbox{cursor:pointer;display:inline-block;line-height:1.25;position:relative}html.theme--documenter-dark .radio input,html.theme--documenter-dark .checkbox input{cursor:pointer}html.theme--documenter-dark .radio:hover,html.theme--documenter-dark .checkbox:hover{color:#8c9b9d}html.theme--documenter-dark .radio[disabled],html.theme--documenter-dark .checkbox[disabled],fieldset[disabled] html.theme--documenter-dark .radio,fieldset[disabled] html.theme--documenter-dark .checkbox,html.theme--documenter-dark .radio input[disabled],html.theme--documenter-dark .checkbox input[disabled]{color:#fff;cursor:not-allowed}html.theme--documenter-dark .radio+.radio{margin-left:.5em}html.theme--documenter-dark .select{display:inline-block;max-width:100%;position:relative;vertical-align:top}html.theme--documenter-dark .select:not(.is-multiple){height:2.5em}html.theme--documenter-dark .select:not(.is-multiple):not(.is-loading)::after{border-color:#1abc9c;right:1.125em;z-index:4}html.theme--documenter-dark .select.is-rounded select,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.select select{border-radius:9999px;padding-left:1em}html.theme--documenter-dark .select select{cursor:pointer;display:block;font-size:1em;max-width:100%;outline:none}html.theme--documenter-dark .select select::-ms-expand{display:none}html.theme--documenter-dark .select select[disabled]:hover,fieldset[disabled] html.theme--documenter-dark .select select:hover{border-color:#282f2f}html.theme--documenter-dark .select select:not([multiple]){padding-right:2.5em}html.theme--documenter-dark .select select[multiple]{height:auto;padding:0}html.theme--documenter-dark .select select[multiple] option{padding:0.5em 1em}html.theme--documenter-dark .select:not(.is-multiple):not(.is-loading):hover::after{border-color:#8c9b9d}html.theme--documenter-dark .select.is-white:not(:hover)::after{border-color:#fff}html.theme--documenter-dark .select.is-white select{border-color:#fff}html.theme--documenter-dark .select.is-white select:hover,html.theme--documenter-dark .select.is-white select.is-hovered{border-color:#f2f2f2}html.theme--documenter-dark .select.is-white select:focus,html.theme--documenter-dark .select.is-white select.is-focused,html.theme--documenter-dark .select.is-white select:active,html.theme--documenter-dark .select.is-white select.is-active{box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}html.theme--documenter-dark .select.is-black:not(:hover)::after{border-color:#0a0a0a}html.theme--documenter-dark .select.is-black select{border-color:#0a0a0a}html.theme--documenter-dark .select.is-black select:hover,html.theme--documenter-dark .select.is-black select.is-hovered{border-color:#000}html.theme--documenter-dark .select.is-black select:focus,html.theme--documenter-dark .select.is-black select.is-focused,html.theme--documenter-dark .select.is-black select:active,html.theme--documenter-dark .select.is-black select.is-active{box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}html.theme--documenter-dark .select.is-light:not(:hover)::after{border-color:#ecf0f1}html.theme--documenter-dark .select.is-light select{border-color:#ecf0f1}html.theme--documenter-dark .select.is-light select:hover,html.theme--documenter-dark .select.is-light select.is-hovered{border-color:#dde4e6}html.theme--documenter-dark .select.is-light select:focus,html.theme--documenter-dark .select.is-light select.is-focused,html.theme--documenter-dark .select.is-light select:active,html.theme--documenter-dark .select.is-light select.is-active{box-shadow:0 0 0 0.125em rgba(236,240,241,0.25)}html.theme--documenter-dark .select.is-dark:not(:hover)::after,html.theme--documenter-dark .content kbd.select:not(:hover)::after{border-color:#282f2f}html.theme--documenter-dark .select.is-dark select,html.theme--documenter-dark .content kbd.select select{border-color:#282f2f}html.theme--documenter-dark .select.is-dark select:hover,html.theme--documenter-dark .content kbd.select select:hover,html.theme--documenter-dark .select.is-dark select.is-hovered,html.theme--documenter-dark .content kbd.select select.is-hovered{border-color:#1d2122}html.theme--documenter-dark .select.is-dark select:focus,html.theme--documenter-dark .content kbd.select select:focus,html.theme--documenter-dark .select.is-dark select.is-focused,html.theme--documenter-dark .content kbd.select select.is-focused,html.theme--documenter-dark .select.is-dark select:active,html.theme--documenter-dark .content kbd.select select:active,html.theme--documenter-dark .select.is-dark select.is-active,html.theme--documenter-dark .content kbd.select select.is-active{box-shadow:0 0 0 0.125em rgba(40,47,47,0.25)}html.theme--documenter-dark .select.is-primary:not(:hover)::after,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink:not(:hover)::after{border-color:#375a7f}html.theme--documenter-dark .select.is-primary select,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select{border-color:#375a7f}html.theme--documenter-dark .select.is-primary select:hover,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select:hover,html.theme--documenter-dark .select.is-primary select.is-hovered,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select.is-hovered{border-color:#2f4d6d}html.theme--documenter-dark .select.is-primary select:focus,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select:focus,html.theme--documenter-dark .select.is-primary select.is-focused,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select.is-focused,html.theme--documenter-dark .select.is-primary select:active,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select:active,html.theme--documenter-dark .select.is-primary select.is-active,html.theme--documenter-dark .docstring>section>a.select.docs-sourcelink select.is-active{box-shadow:0 0 0 0.125em rgba(55,90,127,0.25)}html.theme--documenter-dark .select.is-link:not(:hover)::after{border-color:#1abc9c}html.theme--documenter-dark .select.is-link select{border-color:#1abc9c}html.theme--documenter-dark .select.is-link select:hover,html.theme--documenter-dark .select.is-link select.is-hovered{border-color:#17a689}html.theme--documenter-dark .select.is-link select:focus,html.theme--documenter-dark .select.is-link select.is-focused,html.theme--documenter-dark .select.is-link select:active,html.theme--documenter-dark .select.is-link select.is-active{box-shadow:0 0 0 0.125em rgba(26,188,156,0.25)}html.theme--documenter-dark .select.is-info:not(:hover)::after{border-color:#024c7d}html.theme--documenter-dark .select.is-info select{border-color:#024c7d}html.theme--documenter-dark .select.is-info select:hover,html.theme--documenter-dark .select.is-info select.is-hovered{border-color:#023d64}html.theme--documenter-dark .select.is-info select:focus,html.theme--documenter-dark .select.is-info select.is-focused,html.theme--documenter-dark .select.is-info select:active,html.theme--documenter-dark .select.is-info select.is-active{box-shadow:0 0 0 0.125em rgba(2,76,125,0.25)}html.theme--documenter-dark .select.is-success:not(:hover)::after{border-color:#008438}html.theme--documenter-dark .select.is-success select{border-color:#008438}html.theme--documenter-dark .select.is-success select:hover,html.theme--documenter-dark .select.is-success select.is-hovered{border-color:#006b2d}html.theme--documenter-dark .select.is-success select:focus,html.theme--documenter-dark .select.is-success select.is-focused,html.theme--documenter-dark .select.is-success select:active,html.theme--documenter-dark .select.is-success select.is-active{box-shadow:0 0 0 0.125em rgba(0,132,56,0.25)}html.theme--documenter-dark .select.is-warning:not(:hover)::after{border-color:#ad8100}html.theme--documenter-dark .select.is-warning select{border-color:#ad8100}html.theme--documenter-dark .select.is-warning select:hover,html.theme--documenter-dark .select.is-warning select.is-hovered{border-color:#946e00}html.theme--documenter-dark .select.is-warning select:focus,html.theme--documenter-dark .select.is-warning select.is-focused,html.theme--documenter-dark .select.is-warning select:active,html.theme--documenter-dark .select.is-warning select.is-active{box-shadow:0 0 0 0.125em rgba(173,129,0,0.25)}html.theme--documenter-dark .select.is-danger:not(:hover)::after{border-color:#9e1b0d}html.theme--documenter-dark .select.is-danger select{border-color:#9e1b0d}html.theme--documenter-dark .select.is-danger select:hover,html.theme--documenter-dark .select.is-danger select.is-hovered{border-color:#86170b}html.theme--documenter-dark .select.is-danger select:focus,html.theme--documenter-dark .select.is-danger select.is-focused,html.theme--documenter-dark .select.is-danger select:active,html.theme--documenter-dark .select.is-danger select.is-active{box-shadow:0 0 0 0.125em rgba(158,27,13,0.25)}html.theme--documenter-dark .select.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.select{border-radius:3px;font-size:.75rem}html.theme--documenter-dark .select.is-medium{font-size:1.25rem}html.theme--documenter-dark .select.is-large{font-size:1.5rem}html.theme--documenter-dark .select.is-disabled::after{border-color:#fff !important;opacity:0.5}html.theme--documenter-dark .select.is-fullwidth{width:100%}html.theme--documenter-dark .select.is-fullwidth select{width:100%}html.theme--documenter-dark .select.is-loading::after{margin-top:0;position:absolute;right:.625em;top:0.625em;transform:none}html.theme--documenter-dark .select.is-loading.is-small:after,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-loading:after{font-size:.75rem}html.theme--documenter-dark .select.is-loading.is-medium:after{font-size:1.25rem}html.theme--documenter-dark .select.is-loading.is-large:after{font-size:1.5rem}html.theme--documenter-dark .file{align-items:stretch;display:flex;justify-content:flex-start;position:relative}html.theme--documenter-dark .file.is-white .file-cta{background-color:#fff;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .file.is-white:hover .file-cta,html.theme--documenter-dark .file.is-white.is-hovered .file-cta{background-color:#f9f9f9;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .file.is-white:focus .file-cta,html.theme--documenter-dark .file.is-white.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(255,255,255,0.25);color:#0a0a0a}html.theme--documenter-dark .file.is-white:active .file-cta,html.theme--documenter-dark .file.is-white.is-active .file-cta{background-color:#f2f2f2;border-color:transparent;color:#0a0a0a}html.theme--documenter-dark .file.is-black .file-cta{background-color:#0a0a0a;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-black:hover .file-cta,html.theme--documenter-dark .file.is-black.is-hovered .file-cta{background-color:#040404;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-black:focus .file-cta,html.theme--documenter-dark .file.is-black.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(10,10,10,0.25);color:#fff}html.theme--documenter-dark .file.is-black:active .file-cta,html.theme--documenter-dark .file.is-black.is-active .file-cta{background-color:#000;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-light .file-cta{background-color:#ecf0f1;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .file.is-light:hover .file-cta,html.theme--documenter-dark .file.is-light.is-hovered .file-cta{background-color:#e5eaec;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .file.is-light:focus .file-cta,html.theme--documenter-dark .file.is-light.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(236,240,241,0.25);color:rgba(0,0,0,0.7)}html.theme--documenter-dark .file.is-light:active .file-cta,html.theme--documenter-dark .file.is-light.is-active .file-cta{background-color:#dde4e6;border-color:transparent;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .file.is-dark .file-cta,html.theme--documenter-dark .content kbd.file .file-cta{background-color:#282f2f;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-dark:hover .file-cta,html.theme--documenter-dark .content kbd.file:hover .file-cta,html.theme--documenter-dark .file.is-dark.is-hovered .file-cta,html.theme--documenter-dark .content kbd.file.is-hovered .file-cta{background-color:#232829;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-dark:focus .file-cta,html.theme--documenter-dark .content kbd.file:focus .file-cta,html.theme--documenter-dark .file.is-dark.is-focused .file-cta,html.theme--documenter-dark .content kbd.file.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(40,47,47,0.25);color:#fff}html.theme--documenter-dark .file.is-dark:active .file-cta,html.theme--documenter-dark .content kbd.file:active .file-cta,html.theme--documenter-dark .file.is-dark.is-active .file-cta,html.theme--documenter-dark .content kbd.file.is-active .file-cta{background-color:#1d2122;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-primary .file-cta,html.theme--documenter-dark .docstring>section>a.file.docs-sourcelink .file-cta{background-color:#375a7f;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-primary:hover .file-cta,html.theme--documenter-dark .docstring>section>a.file.docs-sourcelink:hover .file-cta,html.theme--documenter-dark .file.is-primary.is-hovered .file-cta,html.theme--documenter-dark .docstring>section>a.file.is-hovered.docs-sourcelink .file-cta{background-color:#335476;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-primary:focus .file-cta,html.theme--documenter-dark .docstring>section>a.file.docs-sourcelink:focus .file-cta,html.theme--documenter-dark .file.is-primary.is-focused .file-cta,html.theme--documenter-dark .docstring>section>a.file.is-focused.docs-sourcelink .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(55,90,127,0.25);color:#fff}html.theme--documenter-dark .file.is-primary:active .file-cta,html.theme--documenter-dark .docstring>section>a.file.docs-sourcelink:active .file-cta,html.theme--documenter-dark .file.is-primary.is-active .file-cta,html.theme--documenter-dark .docstring>section>a.file.is-active.docs-sourcelink .file-cta{background-color:#2f4d6d;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-link .file-cta{background-color:#1abc9c;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-link:hover .file-cta,html.theme--documenter-dark .file.is-link.is-hovered .file-cta{background-color:#18b193;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-link:focus .file-cta,html.theme--documenter-dark .file.is-link.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(26,188,156,0.25);color:#fff}html.theme--documenter-dark .file.is-link:active .file-cta,html.theme--documenter-dark .file.is-link.is-active .file-cta{background-color:#17a689;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-info .file-cta{background-color:#024c7d;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-info:hover .file-cta,html.theme--documenter-dark .file.is-info.is-hovered .file-cta{background-color:#024470;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-info:focus .file-cta,html.theme--documenter-dark .file.is-info.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(2,76,125,0.25);color:#fff}html.theme--documenter-dark .file.is-info:active .file-cta,html.theme--documenter-dark .file.is-info.is-active .file-cta{background-color:#023d64;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-success .file-cta{background-color:#008438;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-success:hover .file-cta,html.theme--documenter-dark .file.is-success.is-hovered .file-cta{background-color:#073;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-success:focus .file-cta,html.theme--documenter-dark .file.is-success.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(0,132,56,0.25);color:#fff}html.theme--documenter-dark .file.is-success:active .file-cta,html.theme--documenter-dark .file.is-success.is-active .file-cta{background-color:#006b2d;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-warning .file-cta{background-color:#ad8100;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-warning:hover .file-cta,html.theme--documenter-dark .file.is-warning.is-hovered .file-cta{background-color:#a07700;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-warning:focus .file-cta,html.theme--documenter-dark .file.is-warning.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(173,129,0,0.25);color:#fff}html.theme--documenter-dark .file.is-warning:active .file-cta,html.theme--documenter-dark .file.is-warning.is-active .file-cta{background-color:#946e00;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-danger .file-cta{background-color:#9e1b0d;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-danger:hover .file-cta,html.theme--documenter-dark .file.is-danger.is-hovered .file-cta{background-color:#92190c;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-danger:focus .file-cta,html.theme--documenter-dark .file.is-danger.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(158,27,13,0.25);color:#fff}html.theme--documenter-dark .file.is-danger:active .file-cta,html.theme--documenter-dark .file.is-danger.is-active .file-cta{background-color:#86170b;border-color:transparent;color:#fff}html.theme--documenter-dark .file.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.file{font-size:.75rem}html.theme--documenter-dark .file.is-normal{font-size:1rem}html.theme--documenter-dark .file.is-medium{font-size:1.25rem}html.theme--documenter-dark .file.is-medium .file-icon .fa{font-size:21px}html.theme--documenter-dark .file.is-large{font-size:1.5rem}html.theme--documenter-dark .file.is-large .file-icon .fa{font-size:28px}html.theme--documenter-dark .file.has-name .file-cta{border-bottom-right-radius:0;border-top-right-radius:0}html.theme--documenter-dark .file.has-name .file-name{border-bottom-left-radius:0;border-top-left-radius:0}html.theme--documenter-dark .file.has-name.is-empty .file-cta{border-radius:.4em}html.theme--documenter-dark .file.has-name.is-empty .file-name{display:none}html.theme--documenter-dark .file.is-boxed .file-label{flex-direction:column}html.theme--documenter-dark .file.is-boxed .file-cta{flex-direction:column;height:auto;padding:1em 3em}html.theme--documenter-dark .file.is-boxed .file-name{border-width:0 1px 1px}html.theme--documenter-dark .file.is-boxed .file-icon{height:1.5em;width:1.5em}html.theme--documenter-dark .file.is-boxed .file-icon .fa{font-size:21px}html.theme--documenter-dark .file.is-boxed.is-small .file-icon .fa,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-boxed .file-icon .fa{font-size:14px}html.theme--documenter-dark .file.is-boxed.is-medium .file-icon .fa{font-size:28px}html.theme--documenter-dark .file.is-boxed.is-large .file-icon .fa{font-size:35px}html.theme--documenter-dark .file.is-boxed.has-name .file-cta{border-radius:.4em .4em 0 0}html.theme--documenter-dark .file.is-boxed.has-name .file-name{border-radius:0 0 .4em .4em;border-width:0 1px 1px}html.theme--documenter-dark .file.is-centered{justify-content:center}html.theme--documenter-dark .file.is-fullwidth .file-label{width:100%}html.theme--documenter-dark .file.is-fullwidth .file-name{flex-grow:1;max-width:none}html.theme--documenter-dark .file.is-right{justify-content:flex-end}html.theme--documenter-dark .file.is-right .file-cta{border-radius:0 .4em .4em 0}html.theme--documenter-dark .file.is-right .file-name{border-radius:.4em 0 0 .4em;border-width:1px 0 1px 1px;order:-1}html.theme--documenter-dark .file-label{align-items:stretch;display:flex;cursor:pointer;justify-content:flex-start;overflow:hidden;position:relative}html.theme--documenter-dark .file-label:hover .file-cta{background-color:#232829;color:#f2f2f2}html.theme--documenter-dark .file-label:hover .file-name{border-color:#596668}html.theme--documenter-dark .file-label:active .file-cta{background-color:#1d2122;color:#f2f2f2}html.theme--documenter-dark .file-label:active .file-name{border-color:#535f61}html.theme--documenter-dark .file-input{height:100%;left:0;opacity:0;outline:none;position:absolute;top:0;width:100%}html.theme--documenter-dark .file-cta,html.theme--documenter-dark .file-name{border-color:#5e6d6f;border-radius:.4em;font-size:1em;padding-left:1em;padding-right:1em;white-space:nowrap}html.theme--documenter-dark .file-cta{background-color:#282f2f;color:#fff}html.theme--documenter-dark .file-name{border-color:#5e6d6f;border-style:solid;border-width:1px 1px 1px 0;display:block;max-width:16em;overflow:hidden;text-align:inherit;text-overflow:ellipsis}html.theme--documenter-dark .file-icon{align-items:center;display:flex;height:1em;justify-content:center;margin-right:.5em;width:1em}html.theme--documenter-dark .file-icon .fa{font-size:14px}html.theme--documenter-dark .label{color:#f2f2f2;display:block;font-size:1rem;font-weight:700}html.theme--documenter-dark .label:not(:last-child){margin-bottom:0.5em}html.theme--documenter-dark .label.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.label{font-size:.75rem}html.theme--documenter-dark .label.is-medium{font-size:1.25rem}html.theme--documenter-dark .label.is-large{font-size:1.5rem}html.theme--documenter-dark .help{display:block;font-size:.75rem;margin-top:0.25rem}html.theme--documenter-dark .help.is-white{color:#fff}html.theme--documenter-dark .help.is-black{color:#0a0a0a}html.theme--documenter-dark .help.is-light{color:#ecf0f1}html.theme--documenter-dark .help.is-dark,html.theme--documenter-dark .content kbd.help{color:#282f2f}html.theme--documenter-dark .help.is-primary,html.theme--documenter-dark .docstring>section>a.help.docs-sourcelink{color:#375a7f}html.theme--documenter-dark .help.is-link{color:#1abc9c}html.theme--documenter-dark .help.is-info{color:#024c7d}html.theme--documenter-dark .help.is-success{color:#008438}html.theme--documenter-dark .help.is-warning{color:#ad8100}html.theme--documenter-dark .help.is-danger{color:#9e1b0d}html.theme--documenter-dark .field:not(:last-child){margin-bottom:0.75rem}html.theme--documenter-dark .field.has-addons{display:flex;justify-content:flex-start}html.theme--documenter-dark .field.has-addons .control:not(:last-child){margin-right:-1px}html.theme--documenter-dark .field.has-addons .control:not(:first-child):not(:last-child) .button,html.theme--documenter-dark .field.has-addons .control:not(:first-child):not(:last-child) .input,html.theme--documenter-dark .field.has-addons .control:not(:first-child):not(:last-child) #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control:not(:first-child):not(:last-child) form.docs-search>input,html.theme--documenter-dark .field.has-addons .control:not(:first-child):not(:last-child) .select select{border-radius:0}html.theme--documenter-dark .field.has-addons .control:first-child:not(:only-child) .button,html.theme--documenter-dark .field.has-addons .control:first-child:not(:only-child) .input,html.theme--documenter-dark .field.has-addons .control:first-child:not(:only-child) #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control:first-child:not(:only-child) form.docs-search>input,html.theme--documenter-dark .field.has-addons .control:first-child:not(:only-child) .select select{border-bottom-right-radius:0;border-top-right-radius:0}html.theme--documenter-dark .field.has-addons .control:last-child:not(:only-child) .button,html.theme--documenter-dark .field.has-addons .control:last-child:not(:only-child) .input,html.theme--documenter-dark .field.has-addons .control:last-child:not(:only-child) #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control:last-child:not(:only-child) form.docs-search>input,html.theme--documenter-dark .field.has-addons .control:last-child:not(:only-child) .select select{border-bottom-left-radius:0;border-top-left-radius:0}html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .button.is-hovered:not([disabled]),html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .input.is-hovered:not([disabled]),html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-hovered:not([disabled]),html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-hovered:not([disabled]),html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .select select.is-hovered:not([disabled]){z-index:2}html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):focus,html.theme--documenter-dark .field.has-addons .control .button.is-focused:not([disabled]),html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):active,html.theme--documenter-dark .field.has-addons .control .button.is-active:not([disabled]),html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):focus,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):focus,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):focus,html.theme--documenter-dark .field.has-addons .control .input.is-focused:not([disabled]),html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-focused:not([disabled]),html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-focused:not([disabled]),html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):active,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):active,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):active,html.theme--documenter-dark .field.has-addons .control .input.is-active:not([disabled]),html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-active:not([disabled]),html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-active:not([disabled]),html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):focus,html.theme--documenter-dark .field.has-addons .control .select select.is-focused:not([disabled]),html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):active,html.theme--documenter-dark .field.has-addons .control .select select.is-active:not([disabled]){z-index:3}html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):focus:hover,html.theme--documenter-dark .field.has-addons .control .button.is-focused:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .button:not([disabled]):active:hover,html.theme--documenter-dark .field.has-addons .control .button.is-active:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):focus:hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):focus:hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):focus:hover,html.theme--documenter-dark .field.has-addons .control .input.is-focused:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-focused:not([disabled]):hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-focused:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .input:not([disabled]):active:hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):active:hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):active:hover,html.theme--documenter-dark .field.has-addons .control .input.is-active:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-active:not([disabled]):hover,html.theme--documenter-dark #documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-active:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):focus:hover,html.theme--documenter-dark .field.has-addons .control .select select.is-focused:not([disabled]):hover,html.theme--documenter-dark .field.has-addons .control .select select:not([disabled]):active:hover,html.theme--documenter-dark .field.has-addons .control .select select.is-active:not([disabled]):hover{z-index:4}html.theme--documenter-dark .field.has-addons .control.is-expanded{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .field.has-addons.has-addons-centered{justify-content:center}html.theme--documenter-dark .field.has-addons.has-addons-right{justify-content:flex-end}html.theme--documenter-dark .field.has-addons.has-addons-fullwidth .control{flex-grow:1;flex-shrink:0}html.theme--documenter-dark .field.is-grouped{display:flex;justify-content:flex-start}html.theme--documenter-dark .field.is-grouped>.control{flex-shrink:0}html.theme--documenter-dark .field.is-grouped>.control:not(:last-child){margin-bottom:0;margin-right:.75rem}html.theme--documenter-dark .field.is-grouped>.control.is-expanded{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .field.is-grouped.is-grouped-centered{justify-content:center}html.theme--documenter-dark .field.is-grouped.is-grouped-right{justify-content:flex-end}html.theme--documenter-dark .field.is-grouped.is-grouped-multiline{flex-wrap:wrap}html.theme--documenter-dark .field.is-grouped.is-grouped-multiline>.control:last-child,html.theme--documenter-dark .field.is-grouped.is-grouped-multiline>.control:not(:last-child){margin-bottom:0.75rem}html.theme--documenter-dark .field.is-grouped.is-grouped-multiline:last-child{margin-bottom:-0.75rem}html.theme--documenter-dark .field.is-grouped.is-grouped-multiline:not(:last-child){margin-bottom:0}@media screen and (min-width: 769px),print{html.theme--documenter-dark .field.is-horizontal{display:flex}}html.theme--documenter-dark .field-label .label{font-size:inherit}@media screen and (max-width: 768px){html.theme--documenter-dark .field-label{margin-bottom:0.5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .field-label{flex-basis:0;flex-grow:1;flex-shrink:0;margin-right:1.5rem;text-align:right}html.theme--documenter-dark .field-label.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.field-label{font-size:.75rem;padding-top:0.375em}html.theme--documenter-dark .field-label.is-normal{padding-top:0.375em}html.theme--documenter-dark .field-label.is-medium{font-size:1.25rem;padding-top:0.375em}html.theme--documenter-dark .field-label.is-large{font-size:1.5rem;padding-top:0.375em}}html.theme--documenter-dark .field-body .field .field{margin-bottom:0}@media screen and (min-width: 769px),print{html.theme--documenter-dark .field-body{display:flex;flex-basis:0;flex-grow:5;flex-shrink:1}html.theme--documenter-dark .field-body .field{margin-bottom:0}html.theme--documenter-dark .field-body>.field{flex-shrink:1}html.theme--documenter-dark .field-body>.field:not(.is-narrow){flex-grow:1}html.theme--documenter-dark .field-body>.field:not(:last-child){margin-right:.75rem}}html.theme--documenter-dark .control{box-sizing:border-box;clear:both;font-size:1rem;position:relative;text-align:inherit}html.theme--documenter-dark .control.has-icons-left .input:focus~.icon,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input:focus~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input:focus~.icon,html.theme--documenter-dark .control.has-icons-left .select:focus~.icon,html.theme--documenter-dark .control.has-icons-right .input:focus~.icon,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input:focus~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input:focus~.icon,html.theme--documenter-dark .control.has-icons-right .select:focus~.icon{color:#282f2f}html.theme--documenter-dark .control.has-icons-left .input.is-small~.icon,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input~.icon,html.theme--documenter-dark .control.has-icons-left .select.is-small~.icon,html.theme--documenter-dark .control.has-icons-right .input.is-small~.icon,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input~.icon,html.theme--documenter-dark .control.has-icons-right .select.is-small~.icon{font-size:.75rem}html.theme--documenter-dark .control.has-icons-left .input.is-medium~.icon,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input.is-medium~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input.is-medium~.icon,html.theme--documenter-dark .control.has-icons-left .select.is-medium~.icon,html.theme--documenter-dark .control.has-icons-right .input.is-medium~.icon,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input.is-medium~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input.is-medium~.icon,html.theme--documenter-dark .control.has-icons-right .select.is-medium~.icon{font-size:1.25rem}html.theme--documenter-dark .control.has-icons-left .input.is-large~.icon,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input.is-large~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input.is-large~.icon,html.theme--documenter-dark .control.has-icons-left .select.is-large~.icon,html.theme--documenter-dark .control.has-icons-right .input.is-large~.icon,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input.is-large~.icon,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input.is-large~.icon,html.theme--documenter-dark .control.has-icons-right .select.is-large~.icon{font-size:1.5rem}html.theme--documenter-dark .control.has-icons-left .icon,html.theme--documenter-dark .control.has-icons-right .icon{color:#5e6d6f;height:2.5em;pointer-events:none;position:absolute;top:0;width:2.5em;z-index:4}html.theme--documenter-dark .control.has-icons-left .input,html.theme--documenter-dark .control.has-icons-left #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-left form.docs-search>input,html.theme--documenter-dark .control.has-icons-left .select select{padding-left:2.5em}html.theme--documenter-dark .control.has-icons-left .icon.is-left{left:0}html.theme--documenter-dark .control.has-icons-right .input,html.theme--documenter-dark .control.has-icons-right #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .control.has-icons-right form.docs-search>input,html.theme--documenter-dark .control.has-icons-right .select select{padding-right:2.5em}html.theme--documenter-dark .control.has-icons-right .icon.is-right{right:0}html.theme--documenter-dark .control.is-loading::after{position:absolute !important;right:.625em;top:0.625em;z-index:4}html.theme--documenter-dark .control.is-loading.is-small:after,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.is-loading:after{font-size:.75rem}html.theme--documenter-dark .control.is-loading.is-medium:after{font-size:1.25rem}html.theme--documenter-dark .control.is-loading.is-large:after{font-size:1.5rem}html.theme--documenter-dark .breadcrumb{font-size:1rem;white-space:nowrap}html.theme--documenter-dark .breadcrumb a{align-items:center;color:#1abc9c;display:flex;justify-content:center;padding:0 .75em}html.theme--documenter-dark .breadcrumb a:hover{color:#1dd2af}html.theme--documenter-dark .breadcrumb li{align-items:center;display:flex}html.theme--documenter-dark .breadcrumb li:first-child a{padding-left:0}html.theme--documenter-dark .breadcrumb li.is-active a{color:#f2f2f2;cursor:default;pointer-events:none}html.theme--documenter-dark .breadcrumb li+li::before{color:#8c9b9d;content:"\0002f"}html.theme--documenter-dark .breadcrumb ul,html.theme--documenter-dark .breadcrumb ol{align-items:flex-start;display:flex;flex-wrap:wrap;justify-content:flex-start}html.theme--documenter-dark .breadcrumb .icon:first-child{margin-right:.5em}html.theme--documenter-dark .breadcrumb .icon:last-child{margin-left:.5em}html.theme--documenter-dark .breadcrumb.is-centered ol,html.theme--documenter-dark .breadcrumb.is-centered ul{justify-content:center}html.theme--documenter-dark .breadcrumb.is-right ol,html.theme--documenter-dark .breadcrumb.is-right ul{justify-content:flex-end}html.theme--documenter-dark .breadcrumb.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.breadcrumb{font-size:.75rem}html.theme--documenter-dark .breadcrumb.is-medium{font-size:1.25rem}html.theme--documenter-dark .breadcrumb.is-large{font-size:1.5rem}html.theme--documenter-dark .breadcrumb.has-arrow-separator li+li::before{content:"\02192"}html.theme--documenter-dark .breadcrumb.has-bullet-separator li+li::before{content:"\02022"}html.theme--documenter-dark .breadcrumb.has-dot-separator li+li::before{content:"\000b7"}html.theme--documenter-dark .breadcrumb.has-succeeds-separator li+li::before{content:"\0227B"}html.theme--documenter-dark .card{background-color:#fff;border-radius:.25rem;box-shadow:#171717;color:#fff;max-width:100%;position:relative}html.theme--documenter-dark .card-footer:first-child,html.theme--documenter-dark .card-content:first-child,html.theme--documenter-dark .card-header:first-child{border-top-left-radius:.25rem;border-top-right-radius:.25rem}html.theme--documenter-dark .card-footer:last-child,html.theme--documenter-dark .card-content:last-child,html.theme--documenter-dark .card-header:last-child{border-bottom-left-radius:.25rem;border-bottom-right-radius:.25rem}html.theme--documenter-dark .card-header{background-color:rgba(0,0,0,0);align-items:stretch;box-shadow:0 0.125em 0.25em rgba(10,10,10,0.1);display:flex}html.theme--documenter-dark .card-header-title{align-items:center;color:#f2f2f2;display:flex;flex-grow:1;font-weight:700;padding:0.75rem 1rem}html.theme--documenter-dark .card-header-title.is-centered{justify-content:center}html.theme--documenter-dark .card-header-icon{-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;color:currentColor;font-family:inherit;font-size:1em;margin:0;padding:0;align-items:center;cursor:pointer;display:flex;justify-content:center;padding:0.75rem 1rem}html.theme--documenter-dark .card-image{display:block;position:relative}html.theme--documenter-dark .card-image:first-child img{border-top-left-radius:.25rem;border-top-right-radius:.25rem}html.theme--documenter-dark .card-image:last-child img{border-bottom-left-radius:.25rem;border-bottom-right-radius:.25rem}html.theme--documenter-dark .card-content{background-color:rgba(0,0,0,0);padding:1.5rem}html.theme--documenter-dark .card-footer{background-color:rgba(0,0,0,0);border-top:1px solid #ededed;align-items:stretch;display:flex}html.theme--documenter-dark .card-footer-item{align-items:center;display:flex;flex-basis:0;flex-grow:1;flex-shrink:0;justify-content:center;padding:.75rem}html.theme--documenter-dark .card-footer-item:not(:last-child){border-right:1px solid #ededed}html.theme--documenter-dark .card .media:not(:last-child){margin-bottom:1.5rem}html.theme--documenter-dark .dropdown{display:inline-flex;position:relative;vertical-align:top}html.theme--documenter-dark .dropdown.is-active .dropdown-menu,html.theme--documenter-dark .dropdown.is-hoverable:hover .dropdown-menu{display:block}html.theme--documenter-dark .dropdown.is-right .dropdown-menu{left:auto;right:0}html.theme--documenter-dark .dropdown.is-up .dropdown-menu{bottom:100%;padding-bottom:4px;padding-top:initial;top:auto}html.theme--documenter-dark .dropdown-menu{display:none;left:0;min-width:12rem;padding-top:4px;position:absolute;top:100%;z-index:20}html.theme--documenter-dark .dropdown-content{background-color:#282f2f;border-radius:.4em;box-shadow:#171717;padding-bottom:.5rem;padding-top:.5rem}html.theme--documenter-dark .dropdown-item{color:#fff;display:block;font-size:0.875rem;line-height:1.5;padding:0.375rem 1rem;position:relative}html.theme--documenter-dark a.dropdown-item,html.theme--documenter-dark button.dropdown-item{padding-right:3rem;text-align:inherit;white-space:nowrap;width:100%}html.theme--documenter-dark a.dropdown-item:hover,html.theme--documenter-dark button.dropdown-item:hover{background-color:#282f2f;color:#0a0a0a}html.theme--documenter-dark a.dropdown-item.is-active,html.theme--documenter-dark button.dropdown-item.is-active{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .dropdown-divider{background-color:#ededed;border:none;display:block;height:1px;margin:0.5rem 0}html.theme--documenter-dark .level{align-items:center;justify-content:space-between}html.theme--documenter-dark .level code{border-radius:.4em}html.theme--documenter-dark .level img{display:inline-block;vertical-align:top}html.theme--documenter-dark .level.is-mobile{display:flex}html.theme--documenter-dark .level.is-mobile .level-left,html.theme--documenter-dark .level.is-mobile .level-right{display:flex}html.theme--documenter-dark .level.is-mobile .level-left+.level-right{margin-top:0}html.theme--documenter-dark .level.is-mobile .level-item:not(:last-child){margin-bottom:0;margin-right:.75rem}html.theme--documenter-dark .level.is-mobile .level-item:not(.is-narrow){flex-grow:1}@media screen and (min-width: 769px),print{html.theme--documenter-dark .level{display:flex}html.theme--documenter-dark .level>.level-item:not(.is-narrow){flex-grow:1}}html.theme--documenter-dark .level-item{align-items:center;display:flex;flex-basis:auto;flex-grow:0;flex-shrink:0;justify-content:center}html.theme--documenter-dark .level-item .title,html.theme--documenter-dark .level-item .subtitle{margin-bottom:0}@media screen and (max-width: 768px){html.theme--documenter-dark .level-item:not(:last-child){margin-bottom:.75rem}}html.theme--documenter-dark .level-left,html.theme--documenter-dark .level-right{flex-basis:auto;flex-grow:0;flex-shrink:0}html.theme--documenter-dark .level-left .level-item.is-flexible,html.theme--documenter-dark .level-right .level-item.is-flexible{flex-grow:1}@media screen and (min-width: 769px),print{html.theme--documenter-dark .level-left .level-item:not(:last-child),html.theme--documenter-dark .level-right .level-item:not(:last-child){margin-right:.75rem}}html.theme--documenter-dark .level-left{align-items:center;justify-content:flex-start}@media screen and (max-width: 768px){html.theme--documenter-dark .level-left+.level-right{margin-top:1.5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .level-left{display:flex}}html.theme--documenter-dark .level-right{align-items:center;justify-content:flex-end}@media screen and (min-width: 769px),print{html.theme--documenter-dark .level-right{display:flex}}html.theme--documenter-dark .media{align-items:flex-start;display:flex;text-align:inherit}html.theme--documenter-dark .media .content:not(:last-child){margin-bottom:.75rem}html.theme--documenter-dark .media .media{border-top:1px solid rgba(94,109,111,0.5);display:flex;padding-top:.75rem}html.theme--documenter-dark .media .media .content:not(:last-child),html.theme--documenter-dark .media .media .control:not(:last-child){margin-bottom:.5rem}html.theme--documenter-dark .media .media .media{padding-top:.5rem}html.theme--documenter-dark .media .media .media+.media{margin-top:.5rem}html.theme--documenter-dark .media+.media{border-top:1px solid rgba(94,109,111,0.5);margin-top:1rem;padding-top:1rem}html.theme--documenter-dark .media.is-large+.media{margin-top:1.5rem;padding-top:1.5rem}html.theme--documenter-dark .media-left,html.theme--documenter-dark .media-right{flex-basis:auto;flex-grow:0;flex-shrink:0}html.theme--documenter-dark .media-left{margin-right:1rem}html.theme--documenter-dark .media-right{margin-left:1rem}html.theme--documenter-dark .media-content{flex-basis:auto;flex-grow:1;flex-shrink:1;text-align:inherit}@media screen and (max-width: 768px){html.theme--documenter-dark .media-content{overflow-x:auto}}html.theme--documenter-dark .menu{font-size:1rem}html.theme--documenter-dark .menu.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.menu{font-size:.75rem}html.theme--documenter-dark .menu.is-medium{font-size:1.25rem}html.theme--documenter-dark .menu.is-large{font-size:1.5rem}html.theme--documenter-dark .menu-list{line-height:1.25}html.theme--documenter-dark .menu-list a{border-radius:3px;color:#fff;display:block;padding:0.5em 0.75em}html.theme--documenter-dark .menu-list a:hover{background-color:#282f2f;color:#f2f2f2}html.theme--documenter-dark .menu-list a.is-active{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .menu-list li ul{border-left:1px solid #5e6d6f;margin:.75em;padding-left:.75em}html.theme--documenter-dark .menu-label{color:#fff;font-size:.75em;letter-spacing:.1em;text-transform:uppercase}html.theme--documenter-dark .menu-label:not(:first-child){margin-top:1em}html.theme--documenter-dark .menu-label:not(:last-child){margin-bottom:1em}html.theme--documenter-dark .message{background-color:#282f2f;border-radius:.4em;font-size:1rem}html.theme--documenter-dark .message strong{color:currentColor}html.theme--documenter-dark .message a:not(.button):not(.tag):not(.dropdown-item){color:currentColor;text-decoration:underline}html.theme--documenter-dark .message.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.message{font-size:.75rem}html.theme--documenter-dark .message.is-medium{font-size:1.25rem}html.theme--documenter-dark .message.is-large{font-size:1.5rem}html.theme--documenter-dark .message.is-white{background-color:#fff}html.theme--documenter-dark .message.is-white .message-header{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .message.is-white .message-body{border-color:#fff}html.theme--documenter-dark .message.is-black{background-color:#fafafa}html.theme--documenter-dark .message.is-black .message-header{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .message.is-black .message-body{border-color:#0a0a0a}html.theme--documenter-dark .message.is-light{background-color:#f9fafb}html.theme--documenter-dark .message.is-light .message-header{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .message.is-light .message-body{border-color:#ecf0f1}html.theme--documenter-dark .message.is-dark,html.theme--documenter-dark .content kbd.message{background-color:#f9fafa}html.theme--documenter-dark .message.is-dark .message-header,html.theme--documenter-dark .content kbd.message .message-header{background-color:#282f2f;color:#fff}html.theme--documenter-dark .message.is-dark .message-body,html.theme--documenter-dark .content kbd.message .message-body{border-color:#282f2f}html.theme--documenter-dark .message.is-primary,html.theme--documenter-dark .docstring>section>a.message.docs-sourcelink{background-color:#f1f5f9}html.theme--documenter-dark .message.is-primary .message-header,html.theme--documenter-dark .docstring>section>a.message.docs-sourcelink .message-header{background-color:#375a7f;color:#fff}html.theme--documenter-dark .message.is-primary .message-body,html.theme--documenter-dark .docstring>section>a.message.docs-sourcelink .message-body{border-color:#375a7f;color:#4d7eb2}html.theme--documenter-dark .message.is-link{background-color:#edfdf9}html.theme--documenter-dark .message.is-link .message-header{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .message.is-link .message-body{border-color:#1abc9c;color:#15987e}html.theme--documenter-dark .message.is-info{background-color:#ebf7ff}html.theme--documenter-dark .message.is-info .message-header{background-color:#024c7d;color:#fff}html.theme--documenter-dark .message.is-info .message-body{border-color:#024c7d;color:#0e9dfb}html.theme--documenter-dark .message.is-success{background-color:#ebfff3}html.theme--documenter-dark .message.is-success .message-header{background-color:#008438;color:#fff}html.theme--documenter-dark .message.is-success .message-body{border-color:#008438;color:#00eb64}html.theme--documenter-dark .message.is-warning{background-color:#fffaeb}html.theme--documenter-dark .message.is-warning .message-header{background-color:#ad8100;color:#fff}html.theme--documenter-dark .message.is-warning .message-body{border-color:#ad8100;color:#d19c00}html.theme--documenter-dark .message.is-danger{background-color:#fdeeec}html.theme--documenter-dark .message.is-danger .message-header{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .message.is-danger .message-body{border-color:#9e1b0d;color:#ec311d}html.theme--documenter-dark .message-header{align-items:center;background-color:#fff;border-radius:.4em .4em 0 0;color:rgba(0,0,0,0.7);display:flex;font-weight:700;justify-content:space-between;line-height:1.25;padding:0.75em 1em;position:relative}html.theme--documenter-dark .message-header .delete{flex-grow:0;flex-shrink:0;margin-left:.75em}html.theme--documenter-dark .message-header+.message-body{border-width:0;border-top-left-radius:0;border-top-right-radius:0}html.theme--documenter-dark .message-body{border-color:#5e6d6f;border-radius:.4em;border-style:solid;border-width:0 0 0 4px;color:#fff;padding:1.25em 1.5em}html.theme--documenter-dark .message-body code,html.theme--documenter-dark .message-body pre{background-color:#fff}html.theme--documenter-dark .message-body pre code{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .modal{align-items:center;display:none;flex-direction:column;justify-content:center;overflow:hidden;position:fixed;z-index:40}html.theme--documenter-dark .modal.is-active{display:flex}html.theme--documenter-dark .modal-background{background-color:rgba(10,10,10,0.86)}html.theme--documenter-dark .modal-content,html.theme--documenter-dark .modal-card{margin:0 20px;max-height:calc(100vh - 160px);overflow:auto;position:relative;width:100%}@media screen and (min-width: 769px){html.theme--documenter-dark .modal-content,html.theme--documenter-dark .modal-card{margin:0 auto;max-height:calc(100vh - 40px);width:640px}}html.theme--documenter-dark .modal-close{background:none;height:40px;position:fixed;right:20px;top:20px;width:40px}html.theme--documenter-dark .modal-card{display:flex;flex-direction:column;max-height:calc(100vh - 40px);overflow:hidden;-ms-overflow-y:visible}html.theme--documenter-dark .modal-card-head,html.theme--documenter-dark .modal-card-foot{align-items:center;background-color:#282f2f;display:flex;flex-shrink:0;justify-content:flex-start;padding:20px;position:relative}html.theme--documenter-dark .modal-card-head{border-bottom:1px solid #5e6d6f;border-top-left-radius:8px;border-top-right-radius:8px}html.theme--documenter-dark .modal-card-title{color:#f2f2f2;flex-grow:1;flex-shrink:0;font-size:1.5rem;line-height:1}html.theme--documenter-dark .modal-card-foot{border-bottom-left-radius:8px;border-bottom-right-radius:8px;border-top:1px solid #5e6d6f}html.theme--documenter-dark .modal-card-foot .button:not(:last-child){margin-right:.5em}html.theme--documenter-dark .modal-card-body{-webkit-overflow-scrolling:touch;background-color:#fff;flex-grow:1;flex-shrink:1;overflow:auto;padding:20px}html.theme--documenter-dark .navbar{background-color:#375a7f;min-height:4rem;position:relative;z-index:30}html.theme--documenter-dark .navbar.is-white{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link{color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-white .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-white .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-brand .navbar-link::after{border-color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-burger{color:#0a0a0a}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-white .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-white .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link{color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-white .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-white .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-white .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-white .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-white .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-white .navbar-end .navbar-link::after{border-color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-white .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-white .navbar-item.has-dropdown.is-active .navbar-link{background-color:#f2f2f2;color:#0a0a0a}html.theme--documenter-dark .navbar.is-white .navbar-dropdown a.navbar-item.is-active{background-color:#fff;color:#0a0a0a}}html.theme--documenter-dark .navbar.is-black{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-black .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-black .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link.is-active{background-color:#000;color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-black .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-black .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-black .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-black .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-black .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-black .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-black .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link.is-active{background-color:#000;color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-black .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-black .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-black .navbar-item.has-dropdown.is-active .navbar-link{background-color:#000;color:#fff}html.theme--documenter-dark .navbar.is-black .navbar-dropdown a.navbar-item.is-active{background-color:#0a0a0a;color:#fff}}html.theme--documenter-dark .navbar.is-light{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-light .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-light .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link.is-active{background-color:#dde4e6;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-brand .navbar-link::after{border-color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-burger{color:rgba(0,0,0,0.7)}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-light .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-light .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-light .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-light .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-light .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-light .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-light .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link.is-active{background-color:#dde4e6;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-light .navbar-end .navbar-link::after{border-color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-light .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-light .navbar-item.has-dropdown.is-active .navbar-link{background-color:#dde4e6;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .navbar.is-light .navbar-dropdown a.navbar-item.is-active{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}}html.theme--documenter-dark .navbar.is-dark,html.theme--documenter-dark .content kbd.navbar{background-color:#282f2f;color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-brand>.navbar-item,html.theme--documenter-dark .content kbd.navbar .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .content kbd.navbar .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-dark .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .content kbd.navbar .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-dark .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link:focus,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link:hover,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link.is-active{background-color:#1d2122;color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-brand .navbar-link::after,html.theme--documenter-dark .content kbd.navbar .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-burger,html.theme--documenter-dark .content kbd.navbar .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-dark .navbar-start>.navbar-item,html.theme--documenter-dark .content kbd.navbar .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-dark .navbar-end>.navbar-item,html.theme--documenter-dark .content kbd.navbar .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .content kbd.navbar .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-dark .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .content kbd.navbar .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-dark .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link:focus,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link:hover,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-dark .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .content kbd.navbar .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-dark .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .content kbd.navbar .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-dark .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link:focus,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link:hover,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link.is-active{background-color:#1d2122;color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-start .navbar-link::after,html.theme--documenter-dark .content kbd.navbar .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-dark .navbar-end .navbar-link::after,html.theme--documenter-dark .content kbd.navbar .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-dark .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-dark .navbar-item.has-dropdown.is-active .navbar-link,html.theme--documenter-dark .content kbd.navbar .navbar-item.has-dropdown.is-active .navbar-link{background-color:#1d2122;color:#fff}html.theme--documenter-dark .navbar.is-dark .navbar-dropdown a.navbar-item.is-active,html.theme--documenter-dark .content kbd.navbar .navbar-dropdown a.navbar-item.is-active{background-color:#282f2f;color:#fff}}html.theme--documenter-dark .navbar.is-primary,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink{background-color:#375a7f;color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-brand>.navbar-item,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-primary .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-primary .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link.is-active{background-color:#2f4d6d;color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-brand .navbar-link::after,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-burger,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-primary .navbar-start>.navbar-item,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-primary .navbar-end>.navbar-item,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-primary .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-primary .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-primary .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-primary .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-primary .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link:focus,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link:hover,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link.is-active{background-color:#2f4d6d;color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-start .navbar-link::after,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-primary .navbar-end .navbar-link::after,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-primary .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-primary .navbar-item.has-dropdown.is-active .navbar-link,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown.is-active .navbar-link{background-color:#2f4d6d;color:#fff}html.theme--documenter-dark .navbar.is-primary .navbar-dropdown a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-dropdown a.navbar-item.is-active{background-color:#375a7f;color:#fff}}html.theme--documenter-dark .navbar.is-link{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-link .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-link .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link.is-active{background-color:#17a689;color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-link .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-link .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-link .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-link .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-link .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-link .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-link .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link.is-active{background-color:#17a689;color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-link .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-link .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-link .navbar-item.has-dropdown.is-active .navbar-link{background-color:#17a689;color:#fff}html.theme--documenter-dark .navbar.is-link .navbar-dropdown a.navbar-item.is-active{background-color:#1abc9c;color:#fff}}html.theme--documenter-dark .navbar.is-info{background-color:#024c7d;color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-info .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-info .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link.is-active{background-color:#023d64;color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-info .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-info .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-info .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-info .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-info .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-info .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-info .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link.is-active{background-color:#023d64;color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-info .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-info .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-info .navbar-item.has-dropdown.is-active .navbar-link{background-color:#023d64;color:#fff}html.theme--documenter-dark .navbar.is-info .navbar-dropdown a.navbar-item.is-active{background-color:#024c7d;color:#fff}}html.theme--documenter-dark .navbar.is-success{background-color:#008438;color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-success .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-success .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link.is-active{background-color:#006b2d;color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-success .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-success .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-success .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-success .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-success .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-success .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-success .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link.is-active{background-color:#006b2d;color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-success .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-success .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-success .navbar-item.has-dropdown.is-active .navbar-link{background-color:#006b2d;color:#fff}html.theme--documenter-dark .navbar.is-success .navbar-dropdown a.navbar-item.is-active{background-color:#008438;color:#fff}}html.theme--documenter-dark .navbar.is-warning{background-color:#ad8100;color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-warning .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-warning .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link.is-active{background-color:#946e00;color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-warning .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-warning .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-warning .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-warning .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-warning .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-warning .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-warning .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link.is-active{background-color:#946e00;color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-warning .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-warning .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-warning .navbar-item.has-dropdown.is-active .navbar-link{background-color:#946e00;color:#fff}html.theme--documenter-dark .navbar.is-warning .navbar-dropdown a.navbar-item.is-active{background-color:#ad8100;color:#fff}}html.theme--documenter-dark .navbar.is-danger{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-brand>.navbar-item,html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-brand>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-danger .navbar-brand>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-danger .navbar-brand>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link:focus,html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link:hover,html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link.is-active{background-color:#86170b;color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-brand .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-burger{color:#fff}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar.is-danger .navbar-start>.navbar-item,html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link,html.theme--documenter-dark .navbar.is-danger .navbar-end>.navbar-item,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link{color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-start>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-danger .navbar-start>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-danger .navbar-start>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link:focus,html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link:hover,html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link.is-active,html.theme--documenter-dark .navbar.is-danger .navbar-end>a.navbar-item:focus,html.theme--documenter-dark .navbar.is-danger .navbar-end>a.navbar-item:hover,html.theme--documenter-dark .navbar.is-danger .navbar-end>a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link:focus,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link:hover,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link.is-active{background-color:#86170b;color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-start .navbar-link::after,html.theme--documenter-dark .navbar.is-danger .navbar-end .navbar-link::after{border-color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar.is-danger .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar.is-danger .navbar-item.has-dropdown.is-active .navbar-link{background-color:#86170b;color:#fff}html.theme--documenter-dark .navbar.is-danger .navbar-dropdown a.navbar-item.is-active{background-color:#9e1b0d;color:#fff}}html.theme--documenter-dark .navbar>.container{align-items:stretch;display:flex;min-height:4rem;width:100%}html.theme--documenter-dark .navbar.has-shadow{box-shadow:0 2px 0 0 #282f2f}html.theme--documenter-dark .navbar.is-fixed-bottom,html.theme--documenter-dark .navbar.is-fixed-top{left:0;position:fixed;right:0;z-index:30}html.theme--documenter-dark .navbar.is-fixed-bottom{bottom:0}html.theme--documenter-dark .navbar.is-fixed-bottom.has-shadow{box-shadow:0 -2px 0 0 #282f2f}html.theme--documenter-dark .navbar.is-fixed-top{top:0}html.theme--documenter-dark html.has-navbar-fixed-top,html.theme--documenter-dark body.has-navbar-fixed-top{padding-top:4rem}html.theme--documenter-dark html.has-navbar-fixed-bottom,html.theme--documenter-dark body.has-navbar-fixed-bottom{padding-bottom:4rem}html.theme--documenter-dark .navbar-brand,html.theme--documenter-dark .navbar-tabs{align-items:stretch;display:flex;flex-shrink:0;min-height:4rem}html.theme--documenter-dark .navbar-brand a.navbar-item:focus,html.theme--documenter-dark .navbar-brand a.navbar-item:hover{background-color:transparent}html.theme--documenter-dark .navbar-tabs{-webkit-overflow-scrolling:touch;max-width:100vw;overflow-x:auto;overflow-y:hidden}html.theme--documenter-dark .navbar-burger{color:#fff;-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;cursor:pointer;display:block;height:4rem;position:relative;width:4rem;margin-left:auto}html.theme--documenter-dark .navbar-burger span{background-color:currentColor;display:block;height:1px;left:calc(50% - 8px);position:absolute;transform-origin:center;transition-duration:86ms;transition-property:background-color, opacity, transform;transition-timing-function:ease-out;width:16px}html.theme--documenter-dark .navbar-burger span:nth-child(1){top:calc(50% - 6px)}html.theme--documenter-dark .navbar-burger span:nth-child(2){top:calc(50% - 1px)}html.theme--documenter-dark .navbar-burger span:nth-child(3){top:calc(50% + 4px)}html.theme--documenter-dark .navbar-burger:hover{background-color:rgba(0,0,0,0.05)}html.theme--documenter-dark .navbar-burger.is-active span:nth-child(1){transform:translateY(5px) rotate(45deg)}html.theme--documenter-dark .navbar-burger.is-active span:nth-child(2){opacity:0}html.theme--documenter-dark .navbar-burger.is-active span:nth-child(3){transform:translateY(-5px) rotate(-45deg)}html.theme--documenter-dark .navbar-menu{display:none}html.theme--documenter-dark .navbar-item,html.theme--documenter-dark .navbar-link{color:#fff;display:block;line-height:1.5;padding:0.5rem 0.75rem;position:relative}html.theme--documenter-dark .navbar-item .icon:only-child,html.theme--documenter-dark .navbar-link .icon:only-child{margin-left:-0.25rem;margin-right:-0.25rem}html.theme--documenter-dark a.navbar-item,html.theme--documenter-dark .navbar-link{cursor:pointer}html.theme--documenter-dark a.navbar-item:focus,html.theme--documenter-dark a.navbar-item:focus-within,html.theme--documenter-dark a.navbar-item:hover,html.theme--documenter-dark a.navbar-item.is-active,html.theme--documenter-dark .navbar-link:focus,html.theme--documenter-dark .navbar-link:focus-within,html.theme--documenter-dark .navbar-link:hover,html.theme--documenter-dark .navbar-link.is-active{background-color:rgba(0,0,0,0);color:#1abc9c}html.theme--documenter-dark .navbar-item{flex-grow:0;flex-shrink:0}html.theme--documenter-dark .navbar-item img{max-height:1.75rem}html.theme--documenter-dark .navbar-item.has-dropdown{padding:0}html.theme--documenter-dark .navbar-item.is-expanded{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .navbar-item.is-tab{border-bottom:1px solid transparent;min-height:4rem;padding-bottom:calc(0.5rem - 1px)}html.theme--documenter-dark .navbar-item.is-tab:focus,html.theme--documenter-dark .navbar-item.is-tab:hover{background-color:rgba(0,0,0,0);border-bottom-color:#1abc9c}html.theme--documenter-dark .navbar-item.is-tab.is-active{background-color:rgba(0,0,0,0);border-bottom-color:#1abc9c;border-bottom-style:solid;border-bottom-width:3px;color:#1abc9c;padding-bottom:calc(0.5rem - 3px)}html.theme--documenter-dark .navbar-content{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .navbar-link:not(.is-arrowless){padding-right:2.5em}html.theme--documenter-dark .navbar-link:not(.is-arrowless)::after{border-color:#fff;margin-top:-0.375em;right:1.125em}html.theme--documenter-dark .navbar-dropdown{font-size:0.875rem;padding-bottom:0.5rem;padding-top:0.5rem}html.theme--documenter-dark .navbar-dropdown .navbar-item{padding-left:1.5rem;padding-right:1.5rem}html.theme--documenter-dark .navbar-divider{background-color:rgba(0,0,0,0.2);border:none;display:none;height:2px;margin:0.5rem 0}@media screen and (max-width: 1055px){html.theme--documenter-dark .navbar>.container{display:block}html.theme--documenter-dark .navbar-brand .navbar-item,html.theme--documenter-dark .navbar-tabs .navbar-item{align-items:center;display:flex}html.theme--documenter-dark .navbar-link::after{display:none}html.theme--documenter-dark .navbar-menu{background-color:#375a7f;box-shadow:0 8px 16px rgba(10,10,10,0.1);padding:0.5rem 0}html.theme--documenter-dark .navbar-menu.is-active{display:block}html.theme--documenter-dark .navbar.is-fixed-bottom-touch,html.theme--documenter-dark .navbar.is-fixed-top-touch{left:0;position:fixed;right:0;z-index:30}html.theme--documenter-dark .navbar.is-fixed-bottom-touch{bottom:0}html.theme--documenter-dark .navbar.is-fixed-bottom-touch.has-shadow{box-shadow:0 -2px 3px rgba(10,10,10,0.1)}html.theme--documenter-dark .navbar.is-fixed-top-touch{top:0}html.theme--documenter-dark .navbar.is-fixed-top .navbar-menu,html.theme--documenter-dark .navbar.is-fixed-top-touch .navbar-menu{-webkit-overflow-scrolling:touch;max-height:calc(100vh - 4rem);overflow:auto}html.theme--documenter-dark html.has-navbar-fixed-top-touch,html.theme--documenter-dark body.has-navbar-fixed-top-touch{padding-top:4rem}html.theme--documenter-dark html.has-navbar-fixed-bottom-touch,html.theme--documenter-dark body.has-navbar-fixed-bottom-touch{padding-bottom:4rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .navbar,html.theme--documenter-dark .navbar-menu,html.theme--documenter-dark .navbar-start,html.theme--documenter-dark .navbar-end{align-items:stretch;display:flex}html.theme--documenter-dark .navbar{min-height:4rem}html.theme--documenter-dark .navbar.is-spaced{padding:1rem 2rem}html.theme--documenter-dark .navbar.is-spaced .navbar-start,html.theme--documenter-dark .navbar.is-spaced .navbar-end{align-items:center}html.theme--documenter-dark .navbar.is-spaced a.navbar-item,html.theme--documenter-dark .navbar.is-spaced .navbar-link{border-radius:.4em}html.theme--documenter-dark .navbar.is-transparent a.navbar-item:focus,html.theme--documenter-dark .navbar.is-transparent a.navbar-item:hover,html.theme--documenter-dark .navbar.is-transparent a.navbar-item.is-active,html.theme--documenter-dark .navbar.is-transparent .navbar-link:focus,html.theme--documenter-dark .navbar.is-transparent .navbar-link:hover,html.theme--documenter-dark .navbar.is-transparent .navbar-link.is-active{background-color:transparent !important}html.theme--documenter-dark .navbar.is-transparent .navbar-item.has-dropdown.is-active .navbar-link,html.theme--documenter-dark .navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:focus .navbar-link,html.theme--documenter-dark .navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:focus-within .navbar-link,html.theme--documenter-dark .navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:hover .navbar-link{background-color:transparent !important}html.theme--documenter-dark .navbar.is-transparent .navbar-dropdown a.navbar-item:focus,html.theme--documenter-dark .navbar.is-transparent .navbar-dropdown a.navbar-item:hover{background-color:rgba(0,0,0,0);color:#dbdee0}html.theme--documenter-dark .navbar.is-transparent .navbar-dropdown a.navbar-item.is-active{background-color:rgba(0,0,0,0);color:#1abc9c}html.theme--documenter-dark .navbar-burger{display:none}html.theme--documenter-dark .navbar-item,html.theme--documenter-dark .navbar-link{align-items:center;display:flex}html.theme--documenter-dark .navbar-item.has-dropdown{align-items:stretch}html.theme--documenter-dark .navbar-item.has-dropdown-up .navbar-link::after{transform:rotate(135deg) translate(0.25em, -0.25em)}html.theme--documenter-dark .navbar-item.has-dropdown-up .navbar-dropdown{border-bottom:1px solid rgba(0,0,0,0.2);border-radius:8px 8px 0 0;border-top:none;bottom:100%;box-shadow:0 -8px 8px rgba(10,10,10,0.1);top:auto}html.theme--documenter-dark .navbar-item.is-active .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:focus .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:focus-within .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:hover .navbar-dropdown{display:block}.navbar.is-spaced html.theme--documenter-dark .navbar-item.is-active .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-active .navbar-dropdown.is-boxed,.navbar.is-spaced html.theme--documenter-dark .navbar-item.is-hoverable:focus .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:focus .navbar-dropdown.is-boxed,.navbar.is-spaced html.theme--documenter-dark .navbar-item.is-hoverable:focus-within .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:focus-within .navbar-dropdown.is-boxed,.navbar.is-spaced html.theme--documenter-dark .navbar-item.is-hoverable:hover .navbar-dropdown,html.theme--documenter-dark .navbar-item.is-hoverable:hover .navbar-dropdown.is-boxed{opacity:1;pointer-events:auto;transform:translateY(0)}html.theme--documenter-dark .navbar-menu{flex-grow:1;flex-shrink:0}html.theme--documenter-dark .navbar-start{justify-content:flex-start;margin-right:auto}html.theme--documenter-dark .navbar-end{justify-content:flex-end;margin-left:auto}html.theme--documenter-dark .navbar-dropdown{background-color:#375a7f;border-bottom-left-radius:8px;border-bottom-right-radius:8px;border-top:1px solid rgba(0,0,0,0.2);box-shadow:0 8px 8px rgba(10,10,10,0.1);display:none;font-size:0.875rem;left:0;min-width:100%;position:absolute;top:100%;z-index:20}html.theme--documenter-dark .navbar-dropdown .navbar-item{padding:0.375rem 1rem;white-space:nowrap}html.theme--documenter-dark .navbar-dropdown a.navbar-item{padding-right:3rem}html.theme--documenter-dark .navbar-dropdown a.navbar-item:focus,html.theme--documenter-dark .navbar-dropdown a.navbar-item:hover{background-color:rgba(0,0,0,0);color:#dbdee0}html.theme--documenter-dark .navbar-dropdown a.navbar-item.is-active{background-color:rgba(0,0,0,0);color:#1abc9c}.navbar.is-spaced html.theme--documenter-dark .navbar-dropdown,html.theme--documenter-dark .navbar-dropdown.is-boxed{border-radius:8px;border-top:none;box-shadow:0 8px 8px rgba(10,10,10,0.1), 0 0 0 1px rgba(10,10,10,0.1);display:block;opacity:0;pointer-events:none;top:calc(100% + (-4px));transform:translateY(-5px);transition-duration:86ms;transition-property:opacity, transform}html.theme--documenter-dark .navbar-dropdown.is-right{left:auto;right:0}html.theme--documenter-dark .navbar-divider{display:block}html.theme--documenter-dark .navbar>.container .navbar-brand,html.theme--documenter-dark .container>.navbar .navbar-brand{margin-left:-.75rem}html.theme--documenter-dark .navbar>.container .navbar-menu,html.theme--documenter-dark .container>.navbar .navbar-menu{margin-right:-.75rem}html.theme--documenter-dark .navbar.is-fixed-bottom-desktop,html.theme--documenter-dark .navbar.is-fixed-top-desktop{left:0;position:fixed;right:0;z-index:30}html.theme--documenter-dark .navbar.is-fixed-bottom-desktop{bottom:0}html.theme--documenter-dark .navbar.is-fixed-bottom-desktop.has-shadow{box-shadow:0 -2px 3px rgba(10,10,10,0.1)}html.theme--documenter-dark .navbar.is-fixed-top-desktop{top:0}html.theme--documenter-dark html.has-navbar-fixed-top-desktop,html.theme--documenter-dark body.has-navbar-fixed-top-desktop{padding-top:4rem}html.theme--documenter-dark html.has-navbar-fixed-bottom-desktop,html.theme--documenter-dark body.has-navbar-fixed-bottom-desktop{padding-bottom:4rem}html.theme--documenter-dark html.has-spaced-navbar-fixed-top,html.theme--documenter-dark body.has-spaced-navbar-fixed-top{padding-top:6rem}html.theme--documenter-dark html.has-spaced-navbar-fixed-bottom,html.theme--documenter-dark body.has-spaced-navbar-fixed-bottom{padding-bottom:6rem}html.theme--documenter-dark a.navbar-item.is-active,html.theme--documenter-dark .navbar-link.is-active{color:#1abc9c}html.theme--documenter-dark a.navbar-item.is-active:not(:focus):not(:hover),html.theme--documenter-dark .navbar-link.is-active:not(:focus):not(:hover){background-color:rgba(0,0,0,0)}html.theme--documenter-dark .navbar-item.has-dropdown:focus .navbar-link,html.theme--documenter-dark .navbar-item.has-dropdown:hover .navbar-link,html.theme--documenter-dark .navbar-item.has-dropdown.is-active .navbar-link{background-color:rgba(0,0,0,0)}}html.theme--documenter-dark .hero.is-fullheight-with-navbar{min-height:calc(100vh - 4rem)}html.theme--documenter-dark .pagination{font-size:1rem;margin:-.25rem}html.theme--documenter-dark .pagination.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.pagination{font-size:.75rem}html.theme--documenter-dark .pagination.is-medium{font-size:1.25rem}html.theme--documenter-dark .pagination.is-large{font-size:1.5rem}html.theme--documenter-dark .pagination.is-rounded .pagination-previous,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.pagination .pagination-previous,html.theme--documenter-dark .pagination.is-rounded .pagination-next,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.pagination .pagination-next{padding-left:1em;padding-right:1em;border-radius:9999px}html.theme--documenter-dark .pagination.is-rounded .pagination-link,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.pagination .pagination-link{border-radius:9999px}html.theme--documenter-dark .pagination,html.theme--documenter-dark .pagination-list{align-items:center;display:flex;justify-content:center;text-align:center}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-ellipsis{font-size:1em;justify-content:center;margin:.25rem;padding-left:.5em;padding-right:.5em;text-align:center}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link{border-color:#5e6d6f;color:#1abc9c;min-width:2.5em}html.theme--documenter-dark .pagination-previous:hover,html.theme--documenter-dark .pagination-next:hover,html.theme--documenter-dark .pagination-link:hover{border-color:#8c9b9d;color:#1dd2af}html.theme--documenter-dark .pagination-previous:focus,html.theme--documenter-dark .pagination-next:focus,html.theme--documenter-dark .pagination-link:focus{border-color:#8c9b9d}html.theme--documenter-dark .pagination-previous:active,html.theme--documenter-dark .pagination-next:active,html.theme--documenter-dark .pagination-link:active{box-shadow:inset 0 1px 2px rgba(10,10,10,0.2)}html.theme--documenter-dark .pagination-previous[disabled],html.theme--documenter-dark .pagination-previous.is-disabled,html.theme--documenter-dark .pagination-next[disabled],html.theme--documenter-dark .pagination-next.is-disabled,html.theme--documenter-dark .pagination-link[disabled],html.theme--documenter-dark .pagination-link.is-disabled{background-color:#5e6d6f;border-color:#5e6d6f;box-shadow:none;color:#fff;opacity:0.5}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next{padding-left:.75em;padding-right:.75em;white-space:nowrap}html.theme--documenter-dark .pagination-link.is-current{background-color:#1abc9c;border-color:#1abc9c;color:#fff}html.theme--documenter-dark .pagination-ellipsis{color:#8c9b9d;pointer-events:none}html.theme--documenter-dark .pagination-list{flex-wrap:wrap}html.theme--documenter-dark .pagination-list li{list-style:none}@media screen and (max-width: 768px){html.theme--documenter-dark .pagination{flex-wrap:wrap}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .pagination-list li{flex-grow:1;flex-shrink:1}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .pagination-list{flex-grow:1;flex-shrink:1;justify-content:flex-start;order:1}html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-ellipsis{margin-bottom:0;margin-top:0}html.theme--documenter-dark .pagination-previous{order:2}html.theme--documenter-dark .pagination-next{order:3}html.theme--documenter-dark .pagination{justify-content:space-between;margin-bottom:0;margin-top:0}html.theme--documenter-dark .pagination.is-centered .pagination-previous{order:1}html.theme--documenter-dark .pagination.is-centered .pagination-list{justify-content:center;order:2}html.theme--documenter-dark .pagination.is-centered .pagination-next{order:3}html.theme--documenter-dark .pagination.is-right .pagination-previous{order:1}html.theme--documenter-dark .pagination.is-right .pagination-next{order:2}html.theme--documenter-dark .pagination.is-right .pagination-list{justify-content:flex-end;order:3}}html.theme--documenter-dark .panel{border-radius:8px;box-shadow:#171717;font-size:1rem}html.theme--documenter-dark .panel:not(:last-child){margin-bottom:1.5rem}html.theme--documenter-dark .panel.is-white .panel-heading{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .panel.is-white .panel-tabs a.is-active{border-bottom-color:#fff}html.theme--documenter-dark .panel.is-white .panel-block.is-active .panel-icon{color:#fff}html.theme--documenter-dark .panel.is-black .panel-heading{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .panel.is-black .panel-tabs a.is-active{border-bottom-color:#0a0a0a}html.theme--documenter-dark .panel.is-black .panel-block.is-active .panel-icon{color:#0a0a0a}html.theme--documenter-dark .panel.is-light .panel-heading{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .panel.is-light .panel-tabs a.is-active{border-bottom-color:#ecf0f1}html.theme--documenter-dark .panel.is-light .panel-block.is-active .panel-icon{color:#ecf0f1}html.theme--documenter-dark .panel.is-dark .panel-heading,html.theme--documenter-dark .content kbd.panel .panel-heading{background-color:#282f2f;color:#fff}html.theme--documenter-dark .panel.is-dark .panel-tabs a.is-active,html.theme--documenter-dark .content kbd.panel .panel-tabs a.is-active{border-bottom-color:#282f2f}html.theme--documenter-dark .panel.is-dark .panel-block.is-active .panel-icon,html.theme--documenter-dark .content kbd.panel .panel-block.is-active .panel-icon{color:#282f2f}html.theme--documenter-dark .panel.is-primary .panel-heading,html.theme--documenter-dark .docstring>section>a.panel.docs-sourcelink .panel-heading{background-color:#375a7f;color:#fff}html.theme--documenter-dark .panel.is-primary .panel-tabs a.is-active,html.theme--documenter-dark .docstring>section>a.panel.docs-sourcelink .panel-tabs a.is-active{border-bottom-color:#375a7f}html.theme--documenter-dark .panel.is-primary .panel-block.is-active .panel-icon,html.theme--documenter-dark .docstring>section>a.panel.docs-sourcelink .panel-block.is-active .panel-icon{color:#375a7f}html.theme--documenter-dark .panel.is-link .panel-heading{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .panel.is-link .panel-tabs a.is-active{border-bottom-color:#1abc9c}html.theme--documenter-dark .panel.is-link .panel-block.is-active .panel-icon{color:#1abc9c}html.theme--documenter-dark .panel.is-info .panel-heading{background-color:#024c7d;color:#fff}html.theme--documenter-dark .panel.is-info .panel-tabs a.is-active{border-bottom-color:#024c7d}html.theme--documenter-dark .panel.is-info .panel-block.is-active .panel-icon{color:#024c7d}html.theme--documenter-dark .panel.is-success .panel-heading{background-color:#008438;color:#fff}html.theme--documenter-dark .panel.is-success .panel-tabs a.is-active{border-bottom-color:#008438}html.theme--documenter-dark .panel.is-success .panel-block.is-active .panel-icon{color:#008438}html.theme--documenter-dark .panel.is-warning .panel-heading{background-color:#ad8100;color:#fff}html.theme--documenter-dark .panel.is-warning .panel-tabs a.is-active{border-bottom-color:#ad8100}html.theme--documenter-dark .panel.is-warning .panel-block.is-active .panel-icon{color:#ad8100}html.theme--documenter-dark .panel.is-danger .panel-heading{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .panel.is-danger .panel-tabs a.is-active{border-bottom-color:#9e1b0d}html.theme--documenter-dark .panel.is-danger .panel-block.is-active .panel-icon{color:#9e1b0d}html.theme--documenter-dark .panel-tabs:not(:last-child),html.theme--documenter-dark .panel-block:not(:last-child){border-bottom:1px solid #ededed}html.theme--documenter-dark .panel-heading{background-color:#343c3d;border-radius:8px 8px 0 0;color:#f2f2f2;font-size:1.25em;font-weight:700;line-height:1.25;padding:0.75em 1em}html.theme--documenter-dark .panel-tabs{align-items:flex-end;display:flex;font-size:.875em;justify-content:center}html.theme--documenter-dark .panel-tabs a{border-bottom:1px solid #5e6d6f;margin-bottom:-1px;padding:0.5em}html.theme--documenter-dark .panel-tabs a.is-active{border-bottom-color:#343c3d;color:#17a689}html.theme--documenter-dark .panel-list a{color:#fff}html.theme--documenter-dark .panel-list a:hover{color:#1abc9c}html.theme--documenter-dark .panel-block{align-items:center;color:#f2f2f2;display:flex;justify-content:flex-start;padding:0.5em 0.75em}html.theme--documenter-dark .panel-block input[type="checkbox"]{margin-right:.75em}html.theme--documenter-dark .panel-block>.control{flex-grow:1;flex-shrink:1;width:100%}html.theme--documenter-dark .panel-block.is-wrapped{flex-wrap:wrap}html.theme--documenter-dark .panel-block.is-active{border-left-color:#1abc9c;color:#17a689}html.theme--documenter-dark .panel-block.is-active .panel-icon{color:#1abc9c}html.theme--documenter-dark .panel-block:last-child{border-bottom-left-radius:8px;border-bottom-right-radius:8px}html.theme--documenter-dark a.panel-block,html.theme--documenter-dark label.panel-block{cursor:pointer}html.theme--documenter-dark a.panel-block:hover,html.theme--documenter-dark label.panel-block:hover{background-color:#282f2f}html.theme--documenter-dark .panel-icon{display:inline-block;font-size:14px;height:1em;line-height:1em;text-align:center;vertical-align:top;width:1em;color:#fff;margin-right:.75em}html.theme--documenter-dark .panel-icon .fa{font-size:inherit;line-height:inherit}html.theme--documenter-dark .tabs{-webkit-overflow-scrolling:touch;align-items:stretch;display:flex;font-size:1rem;justify-content:space-between;overflow:hidden;overflow-x:auto;white-space:nowrap}html.theme--documenter-dark .tabs a{align-items:center;border-bottom-color:#5e6d6f;border-bottom-style:solid;border-bottom-width:1px;color:#fff;display:flex;justify-content:center;margin-bottom:-1px;padding:0.5em 1em;vertical-align:top}html.theme--documenter-dark .tabs a:hover{border-bottom-color:#f2f2f2;color:#f2f2f2}html.theme--documenter-dark .tabs li{display:block}html.theme--documenter-dark .tabs li.is-active a{border-bottom-color:#1abc9c;color:#1abc9c}html.theme--documenter-dark .tabs ul{align-items:center;border-bottom-color:#5e6d6f;border-bottom-style:solid;border-bottom-width:1px;display:flex;flex-grow:1;flex-shrink:0;justify-content:flex-start}html.theme--documenter-dark .tabs ul.is-left{padding-right:0.75em}html.theme--documenter-dark .tabs ul.is-center{flex:none;justify-content:center;padding-left:0.75em;padding-right:0.75em}html.theme--documenter-dark .tabs ul.is-right{justify-content:flex-end;padding-left:0.75em}html.theme--documenter-dark .tabs .icon:first-child{margin-right:.5em}html.theme--documenter-dark .tabs .icon:last-child{margin-left:.5em}html.theme--documenter-dark .tabs.is-centered ul{justify-content:center}html.theme--documenter-dark .tabs.is-right ul{justify-content:flex-end}html.theme--documenter-dark .tabs.is-boxed a{border:1px solid transparent;border-radius:.4em .4em 0 0}html.theme--documenter-dark .tabs.is-boxed a:hover{background-color:#282f2f;border-bottom-color:#5e6d6f}html.theme--documenter-dark .tabs.is-boxed li.is-active a{background-color:#fff;border-color:#5e6d6f;border-bottom-color:rgba(0,0,0,0) !important}html.theme--documenter-dark .tabs.is-fullwidth li{flex-grow:1;flex-shrink:0}html.theme--documenter-dark .tabs.is-toggle a{border-color:#5e6d6f;border-style:solid;border-width:1px;margin-bottom:0;position:relative}html.theme--documenter-dark .tabs.is-toggle a:hover{background-color:#282f2f;border-color:#8c9b9d;z-index:2}html.theme--documenter-dark .tabs.is-toggle li+li{margin-left:-1px}html.theme--documenter-dark .tabs.is-toggle li:first-child a{border-top-left-radius:.4em;border-bottom-left-radius:.4em}html.theme--documenter-dark .tabs.is-toggle li:last-child a{border-top-right-radius:.4em;border-bottom-right-radius:.4em}html.theme--documenter-dark .tabs.is-toggle li.is-active a{background-color:#1abc9c;border-color:#1abc9c;color:#fff;z-index:1}html.theme--documenter-dark .tabs.is-toggle ul{border-bottom:none}html.theme--documenter-dark .tabs.is-toggle.is-toggle-rounded li:first-child a{border-bottom-left-radius:9999px;border-top-left-radius:9999px;padding-left:1.25em}html.theme--documenter-dark .tabs.is-toggle.is-toggle-rounded li:last-child a{border-bottom-right-radius:9999px;border-top-right-radius:9999px;padding-right:1.25em}html.theme--documenter-dark .tabs.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.tabs{font-size:.75rem}html.theme--documenter-dark .tabs.is-medium{font-size:1.25rem}html.theme--documenter-dark .tabs.is-large{font-size:1.5rem}html.theme--documenter-dark .column{display:block;flex-basis:0;flex-grow:1;flex-shrink:1;padding:.75rem}.columns.is-mobile>html.theme--documenter-dark .column.is-narrow{flex:none;width:unset}.columns.is-mobile>html.theme--documenter-dark .column.is-full{flex:none;width:100%}.columns.is-mobile>html.theme--documenter-dark .column.is-three-quarters{flex:none;width:75%}.columns.is-mobile>html.theme--documenter-dark .column.is-two-thirds{flex:none;width:66.6666%}.columns.is-mobile>html.theme--documenter-dark .column.is-half{flex:none;width:50%}.columns.is-mobile>html.theme--documenter-dark .column.is-one-third{flex:none;width:33.3333%}.columns.is-mobile>html.theme--documenter-dark .column.is-one-quarter{flex:none;width:25%}.columns.is-mobile>html.theme--documenter-dark .column.is-one-fifth{flex:none;width:20%}.columns.is-mobile>html.theme--documenter-dark .column.is-two-fifths{flex:none;width:40%}.columns.is-mobile>html.theme--documenter-dark .column.is-three-fifths{flex:none;width:60%}.columns.is-mobile>html.theme--documenter-dark .column.is-four-fifths{flex:none;width:80%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-three-quarters{margin-left:75%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-two-thirds{margin-left:66.6666%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-half{margin-left:50%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-one-third{margin-left:33.3333%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-one-quarter{margin-left:25%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-one-fifth{margin-left:20%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-two-fifths{margin-left:40%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-three-fifths{margin-left:60%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-four-fifths{margin-left:80%}.columns.is-mobile>html.theme--documenter-dark .column.is-0{flex:none;width:0%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-0{margin-left:0%}.columns.is-mobile>html.theme--documenter-dark .column.is-1{flex:none;width:8.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-1{margin-left:8.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-2{flex:none;width:16.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-2{margin-left:16.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-3{flex:none;width:25%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-3{margin-left:25%}.columns.is-mobile>html.theme--documenter-dark .column.is-4{flex:none;width:33.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-4{margin-left:33.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-5{flex:none;width:41.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-5{margin-left:41.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-6{flex:none;width:50%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-6{margin-left:50%}.columns.is-mobile>html.theme--documenter-dark .column.is-7{flex:none;width:58.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-7{margin-left:58.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-8{flex:none;width:66.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-8{margin-left:66.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-9{flex:none;width:75%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-9{margin-left:75%}.columns.is-mobile>html.theme--documenter-dark .column.is-10{flex:none;width:83.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-10{margin-left:83.33333337%}.columns.is-mobile>html.theme--documenter-dark .column.is-11{flex:none;width:91.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-11{margin-left:91.66666674%}.columns.is-mobile>html.theme--documenter-dark .column.is-12{flex:none;width:100%}.columns.is-mobile>html.theme--documenter-dark .column.is-offset-12{margin-left:100%}@media screen and (max-width: 768px){html.theme--documenter-dark .column.is-narrow-mobile{flex:none;width:unset}html.theme--documenter-dark .column.is-full-mobile{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-mobile{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-mobile{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-mobile{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-mobile{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-mobile{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-mobile{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-mobile{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-mobile{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-mobile{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-mobile{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-mobile{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-mobile{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-mobile{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-mobile{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-mobile{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-mobile{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-mobile{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-mobile{margin-left:80%}html.theme--documenter-dark .column.is-0-mobile{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-mobile{margin-left:0%}html.theme--documenter-dark .column.is-1-mobile{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-mobile{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-mobile{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-mobile{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-mobile{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-mobile{margin-left:25%}html.theme--documenter-dark .column.is-4-mobile{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-mobile{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-mobile{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-mobile{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-mobile{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-mobile{margin-left:50%}html.theme--documenter-dark .column.is-7-mobile{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-mobile{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-mobile{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-mobile{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-mobile{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-mobile{margin-left:75%}html.theme--documenter-dark .column.is-10-mobile{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-mobile{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-mobile{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-mobile{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-mobile{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-mobile{margin-left:100%}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .column.is-narrow,html.theme--documenter-dark .column.is-narrow-tablet{flex:none;width:unset}html.theme--documenter-dark .column.is-full,html.theme--documenter-dark .column.is-full-tablet{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters,html.theme--documenter-dark .column.is-three-quarters-tablet{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds,html.theme--documenter-dark .column.is-two-thirds-tablet{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half,html.theme--documenter-dark .column.is-half-tablet{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third,html.theme--documenter-dark .column.is-one-third-tablet{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter,html.theme--documenter-dark .column.is-one-quarter-tablet{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth,html.theme--documenter-dark .column.is-one-fifth-tablet{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths,html.theme--documenter-dark .column.is-two-fifths-tablet{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths,html.theme--documenter-dark .column.is-three-fifths-tablet{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths,html.theme--documenter-dark .column.is-four-fifths-tablet{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters,html.theme--documenter-dark .column.is-offset-three-quarters-tablet{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds,html.theme--documenter-dark .column.is-offset-two-thirds-tablet{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half,html.theme--documenter-dark .column.is-offset-half-tablet{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third,html.theme--documenter-dark .column.is-offset-one-third-tablet{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter,html.theme--documenter-dark .column.is-offset-one-quarter-tablet{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth,html.theme--documenter-dark .column.is-offset-one-fifth-tablet{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths,html.theme--documenter-dark .column.is-offset-two-fifths-tablet{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths,html.theme--documenter-dark .column.is-offset-three-fifths-tablet{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths,html.theme--documenter-dark .column.is-offset-four-fifths-tablet{margin-left:80%}html.theme--documenter-dark .column.is-0,html.theme--documenter-dark .column.is-0-tablet{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0,html.theme--documenter-dark .column.is-offset-0-tablet{margin-left:0%}html.theme--documenter-dark .column.is-1,html.theme--documenter-dark .column.is-1-tablet{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1,html.theme--documenter-dark .column.is-offset-1-tablet{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2,html.theme--documenter-dark .column.is-2-tablet{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2,html.theme--documenter-dark .column.is-offset-2-tablet{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3,html.theme--documenter-dark .column.is-3-tablet{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3,html.theme--documenter-dark .column.is-offset-3-tablet{margin-left:25%}html.theme--documenter-dark .column.is-4,html.theme--documenter-dark .column.is-4-tablet{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4,html.theme--documenter-dark .column.is-offset-4-tablet{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5,html.theme--documenter-dark .column.is-5-tablet{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5,html.theme--documenter-dark .column.is-offset-5-tablet{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6,html.theme--documenter-dark .column.is-6-tablet{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6,html.theme--documenter-dark .column.is-offset-6-tablet{margin-left:50%}html.theme--documenter-dark .column.is-7,html.theme--documenter-dark .column.is-7-tablet{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7,html.theme--documenter-dark .column.is-offset-7-tablet{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8,html.theme--documenter-dark .column.is-8-tablet{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8,html.theme--documenter-dark .column.is-offset-8-tablet{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9,html.theme--documenter-dark .column.is-9-tablet{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9,html.theme--documenter-dark .column.is-offset-9-tablet{margin-left:75%}html.theme--documenter-dark .column.is-10,html.theme--documenter-dark .column.is-10-tablet{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10,html.theme--documenter-dark .column.is-offset-10-tablet{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11,html.theme--documenter-dark .column.is-11-tablet{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11,html.theme--documenter-dark .column.is-offset-11-tablet{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12,html.theme--documenter-dark .column.is-12-tablet{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12,html.theme--documenter-dark .column.is-offset-12-tablet{margin-left:100%}}@media screen and (max-width: 1055px){html.theme--documenter-dark .column.is-narrow-touch{flex:none;width:unset}html.theme--documenter-dark .column.is-full-touch{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-touch{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-touch{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-touch{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-touch{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-touch{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-touch{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-touch{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-touch{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-touch{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-touch{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-touch{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-touch{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-touch{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-touch{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-touch{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-touch{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-touch{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-touch{margin-left:80%}html.theme--documenter-dark .column.is-0-touch{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-touch{margin-left:0%}html.theme--documenter-dark .column.is-1-touch{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-touch{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-touch{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-touch{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-touch{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-touch{margin-left:25%}html.theme--documenter-dark .column.is-4-touch{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-touch{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-touch{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-touch{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-touch{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-touch{margin-left:50%}html.theme--documenter-dark .column.is-7-touch{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-touch{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-touch{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-touch{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-touch{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-touch{margin-left:75%}html.theme--documenter-dark .column.is-10-touch{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-touch{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-touch{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-touch{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-touch{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-touch{margin-left:100%}}@media screen and (min-width: 1056px){html.theme--documenter-dark .column.is-narrow-desktop{flex:none;width:unset}html.theme--documenter-dark .column.is-full-desktop{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-desktop{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-desktop{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-desktop{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-desktop{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-desktop{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-desktop{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-desktop{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-desktop{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-desktop{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-desktop{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-desktop{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-desktop{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-desktop{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-desktop{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-desktop{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-desktop{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-desktop{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-desktop{margin-left:80%}html.theme--documenter-dark .column.is-0-desktop{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-desktop{margin-left:0%}html.theme--documenter-dark .column.is-1-desktop{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-desktop{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-desktop{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-desktop{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-desktop{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-desktop{margin-left:25%}html.theme--documenter-dark .column.is-4-desktop{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-desktop{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-desktop{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-desktop{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-desktop{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-desktop{margin-left:50%}html.theme--documenter-dark .column.is-7-desktop{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-desktop{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-desktop{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-desktop{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-desktop{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-desktop{margin-left:75%}html.theme--documenter-dark .column.is-10-desktop{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-desktop{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-desktop{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-desktop{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-desktop{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-desktop{margin-left:100%}}@media screen and (min-width: 1216px){html.theme--documenter-dark .column.is-narrow-widescreen{flex:none;width:unset}html.theme--documenter-dark .column.is-full-widescreen{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-widescreen{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-widescreen{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-widescreen{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-widescreen{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-widescreen{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-widescreen{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-widescreen{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-widescreen{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-widescreen{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-widescreen{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-widescreen{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-widescreen{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-widescreen{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-widescreen{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-widescreen{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-widescreen{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-widescreen{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-widescreen{margin-left:80%}html.theme--documenter-dark .column.is-0-widescreen{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-widescreen{margin-left:0%}html.theme--documenter-dark .column.is-1-widescreen{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-widescreen{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-widescreen{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-widescreen{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-widescreen{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-widescreen{margin-left:25%}html.theme--documenter-dark .column.is-4-widescreen{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-widescreen{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-widescreen{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-widescreen{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-widescreen{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-widescreen{margin-left:50%}html.theme--documenter-dark .column.is-7-widescreen{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-widescreen{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-widescreen{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-widescreen{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-widescreen{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-widescreen{margin-left:75%}html.theme--documenter-dark .column.is-10-widescreen{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-widescreen{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-widescreen{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-widescreen{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-widescreen{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-widescreen{margin-left:100%}}@media screen and (min-width: 1408px){html.theme--documenter-dark .column.is-narrow-fullhd{flex:none;width:unset}html.theme--documenter-dark .column.is-full-fullhd{flex:none;width:100%}html.theme--documenter-dark .column.is-three-quarters-fullhd{flex:none;width:75%}html.theme--documenter-dark .column.is-two-thirds-fullhd{flex:none;width:66.6666%}html.theme--documenter-dark .column.is-half-fullhd{flex:none;width:50%}html.theme--documenter-dark .column.is-one-third-fullhd{flex:none;width:33.3333%}html.theme--documenter-dark .column.is-one-quarter-fullhd{flex:none;width:25%}html.theme--documenter-dark .column.is-one-fifth-fullhd{flex:none;width:20%}html.theme--documenter-dark .column.is-two-fifths-fullhd{flex:none;width:40%}html.theme--documenter-dark .column.is-three-fifths-fullhd{flex:none;width:60%}html.theme--documenter-dark .column.is-four-fifths-fullhd{flex:none;width:80%}html.theme--documenter-dark .column.is-offset-three-quarters-fullhd{margin-left:75%}html.theme--documenter-dark .column.is-offset-two-thirds-fullhd{margin-left:66.6666%}html.theme--documenter-dark .column.is-offset-half-fullhd{margin-left:50%}html.theme--documenter-dark .column.is-offset-one-third-fullhd{margin-left:33.3333%}html.theme--documenter-dark .column.is-offset-one-quarter-fullhd{margin-left:25%}html.theme--documenter-dark .column.is-offset-one-fifth-fullhd{margin-left:20%}html.theme--documenter-dark .column.is-offset-two-fifths-fullhd{margin-left:40%}html.theme--documenter-dark .column.is-offset-three-fifths-fullhd{margin-left:60%}html.theme--documenter-dark .column.is-offset-four-fifths-fullhd{margin-left:80%}html.theme--documenter-dark .column.is-0-fullhd{flex:none;width:0%}html.theme--documenter-dark .column.is-offset-0-fullhd{margin-left:0%}html.theme--documenter-dark .column.is-1-fullhd{flex:none;width:8.33333337%}html.theme--documenter-dark .column.is-offset-1-fullhd{margin-left:8.33333337%}html.theme--documenter-dark .column.is-2-fullhd{flex:none;width:16.66666674%}html.theme--documenter-dark .column.is-offset-2-fullhd{margin-left:16.66666674%}html.theme--documenter-dark .column.is-3-fullhd{flex:none;width:25%}html.theme--documenter-dark .column.is-offset-3-fullhd{margin-left:25%}html.theme--documenter-dark .column.is-4-fullhd{flex:none;width:33.33333337%}html.theme--documenter-dark .column.is-offset-4-fullhd{margin-left:33.33333337%}html.theme--documenter-dark .column.is-5-fullhd{flex:none;width:41.66666674%}html.theme--documenter-dark .column.is-offset-5-fullhd{margin-left:41.66666674%}html.theme--documenter-dark .column.is-6-fullhd{flex:none;width:50%}html.theme--documenter-dark .column.is-offset-6-fullhd{margin-left:50%}html.theme--documenter-dark .column.is-7-fullhd{flex:none;width:58.33333337%}html.theme--documenter-dark .column.is-offset-7-fullhd{margin-left:58.33333337%}html.theme--documenter-dark .column.is-8-fullhd{flex:none;width:66.66666674%}html.theme--documenter-dark .column.is-offset-8-fullhd{margin-left:66.66666674%}html.theme--documenter-dark .column.is-9-fullhd{flex:none;width:75%}html.theme--documenter-dark .column.is-offset-9-fullhd{margin-left:75%}html.theme--documenter-dark .column.is-10-fullhd{flex:none;width:83.33333337%}html.theme--documenter-dark .column.is-offset-10-fullhd{margin-left:83.33333337%}html.theme--documenter-dark .column.is-11-fullhd{flex:none;width:91.66666674%}html.theme--documenter-dark .column.is-offset-11-fullhd{margin-left:91.66666674%}html.theme--documenter-dark .column.is-12-fullhd{flex:none;width:100%}html.theme--documenter-dark .column.is-offset-12-fullhd{margin-left:100%}}html.theme--documenter-dark .columns{margin-left:-.75rem;margin-right:-.75rem;margin-top:-.75rem}html.theme--documenter-dark .columns:last-child{margin-bottom:-.75rem}html.theme--documenter-dark .columns:not(:last-child){margin-bottom:calc(1.5rem - .75rem)}html.theme--documenter-dark .columns.is-centered{justify-content:center}html.theme--documenter-dark .columns.is-gapless{margin-left:0;margin-right:0;margin-top:0}html.theme--documenter-dark .columns.is-gapless>.column{margin:0;padding:0 !important}html.theme--documenter-dark .columns.is-gapless:not(:last-child){margin-bottom:1.5rem}html.theme--documenter-dark .columns.is-gapless:last-child{margin-bottom:0}html.theme--documenter-dark .columns.is-mobile{display:flex}html.theme--documenter-dark .columns.is-multiline{flex-wrap:wrap}html.theme--documenter-dark .columns.is-vcentered{align-items:center}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns:not(.is-desktop){display:flex}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-desktop{display:flex}}html.theme--documenter-dark .columns.is-variable{--columnGap: 0.75rem;margin-left:calc(-1 * var(--columnGap));margin-right:calc(-1 * var(--columnGap))}html.theme--documenter-dark .columns.is-variable>.column{padding-left:var(--columnGap);padding-right:var(--columnGap)}html.theme--documenter-dark .columns.is-variable.is-0{--columnGap: 0rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-0-mobile{--columnGap: 0rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-0-tablet{--columnGap: 0rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-0-tablet-only{--columnGap: 0rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-0-touch{--columnGap: 0rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-0-desktop{--columnGap: 0rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-0-desktop-only{--columnGap: 0rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-0-widescreen{--columnGap: 0rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-0-widescreen-only{--columnGap: 0rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-0-fullhd{--columnGap: 0rem}}html.theme--documenter-dark .columns.is-variable.is-1{--columnGap: .25rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-1-mobile{--columnGap: .25rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-1-tablet{--columnGap: .25rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-1-tablet-only{--columnGap: .25rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-1-touch{--columnGap: .25rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-1-desktop{--columnGap: .25rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-1-desktop-only{--columnGap: .25rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-1-widescreen{--columnGap: .25rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-1-widescreen-only{--columnGap: .25rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-1-fullhd{--columnGap: .25rem}}html.theme--documenter-dark .columns.is-variable.is-2{--columnGap: .5rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-2-mobile{--columnGap: .5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-2-tablet{--columnGap: .5rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-2-tablet-only{--columnGap: .5rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-2-touch{--columnGap: .5rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-2-desktop{--columnGap: .5rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-2-desktop-only{--columnGap: .5rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-2-widescreen{--columnGap: .5rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-2-widescreen-only{--columnGap: .5rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-2-fullhd{--columnGap: .5rem}}html.theme--documenter-dark .columns.is-variable.is-3{--columnGap: .75rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-3-mobile{--columnGap: .75rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-3-tablet{--columnGap: .75rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-3-tablet-only{--columnGap: .75rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-3-touch{--columnGap: .75rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-3-desktop{--columnGap: .75rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-3-desktop-only{--columnGap: .75rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-3-widescreen{--columnGap: .75rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-3-widescreen-only{--columnGap: .75rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-3-fullhd{--columnGap: .75rem}}html.theme--documenter-dark .columns.is-variable.is-4{--columnGap: 1rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-4-mobile{--columnGap: 1rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-4-tablet{--columnGap: 1rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-4-tablet-only{--columnGap: 1rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-4-touch{--columnGap: 1rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-4-desktop{--columnGap: 1rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-4-desktop-only{--columnGap: 1rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-4-widescreen{--columnGap: 1rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-4-widescreen-only{--columnGap: 1rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-4-fullhd{--columnGap: 1rem}}html.theme--documenter-dark .columns.is-variable.is-5{--columnGap: 1.25rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-5-mobile{--columnGap: 1.25rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-5-tablet{--columnGap: 1.25rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-5-tablet-only{--columnGap: 1.25rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-5-touch{--columnGap: 1.25rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-5-desktop{--columnGap: 1.25rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-5-desktop-only{--columnGap: 1.25rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-5-widescreen{--columnGap: 1.25rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-5-widescreen-only{--columnGap: 1.25rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-5-fullhd{--columnGap: 1.25rem}}html.theme--documenter-dark .columns.is-variable.is-6{--columnGap: 1.5rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-6-mobile{--columnGap: 1.5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-6-tablet{--columnGap: 1.5rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-6-tablet-only{--columnGap: 1.5rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-6-touch{--columnGap: 1.5rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-6-desktop{--columnGap: 1.5rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-6-desktop-only{--columnGap: 1.5rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-6-widescreen{--columnGap: 1.5rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-6-widescreen-only{--columnGap: 1.5rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-6-fullhd{--columnGap: 1.5rem}}html.theme--documenter-dark .columns.is-variable.is-7{--columnGap: 1.75rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-7-mobile{--columnGap: 1.75rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-7-tablet{--columnGap: 1.75rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-7-tablet-only{--columnGap: 1.75rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-7-touch{--columnGap: 1.75rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-7-desktop{--columnGap: 1.75rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-7-desktop-only{--columnGap: 1.75rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-7-widescreen{--columnGap: 1.75rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-7-widescreen-only{--columnGap: 1.75rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-7-fullhd{--columnGap: 1.75rem}}html.theme--documenter-dark .columns.is-variable.is-8{--columnGap: 2rem}@media screen and (max-width: 768px){html.theme--documenter-dark .columns.is-variable.is-8-mobile{--columnGap: 2rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .columns.is-variable.is-8-tablet{--columnGap: 2rem}}@media screen and (min-width: 769px) and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-8-tablet-only{--columnGap: 2rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark .columns.is-variable.is-8-touch{--columnGap: 2rem}}@media screen and (min-width: 1056px){html.theme--documenter-dark .columns.is-variable.is-8-desktop{--columnGap: 2rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){html.theme--documenter-dark .columns.is-variable.is-8-desktop-only{--columnGap: 2rem}}@media screen and (min-width: 1216px){html.theme--documenter-dark .columns.is-variable.is-8-widescreen{--columnGap: 2rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){html.theme--documenter-dark .columns.is-variable.is-8-widescreen-only{--columnGap: 2rem}}@media screen and (min-width: 1408px){html.theme--documenter-dark .columns.is-variable.is-8-fullhd{--columnGap: 2rem}}html.theme--documenter-dark .tile{align-items:stretch;display:block;flex-basis:0;flex-grow:1;flex-shrink:1;min-height:min-content}html.theme--documenter-dark .tile.is-ancestor{margin-left:-.75rem;margin-right:-.75rem;margin-top:-.75rem}html.theme--documenter-dark .tile.is-ancestor:last-child{margin-bottom:-.75rem}html.theme--documenter-dark .tile.is-ancestor:not(:last-child){margin-bottom:.75rem}html.theme--documenter-dark .tile.is-child{margin:0 !important}html.theme--documenter-dark .tile.is-parent{padding:.75rem}html.theme--documenter-dark .tile.is-vertical{flex-direction:column}html.theme--documenter-dark .tile.is-vertical>.tile.is-child:not(:last-child){margin-bottom:1.5rem !important}@media screen and (min-width: 769px),print{html.theme--documenter-dark .tile:not(.is-child){display:flex}html.theme--documenter-dark .tile.is-1{flex:none;width:8.33333337%}html.theme--documenter-dark .tile.is-2{flex:none;width:16.66666674%}html.theme--documenter-dark .tile.is-3{flex:none;width:25%}html.theme--documenter-dark .tile.is-4{flex:none;width:33.33333337%}html.theme--documenter-dark .tile.is-5{flex:none;width:41.66666674%}html.theme--documenter-dark .tile.is-6{flex:none;width:50%}html.theme--documenter-dark .tile.is-7{flex:none;width:58.33333337%}html.theme--documenter-dark .tile.is-8{flex:none;width:66.66666674%}html.theme--documenter-dark .tile.is-9{flex:none;width:75%}html.theme--documenter-dark .tile.is-10{flex:none;width:83.33333337%}html.theme--documenter-dark .tile.is-11{flex:none;width:91.66666674%}html.theme--documenter-dark .tile.is-12{flex:none;width:100%}}html.theme--documenter-dark .hero{align-items:stretch;display:flex;flex-direction:column;justify-content:space-between}html.theme--documenter-dark .hero .navbar{background:none}html.theme--documenter-dark .hero .tabs ul{border-bottom:none}html.theme--documenter-dark .hero.is-white{background-color:#fff;color:#0a0a0a}html.theme--documenter-dark .hero.is-white a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-white strong{color:inherit}html.theme--documenter-dark .hero.is-white .title{color:#0a0a0a}html.theme--documenter-dark .hero.is-white .subtitle{color:rgba(10,10,10,0.9)}html.theme--documenter-dark .hero.is-white .subtitle a:not(.button),html.theme--documenter-dark .hero.is-white .subtitle strong{color:#0a0a0a}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-white .navbar-menu{background-color:#fff}}html.theme--documenter-dark .hero.is-white .navbar-item,html.theme--documenter-dark .hero.is-white .navbar-link{color:rgba(10,10,10,0.7)}html.theme--documenter-dark .hero.is-white a.navbar-item:hover,html.theme--documenter-dark .hero.is-white a.navbar-item.is-active,html.theme--documenter-dark .hero.is-white .navbar-link:hover,html.theme--documenter-dark .hero.is-white .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}html.theme--documenter-dark .hero.is-white .tabs a{color:#0a0a0a;opacity:0.9}html.theme--documenter-dark .hero.is-white .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-white .tabs li.is-active a{color:#fff !important;opacity:1}html.theme--documenter-dark .hero.is-white .tabs.is-boxed a,html.theme--documenter-dark .hero.is-white .tabs.is-toggle a{color:#0a0a0a}html.theme--documenter-dark .hero.is-white .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-white .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-white .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-white .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-white .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-white .tabs.is-toggle li.is-active a:hover{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}html.theme--documenter-dark .hero.is-white.is-bold{background-image:linear-gradient(141deg, #e8e3e4 0%, #fff 71%, #fff 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-white.is-bold .navbar-menu{background-image:linear-gradient(141deg, #e8e3e4 0%, #fff 71%, #fff 100%)}}html.theme--documenter-dark .hero.is-black{background-color:#0a0a0a;color:#fff}html.theme--documenter-dark .hero.is-black a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-black strong{color:inherit}html.theme--documenter-dark .hero.is-black .title{color:#fff}html.theme--documenter-dark .hero.is-black .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-black .subtitle a:not(.button),html.theme--documenter-dark .hero.is-black .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-black .navbar-menu{background-color:#0a0a0a}}html.theme--documenter-dark .hero.is-black .navbar-item,html.theme--documenter-dark .hero.is-black .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-black a.navbar-item:hover,html.theme--documenter-dark .hero.is-black a.navbar-item.is-active,html.theme--documenter-dark .hero.is-black .navbar-link:hover,html.theme--documenter-dark .hero.is-black .navbar-link.is-active{background-color:#000;color:#fff}html.theme--documenter-dark .hero.is-black .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-black .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-black .tabs li.is-active a{color:#0a0a0a !important;opacity:1}html.theme--documenter-dark .hero.is-black .tabs.is-boxed a,html.theme--documenter-dark .hero.is-black .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-black .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-black .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-black .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-black .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-black .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-black .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#0a0a0a}html.theme--documenter-dark .hero.is-black.is-bold{background-image:linear-gradient(141deg, #000 0%, #0a0a0a 71%, #181616 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-black.is-bold .navbar-menu{background-image:linear-gradient(141deg, #000 0%, #0a0a0a 71%, #181616 100%)}}html.theme--documenter-dark .hero.is-light{background-color:#ecf0f1;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-light strong{color:inherit}html.theme--documenter-dark .hero.is-light .title{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light .subtitle{color:rgba(0,0,0,0.9)}html.theme--documenter-dark .hero.is-light .subtitle a:not(.button),html.theme--documenter-dark .hero.is-light .subtitle strong{color:rgba(0,0,0,0.7)}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-light .navbar-menu{background-color:#ecf0f1}}html.theme--documenter-dark .hero.is-light .navbar-item,html.theme--documenter-dark .hero.is-light .navbar-link{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light a.navbar-item:hover,html.theme--documenter-dark .hero.is-light a.navbar-item.is-active,html.theme--documenter-dark .hero.is-light .navbar-link:hover,html.theme--documenter-dark .hero.is-light .navbar-link.is-active{background-color:#dde4e6;color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light .tabs a{color:rgba(0,0,0,0.7);opacity:0.9}html.theme--documenter-dark .hero.is-light .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-light .tabs li.is-active a{color:#ecf0f1 !important;opacity:1}html.theme--documenter-dark .hero.is-light .tabs.is-boxed a,html.theme--documenter-dark .hero.is-light .tabs.is-toggle a{color:rgba(0,0,0,0.7)}html.theme--documenter-dark .hero.is-light .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-light .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-light .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-light .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-light .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-light .tabs.is-toggle li.is-active a:hover{background-color:rgba(0,0,0,0.7);border-color:rgba(0,0,0,0.7);color:#ecf0f1}html.theme--documenter-dark .hero.is-light.is-bold{background-image:linear-gradient(141deg, #cadfe0 0%, #ecf0f1 71%, #fafbfc 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-light.is-bold .navbar-menu{background-image:linear-gradient(141deg, #cadfe0 0%, #ecf0f1 71%, #fafbfc 100%)}}html.theme--documenter-dark .hero.is-dark,html.theme--documenter-dark .content kbd.hero{background-color:#282f2f;color:#fff}html.theme--documenter-dark .hero.is-dark a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .content kbd.hero a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-dark strong,html.theme--documenter-dark .content kbd.hero strong{color:inherit}html.theme--documenter-dark .hero.is-dark .title,html.theme--documenter-dark .content kbd.hero .title{color:#fff}html.theme--documenter-dark .hero.is-dark .subtitle,html.theme--documenter-dark .content kbd.hero .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-dark .subtitle a:not(.button),html.theme--documenter-dark .content kbd.hero .subtitle a:not(.button),html.theme--documenter-dark .hero.is-dark .subtitle strong,html.theme--documenter-dark .content kbd.hero .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-dark .navbar-menu,html.theme--documenter-dark .content kbd.hero .navbar-menu{background-color:#282f2f}}html.theme--documenter-dark .hero.is-dark .navbar-item,html.theme--documenter-dark .content kbd.hero .navbar-item,html.theme--documenter-dark .hero.is-dark .navbar-link,html.theme--documenter-dark .content kbd.hero .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-dark a.navbar-item:hover,html.theme--documenter-dark .content kbd.hero a.navbar-item:hover,html.theme--documenter-dark .hero.is-dark a.navbar-item.is-active,html.theme--documenter-dark .content kbd.hero a.navbar-item.is-active,html.theme--documenter-dark .hero.is-dark .navbar-link:hover,html.theme--documenter-dark .content kbd.hero .navbar-link:hover,html.theme--documenter-dark .hero.is-dark .navbar-link.is-active,html.theme--documenter-dark .content kbd.hero .navbar-link.is-active{background-color:#1d2122;color:#fff}html.theme--documenter-dark .hero.is-dark .tabs a,html.theme--documenter-dark .content kbd.hero .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-dark .tabs a:hover,html.theme--documenter-dark .content kbd.hero .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-dark .tabs li.is-active a,html.theme--documenter-dark .content kbd.hero .tabs li.is-active a{color:#282f2f !important;opacity:1}html.theme--documenter-dark .hero.is-dark .tabs.is-boxed a,html.theme--documenter-dark .content kbd.hero .tabs.is-boxed a,html.theme--documenter-dark .hero.is-dark .tabs.is-toggle a,html.theme--documenter-dark .content kbd.hero .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-dark .tabs.is-boxed a:hover,html.theme--documenter-dark .content kbd.hero .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-dark .tabs.is-toggle a:hover,html.theme--documenter-dark .content kbd.hero .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-dark .tabs.is-boxed li.is-active a,html.theme--documenter-dark .content kbd.hero .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-dark .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-dark .tabs.is-toggle li.is-active a,html.theme--documenter-dark .content kbd.hero .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-dark .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#282f2f}html.theme--documenter-dark .hero.is-dark.is-bold,html.theme--documenter-dark .content kbd.hero.is-bold{background-image:linear-gradient(141deg, #0f1615 0%, #282f2f 71%, #313c40 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-dark.is-bold .navbar-menu,html.theme--documenter-dark .content kbd.hero.is-bold .navbar-menu{background-image:linear-gradient(141deg, #0f1615 0%, #282f2f 71%, #313c40 100%)}}html.theme--documenter-dark .hero.is-primary,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink{background-color:#375a7f;color:#fff}html.theme--documenter-dark .hero.is-primary a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-primary strong,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink strong{color:inherit}html.theme--documenter-dark .hero.is-primary .title,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .title{color:#fff}html.theme--documenter-dark .hero.is-primary .subtitle,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-primary .subtitle a:not(.button),html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .subtitle a:not(.button),html.theme--documenter-dark .hero.is-primary .subtitle strong,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-primary .navbar-menu,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-menu{background-color:#375a7f}}html.theme--documenter-dark .hero.is-primary .navbar-item,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-item,html.theme--documenter-dark .hero.is-primary .navbar-link,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-primary a.navbar-item:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink a.navbar-item:hover,html.theme--documenter-dark .hero.is-primary a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink a.navbar-item.is-active,html.theme--documenter-dark .hero.is-primary .navbar-link:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-link:hover,html.theme--documenter-dark .hero.is-primary .navbar-link.is-active,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar-link.is-active{background-color:#2f4d6d;color:#fff}html.theme--documenter-dark .hero.is-primary .tabs a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-primary .tabs a:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-primary .tabs li.is-active a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs li.is-active a{color:#375a7f !important;opacity:1}html.theme--documenter-dark .hero.is-primary .tabs.is-boxed a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-boxed a,html.theme--documenter-dark .hero.is-primary .tabs.is-toggle a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-primary .tabs.is-boxed a:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-primary .tabs.is-toggle a:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-primary .tabs.is-boxed li.is-active a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-primary .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-primary .tabs.is-toggle li.is-active a,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-primary .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#375a7f}html.theme--documenter-dark .hero.is-primary.is-bold,html.theme--documenter-dark .docstring>section>a.hero.is-bold.docs-sourcelink{background-image:linear-gradient(141deg, #214b62 0%, #375a7f 71%, #3a5796 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-primary.is-bold .navbar-menu,html.theme--documenter-dark .docstring>section>a.hero.is-bold.docs-sourcelink .navbar-menu{background-image:linear-gradient(141deg, #214b62 0%, #375a7f 71%, #3a5796 100%)}}html.theme--documenter-dark .hero.is-link{background-color:#1abc9c;color:#fff}html.theme--documenter-dark .hero.is-link a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-link strong{color:inherit}html.theme--documenter-dark .hero.is-link .title{color:#fff}html.theme--documenter-dark .hero.is-link .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-link .subtitle a:not(.button),html.theme--documenter-dark .hero.is-link .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-link .navbar-menu{background-color:#1abc9c}}html.theme--documenter-dark .hero.is-link .navbar-item,html.theme--documenter-dark .hero.is-link .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-link a.navbar-item:hover,html.theme--documenter-dark .hero.is-link a.navbar-item.is-active,html.theme--documenter-dark .hero.is-link .navbar-link:hover,html.theme--documenter-dark .hero.is-link .navbar-link.is-active{background-color:#17a689;color:#fff}html.theme--documenter-dark .hero.is-link .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-link .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-link .tabs li.is-active a{color:#1abc9c !important;opacity:1}html.theme--documenter-dark .hero.is-link .tabs.is-boxed a,html.theme--documenter-dark .hero.is-link .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-link .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-link .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-link .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-link .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-link .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-link .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#1abc9c}html.theme--documenter-dark .hero.is-link.is-bold{background-image:linear-gradient(141deg, #0c9764 0%, #1abc9c 71%, #17d8d2 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-link.is-bold .navbar-menu{background-image:linear-gradient(141deg, #0c9764 0%, #1abc9c 71%, #17d8d2 100%)}}html.theme--documenter-dark .hero.is-info{background-color:#024c7d;color:#fff}html.theme--documenter-dark .hero.is-info a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-info strong{color:inherit}html.theme--documenter-dark .hero.is-info .title{color:#fff}html.theme--documenter-dark .hero.is-info .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-info .subtitle a:not(.button),html.theme--documenter-dark .hero.is-info .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-info .navbar-menu{background-color:#024c7d}}html.theme--documenter-dark .hero.is-info .navbar-item,html.theme--documenter-dark .hero.is-info .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-info a.navbar-item:hover,html.theme--documenter-dark .hero.is-info a.navbar-item.is-active,html.theme--documenter-dark .hero.is-info .navbar-link:hover,html.theme--documenter-dark .hero.is-info .navbar-link.is-active{background-color:#023d64;color:#fff}html.theme--documenter-dark .hero.is-info .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-info .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-info .tabs li.is-active a{color:#024c7d !important;opacity:1}html.theme--documenter-dark .hero.is-info .tabs.is-boxed a,html.theme--documenter-dark .hero.is-info .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-info .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-info .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-info .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-info .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-info .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-info .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#024c7d}html.theme--documenter-dark .hero.is-info.is-bold{background-image:linear-gradient(141deg, #003a4c 0%, #024c7d 71%, #004299 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-info.is-bold .navbar-menu{background-image:linear-gradient(141deg, #003a4c 0%, #024c7d 71%, #004299 100%)}}html.theme--documenter-dark .hero.is-success{background-color:#008438;color:#fff}html.theme--documenter-dark .hero.is-success a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-success strong{color:inherit}html.theme--documenter-dark .hero.is-success .title{color:#fff}html.theme--documenter-dark .hero.is-success .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-success .subtitle a:not(.button),html.theme--documenter-dark .hero.is-success .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-success .navbar-menu{background-color:#008438}}html.theme--documenter-dark .hero.is-success .navbar-item,html.theme--documenter-dark .hero.is-success .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-success a.navbar-item:hover,html.theme--documenter-dark .hero.is-success a.navbar-item.is-active,html.theme--documenter-dark .hero.is-success .navbar-link:hover,html.theme--documenter-dark .hero.is-success .navbar-link.is-active{background-color:#006b2d;color:#fff}html.theme--documenter-dark .hero.is-success .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-success .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-success .tabs li.is-active a{color:#008438 !important;opacity:1}html.theme--documenter-dark .hero.is-success .tabs.is-boxed a,html.theme--documenter-dark .hero.is-success .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-success .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-success .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-success .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-success .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-success .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-success .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#008438}html.theme--documenter-dark .hero.is-success.is-bold{background-image:linear-gradient(141deg, #005115 0%, #008438 71%, #009e5d 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-success.is-bold .navbar-menu{background-image:linear-gradient(141deg, #005115 0%, #008438 71%, #009e5d 100%)}}html.theme--documenter-dark .hero.is-warning{background-color:#ad8100;color:#fff}html.theme--documenter-dark .hero.is-warning a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-warning strong{color:inherit}html.theme--documenter-dark .hero.is-warning .title{color:#fff}html.theme--documenter-dark .hero.is-warning .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-warning .subtitle a:not(.button),html.theme--documenter-dark .hero.is-warning .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-warning .navbar-menu{background-color:#ad8100}}html.theme--documenter-dark .hero.is-warning .navbar-item,html.theme--documenter-dark .hero.is-warning .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-warning a.navbar-item:hover,html.theme--documenter-dark .hero.is-warning a.navbar-item.is-active,html.theme--documenter-dark .hero.is-warning .navbar-link:hover,html.theme--documenter-dark .hero.is-warning .navbar-link.is-active{background-color:#946e00;color:#fff}html.theme--documenter-dark .hero.is-warning .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-warning .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-warning .tabs li.is-active a{color:#ad8100 !important;opacity:1}html.theme--documenter-dark .hero.is-warning .tabs.is-boxed a,html.theme--documenter-dark .hero.is-warning .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-warning .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-warning .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-warning .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-warning .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-warning .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-warning .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#ad8100}html.theme--documenter-dark .hero.is-warning.is-bold{background-image:linear-gradient(141deg, #7a4700 0%, #ad8100 71%, #c7b500 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-warning.is-bold .navbar-menu{background-image:linear-gradient(141deg, #7a4700 0%, #ad8100 71%, #c7b500 100%)}}html.theme--documenter-dark .hero.is-danger{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .hero.is-danger a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),html.theme--documenter-dark .hero.is-danger strong{color:inherit}html.theme--documenter-dark .hero.is-danger .title{color:#fff}html.theme--documenter-dark .hero.is-danger .subtitle{color:rgba(255,255,255,0.9)}html.theme--documenter-dark .hero.is-danger .subtitle a:not(.button),html.theme--documenter-dark .hero.is-danger .subtitle strong{color:#fff}@media screen and (max-width: 1055px){html.theme--documenter-dark .hero.is-danger .navbar-menu{background-color:#9e1b0d}}html.theme--documenter-dark .hero.is-danger .navbar-item,html.theme--documenter-dark .hero.is-danger .navbar-link{color:rgba(255,255,255,0.7)}html.theme--documenter-dark .hero.is-danger a.navbar-item:hover,html.theme--documenter-dark .hero.is-danger a.navbar-item.is-active,html.theme--documenter-dark .hero.is-danger .navbar-link:hover,html.theme--documenter-dark .hero.is-danger .navbar-link.is-active{background-color:#86170b;color:#fff}html.theme--documenter-dark .hero.is-danger .tabs a{color:#fff;opacity:0.9}html.theme--documenter-dark .hero.is-danger .tabs a:hover{opacity:1}html.theme--documenter-dark .hero.is-danger .tabs li.is-active a{color:#9e1b0d !important;opacity:1}html.theme--documenter-dark .hero.is-danger .tabs.is-boxed a,html.theme--documenter-dark .hero.is-danger .tabs.is-toggle a{color:#fff}html.theme--documenter-dark .hero.is-danger .tabs.is-boxed a:hover,html.theme--documenter-dark .hero.is-danger .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}html.theme--documenter-dark .hero.is-danger .tabs.is-boxed li.is-active a,html.theme--documenter-dark .hero.is-danger .tabs.is-boxed li.is-active a:hover,html.theme--documenter-dark .hero.is-danger .tabs.is-toggle li.is-active a,html.theme--documenter-dark .hero.is-danger .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#9e1b0d}html.theme--documenter-dark .hero.is-danger.is-bold{background-image:linear-gradient(141deg, #75030b 0%, #9e1b0d 71%, #ba380a 100%)}@media screen and (max-width: 768px){html.theme--documenter-dark .hero.is-danger.is-bold .navbar-menu{background-image:linear-gradient(141deg, #75030b 0%, #9e1b0d 71%, #ba380a 100%)}}html.theme--documenter-dark .hero.is-small .hero-body,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.hero .hero-body{padding:1.5rem}@media screen and (min-width: 769px),print{html.theme--documenter-dark .hero.is-medium .hero-body{padding:9rem 4.5rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .hero.is-large .hero-body{padding:18rem 6rem}}html.theme--documenter-dark .hero.is-halfheight .hero-body,html.theme--documenter-dark .hero.is-fullheight .hero-body,html.theme--documenter-dark .hero.is-fullheight-with-navbar .hero-body{align-items:center;display:flex}html.theme--documenter-dark .hero.is-halfheight .hero-body>.container,html.theme--documenter-dark .hero.is-fullheight .hero-body>.container,html.theme--documenter-dark .hero.is-fullheight-with-navbar .hero-body>.container{flex-grow:1;flex-shrink:1}html.theme--documenter-dark .hero.is-halfheight{min-height:50vh}html.theme--documenter-dark .hero.is-fullheight{min-height:100vh}html.theme--documenter-dark .hero-video{overflow:hidden}html.theme--documenter-dark .hero-video video{left:50%;min-height:100%;min-width:100%;position:absolute;top:50%;transform:translate3d(-50%, -50%, 0)}html.theme--documenter-dark .hero-video.is-transparent{opacity:0.3}@media screen and (max-width: 768px){html.theme--documenter-dark .hero-video{display:none}}html.theme--documenter-dark .hero-buttons{margin-top:1.5rem}@media screen and (max-width: 768px){html.theme--documenter-dark .hero-buttons .button{display:flex}html.theme--documenter-dark .hero-buttons .button:not(:last-child){margin-bottom:0.75rem}}@media screen and (min-width: 769px),print{html.theme--documenter-dark .hero-buttons{display:flex;justify-content:center}html.theme--documenter-dark .hero-buttons .button:not(:last-child){margin-right:1.5rem}}html.theme--documenter-dark .hero-head,html.theme--documenter-dark .hero-foot{flex-grow:0;flex-shrink:0}html.theme--documenter-dark .hero-body{flex-grow:1;flex-shrink:0;padding:3rem 1.5rem}@media screen and (min-width: 769px),print{html.theme--documenter-dark .hero-body{padding:3rem 3rem}}html.theme--documenter-dark .section{padding:3rem 1.5rem}@media screen and (min-width: 1056px){html.theme--documenter-dark .section{padding:3rem 3rem}html.theme--documenter-dark .section.is-medium{padding:9rem 4.5rem}html.theme--documenter-dark .section.is-large{padding:18rem 6rem}}html.theme--documenter-dark .footer{background-color:#282f2f;padding:3rem 1.5rem 6rem}html.theme--documenter-dark hr{height:1px}html.theme--documenter-dark h6{text-transform:uppercase;letter-spacing:0.5px}html.theme--documenter-dark .hero{background-color:#343c3d}html.theme--documenter-dark a{transition:all 200ms ease}html.theme--documenter-dark .button{transition:all 200ms ease;border-width:1px;color:#fff}html.theme--documenter-dark .button.is-active,html.theme--documenter-dark .button.is-focused,html.theme--documenter-dark .button:active,html.theme--documenter-dark .button:focus{box-shadow:0 0 0 2px rgba(140,155,157,0.5)}html.theme--documenter-dark .button.is-white.is-hovered,html.theme--documenter-dark .button.is-white:hover{background-color:#fff}html.theme--documenter-dark .button.is-white.is-active,html.theme--documenter-dark .button.is-white.is-focused,html.theme--documenter-dark .button.is-white:active,html.theme--documenter-dark .button.is-white:focus{border-color:#fff;box-shadow:0 0 0 2px rgba(255,255,255,0.5)}html.theme--documenter-dark .button.is-black.is-hovered,html.theme--documenter-dark .button.is-black:hover{background-color:#1d1d1d}html.theme--documenter-dark .button.is-black.is-active,html.theme--documenter-dark .button.is-black.is-focused,html.theme--documenter-dark .button.is-black:active,html.theme--documenter-dark .button.is-black:focus{border-color:#0a0a0a;box-shadow:0 0 0 2px rgba(10,10,10,0.5)}html.theme--documenter-dark .button.is-light.is-hovered,html.theme--documenter-dark .button.is-light:hover{background-color:#fff}html.theme--documenter-dark .button.is-light.is-active,html.theme--documenter-dark .button.is-light.is-focused,html.theme--documenter-dark .button.is-light:active,html.theme--documenter-dark .button.is-light:focus{border-color:#ecf0f1;box-shadow:0 0 0 2px rgba(236,240,241,0.5)}html.theme--documenter-dark .button.is-dark.is-hovered,html.theme--documenter-dark .content kbd.button.is-hovered,html.theme--documenter-dark .button.is-dark:hover,html.theme--documenter-dark .content kbd.button:hover{background-color:#3a4344}html.theme--documenter-dark .button.is-dark.is-active,html.theme--documenter-dark .content kbd.button.is-active,html.theme--documenter-dark .button.is-dark.is-focused,html.theme--documenter-dark .content kbd.button.is-focused,html.theme--documenter-dark .button.is-dark:active,html.theme--documenter-dark .content kbd.button:active,html.theme--documenter-dark .button.is-dark:focus,html.theme--documenter-dark .content kbd.button:focus{border-color:#282f2f;box-shadow:0 0 0 2px rgba(40,47,47,0.5)}html.theme--documenter-dark .button.is-primary.is-hovered,html.theme--documenter-dark .docstring>section>a.button.is-hovered.docs-sourcelink,html.theme--documenter-dark .button.is-primary:hover,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:hover{background-color:#436d9a}html.theme--documenter-dark .button.is-primary.is-active,html.theme--documenter-dark .docstring>section>a.button.is-active.docs-sourcelink,html.theme--documenter-dark .button.is-primary.is-focused,html.theme--documenter-dark .docstring>section>a.button.is-focused.docs-sourcelink,html.theme--documenter-dark .button.is-primary:active,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:active,html.theme--documenter-dark .button.is-primary:focus,html.theme--documenter-dark .docstring>section>a.button.docs-sourcelink:focus{border-color:#375a7f;box-shadow:0 0 0 2px rgba(55,90,127,0.5)}html.theme--documenter-dark .button.is-link.is-hovered,html.theme--documenter-dark .button.is-link:hover{background-color:#1fdeb8}html.theme--documenter-dark .button.is-link.is-active,html.theme--documenter-dark .button.is-link.is-focused,html.theme--documenter-dark .button.is-link:active,html.theme--documenter-dark .button.is-link:focus{border-color:#1abc9c;box-shadow:0 0 0 2px rgba(26,188,156,0.5)}html.theme--documenter-dark .button.is-info.is-hovered,html.theme--documenter-dark .button.is-info:hover{background-color:#0363a3}html.theme--documenter-dark .button.is-info.is-active,html.theme--documenter-dark .button.is-info.is-focused,html.theme--documenter-dark .button.is-info:active,html.theme--documenter-dark .button.is-info:focus{border-color:#024c7d;box-shadow:0 0 0 2px rgba(2,76,125,0.5)}html.theme--documenter-dark .button.is-success.is-hovered,html.theme--documenter-dark .button.is-success:hover{background-color:#00aa48}html.theme--documenter-dark .button.is-success.is-active,html.theme--documenter-dark .button.is-success.is-focused,html.theme--documenter-dark .button.is-success:active,html.theme--documenter-dark .button.is-success:focus{border-color:#008438;box-shadow:0 0 0 2px rgba(0,132,56,0.5)}html.theme--documenter-dark .button.is-warning.is-hovered,html.theme--documenter-dark .button.is-warning:hover{background-color:#d39e00}html.theme--documenter-dark .button.is-warning.is-active,html.theme--documenter-dark .button.is-warning.is-focused,html.theme--documenter-dark .button.is-warning:active,html.theme--documenter-dark .button.is-warning:focus{border-color:#ad8100;box-shadow:0 0 0 2px rgba(173,129,0,0.5)}html.theme--documenter-dark .button.is-danger.is-hovered,html.theme--documenter-dark .button.is-danger:hover{background-color:#c12110}html.theme--documenter-dark .button.is-danger.is-active,html.theme--documenter-dark .button.is-danger.is-focused,html.theme--documenter-dark .button.is-danger:active,html.theme--documenter-dark .button.is-danger:focus{border-color:#9e1b0d;box-shadow:0 0 0 2px rgba(158,27,13,0.5)}html.theme--documenter-dark .label{color:#dbdee0}html.theme--documenter-dark .button,html.theme--documenter-dark .control.has-icons-left .icon,html.theme--documenter-dark .control.has-icons-right .icon,html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark .pagination-ellipsis,html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-previous,html.theme--documenter-dark .select,html.theme--documenter-dark .select select,html.theme--documenter-dark .textarea{height:2.5em}html.theme--documenter-dark .input,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark .textarea{transition:all 200ms ease;box-shadow:none;border-width:1px;padding-left:1em;padding-right:1em}html.theme--documenter-dark .select:after,html.theme--documenter-dark .select select{border-width:1px}html.theme--documenter-dark .control.has-addons .button,html.theme--documenter-dark .control.has-addons .input,html.theme--documenter-dark .control.has-addons #documenter .docs-sidebar form.docs-search>input,html.theme--documenter-dark #documenter .docs-sidebar .control.has-addons form.docs-search>input,html.theme--documenter-dark .control.has-addons .select{margin-right:-1px}html.theme--documenter-dark .notification{background-color:#343c3d}html.theme--documenter-dark .card{box-shadow:none;border:1px solid #343c3d;background-color:#282f2f;border-radius:.4em}html.theme--documenter-dark .card .card-image img{border-radius:.4em .4em 0 0}html.theme--documenter-dark .card .card-header{box-shadow:none;background-color:rgba(18,18,18,0.2);border-radius:.4em .4em 0 0}html.theme--documenter-dark .card .card-footer{background-color:rgba(18,18,18,0.2)}html.theme--documenter-dark .card .card-footer,html.theme--documenter-dark .card .card-footer-item{border-width:1px;border-color:#343c3d}html.theme--documenter-dark .notification.is-white a:not(.button){color:#0a0a0a;text-decoration:underline}html.theme--documenter-dark .notification.is-black a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-light a:not(.button){color:rgba(0,0,0,0.7);text-decoration:underline}html.theme--documenter-dark .notification.is-dark a:not(.button),html.theme--documenter-dark .content kbd.notification a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-primary a:not(.button),html.theme--documenter-dark .docstring>section>a.notification.docs-sourcelink a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-link a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-info a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-success a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-warning a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .notification.is-danger a:not(.button){color:#fff;text-decoration:underline}html.theme--documenter-dark .tag,html.theme--documenter-dark .content kbd,html.theme--documenter-dark .docstring>section>a.docs-sourcelink{border-radius:.4em}html.theme--documenter-dark .menu-list a{transition:all 300ms ease}html.theme--documenter-dark .modal-card-body{background-color:#282f2f}html.theme--documenter-dark .modal-card-foot,html.theme--documenter-dark .modal-card-head{border-color:#343c3d}html.theme--documenter-dark .message-header{font-weight:700;background-color:#343c3d;color:#fff}html.theme--documenter-dark .message-body{border-width:1px;border-color:#343c3d}html.theme--documenter-dark .navbar{border-radius:.4em}html.theme--documenter-dark .navbar.is-transparent{background:none}html.theme--documenter-dark .navbar.is-primary .navbar-dropdown a.navbar-item.is-active,html.theme--documenter-dark .docstring>section>a.navbar.docs-sourcelink .navbar-dropdown a.navbar-item.is-active{background-color:#1abc9c}@media screen and (max-width: 1055px){html.theme--documenter-dark .navbar .navbar-menu{background-color:#375a7f;border-radius:0 0 .4em .4em}}html.theme--documenter-dark .hero .navbar,html.theme--documenter-dark body>.navbar{border-radius:0}html.theme--documenter-dark .pagination-link,html.theme--documenter-dark .pagination-next,html.theme--documenter-dark .pagination-previous{border-width:1px}html.theme--documenter-dark .panel-block,html.theme--documenter-dark .panel-heading,html.theme--documenter-dark .panel-tabs{border-width:1px}html.theme--documenter-dark .panel-block:first-child,html.theme--documenter-dark .panel-heading:first-child,html.theme--documenter-dark .panel-tabs:first-child{border-top-width:1px}html.theme--documenter-dark .panel-heading{font-weight:700}html.theme--documenter-dark .panel-tabs a{border-width:1px;margin-bottom:-1px}html.theme--documenter-dark .panel-tabs a.is-active{border-bottom-color:#17a689}html.theme--documenter-dark .panel-block:hover{color:#1dd2af}html.theme--documenter-dark .panel-block:hover .panel-icon{color:#1dd2af}html.theme--documenter-dark .panel-block.is-active .panel-icon{color:#17a689}html.theme--documenter-dark .tabs a{border-bottom-width:1px;margin-bottom:-1px}html.theme--documenter-dark .tabs ul{border-bottom-width:1px}html.theme--documenter-dark .tabs.is-boxed a{border-width:1px}html.theme--documenter-dark .tabs.is-boxed li.is-active a{background-color:#1f2424}html.theme--documenter-dark .tabs.is-toggle li a{border-width:1px;margin-bottom:0}html.theme--documenter-dark .tabs.is-toggle li+li{margin-left:-1px}html.theme--documenter-dark .hero.is-white .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-black .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-light .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-dark .navbar .navbar-dropdown .navbar-item:hover,html.theme--documenter-dark .content kbd.hero .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-primary .navbar .navbar-dropdown .navbar-item:hover,html.theme--documenter-dark .docstring>section>a.hero.docs-sourcelink .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-link .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-info .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-success .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-warning .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark .hero.is-danger .navbar .navbar-dropdown .navbar-item:hover{background-color:rgba(0,0,0,0)}html.theme--documenter-dark h1 .docs-heading-anchor,html.theme--documenter-dark h1 .docs-heading-anchor:hover,html.theme--documenter-dark h1 .docs-heading-anchor:visited,html.theme--documenter-dark h2 .docs-heading-anchor,html.theme--documenter-dark h2 .docs-heading-anchor:hover,html.theme--documenter-dark h2 .docs-heading-anchor:visited,html.theme--documenter-dark h3 .docs-heading-anchor,html.theme--documenter-dark h3 .docs-heading-anchor:hover,html.theme--documenter-dark h3 .docs-heading-anchor:visited,html.theme--documenter-dark h4 .docs-heading-anchor,html.theme--documenter-dark h4 .docs-heading-anchor:hover,html.theme--documenter-dark h4 .docs-heading-anchor:visited,html.theme--documenter-dark h5 .docs-heading-anchor,html.theme--documenter-dark h5 .docs-heading-anchor:hover,html.theme--documenter-dark h5 .docs-heading-anchor:visited,html.theme--documenter-dark h6 .docs-heading-anchor,html.theme--documenter-dark h6 .docs-heading-anchor:hover,html.theme--documenter-dark h6 .docs-heading-anchor:visited{color:#f2f2f2}html.theme--documenter-dark h1 .docs-heading-anchor-permalink,html.theme--documenter-dark h2 .docs-heading-anchor-permalink,html.theme--documenter-dark h3 .docs-heading-anchor-permalink,html.theme--documenter-dark h4 .docs-heading-anchor-permalink,html.theme--documenter-dark h5 .docs-heading-anchor-permalink,html.theme--documenter-dark h6 .docs-heading-anchor-permalink{visibility:hidden;vertical-align:middle;margin-left:0.5em;font-size:0.7rem}html.theme--documenter-dark h1 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h2 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h3 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h4 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h5 .docs-heading-anchor-permalink::before,html.theme--documenter-dark h6 .docs-heading-anchor-permalink::before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f0c1"}html.theme--documenter-dark h1:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h2:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h3:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h4:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h5:hover .docs-heading-anchor-permalink,html.theme--documenter-dark h6:hover .docs-heading-anchor-permalink{visibility:visible}html.theme--documenter-dark .docs-light-only{display:none !important}html.theme--documenter-dark pre{position:relative;overflow:hidden}html.theme--documenter-dark pre code,html.theme--documenter-dark pre code.hljs{padding:0 .75rem !important;overflow:auto;display:block}html.theme--documenter-dark pre code:first-of-type,html.theme--documenter-dark pre code.hljs:first-of-type{padding-top:0.5rem !important}html.theme--documenter-dark pre code:last-of-type,html.theme--documenter-dark pre code.hljs:last-of-type{padding-bottom:0.5rem !important}html.theme--documenter-dark pre .copy-button{opacity:0.2;transition:opacity 0.2s;position:absolute;right:0em;top:0em;padding:0.5em;width:2.5em;height:2.5em;background:transparent;border:none;font-family:"Font Awesome 6 Free";color:#fff;cursor:pointer;text-align:center}html.theme--documenter-dark pre .copy-button:focus,html.theme--documenter-dark pre .copy-button:hover{opacity:1;background:rgba(255,255,255,0.1);color:#1abc9c}html.theme--documenter-dark pre .copy-button.success{color:#259a12;opacity:1}html.theme--documenter-dark pre .copy-button.error{color:#cb3c33;opacity:1}html.theme--documenter-dark pre:hover .copy-button{opacity:1}html.theme--documenter-dark .admonition{background-color:#282f2f;border-style:solid;border-width:1px;border-color:#5e6d6f;border-radius:.4em;font-size:1rem}html.theme--documenter-dark .admonition strong{color:currentColor}html.theme--documenter-dark .admonition.is-small,html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input.admonition{font-size:.75rem}html.theme--documenter-dark .admonition.is-medium{font-size:1.25rem}html.theme--documenter-dark .admonition.is-large{font-size:1.5rem}html.theme--documenter-dark .admonition.is-default{background-color:#282f2f;border-color:#5e6d6f}html.theme--documenter-dark .admonition.is-default>.admonition-header{background-color:#5e6d6f;color:#fff}html.theme--documenter-dark .admonition.is-default>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-info{background-color:#282f2f;border-color:#024c7d}html.theme--documenter-dark .admonition.is-info>.admonition-header{background-color:#024c7d;color:#fff}html.theme--documenter-dark .admonition.is-info>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-success{background-color:#282f2f;border-color:#008438}html.theme--documenter-dark .admonition.is-success>.admonition-header{background-color:#008438;color:#fff}html.theme--documenter-dark .admonition.is-success>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-warning{background-color:#282f2f;border-color:#ad8100}html.theme--documenter-dark .admonition.is-warning>.admonition-header{background-color:#ad8100;color:#fff}html.theme--documenter-dark .admonition.is-warning>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-danger{background-color:#282f2f;border-color:#9e1b0d}html.theme--documenter-dark .admonition.is-danger>.admonition-header{background-color:#9e1b0d;color:#fff}html.theme--documenter-dark .admonition.is-danger>.admonition-body{color:#fff}html.theme--documenter-dark .admonition.is-compat{background-color:#282f2f;border-color:#137886}html.theme--documenter-dark .admonition.is-compat>.admonition-header{background-color:#137886;color:#fff}html.theme--documenter-dark .admonition.is-compat>.admonition-body{color:#fff}html.theme--documenter-dark .admonition-header{color:#fff;background-color:#5e6d6f;align-items:center;font-weight:700;justify-content:space-between;line-height:1.25;padding:0.5rem .75rem;position:relative}html.theme--documenter-dark .admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;margin-right:.75rem;content:"\f06a"}html.theme--documenter-dark details.admonition.is-details>.admonition-header{list-style:none}html.theme--documenter-dark details.admonition.is-details>.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f055"}html.theme--documenter-dark details.admonition.is-details[open]>.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f056"}html.theme--documenter-dark .admonition-body{color:#fff;padding:0.5rem .75rem}html.theme--documenter-dark .admonition-body pre{background-color:#282f2f}html.theme--documenter-dark .admonition-body code{background-color:rgba(255,255,255,0.05)}html.theme--documenter-dark .docstring{margin-bottom:1em;background-color:rgba(0,0,0,0);border:1px solid #5e6d6f;box-shadow:none;max-width:100%}html.theme--documenter-dark .docstring>header{cursor:pointer;display:flex;flex-grow:1;align-items:stretch;padding:0.5rem .75rem;background-color:#282f2f;box-shadow:0 0.125em 0.25em rgba(10,10,10,0.1);box-shadow:none;border-bottom:1px solid #5e6d6f}html.theme--documenter-dark .docstring>header code{background-color:transparent}html.theme--documenter-dark .docstring>header .docstring-article-toggle-button{min-width:1.1rem;padding:0.2rem 0.2rem 0.2rem 0}html.theme--documenter-dark .docstring>header .docstring-binding{margin-right:0.3em}html.theme--documenter-dark .docstring>header .docstring-category{margin-left:0.3em}html.theme--documenter-dark .docstring>section{position:relative;padding:.75rem .75rem;border-bottom:1px solid #5e6d6f}html.theme--documenter-dark .docstring>section:last-child{border-bottom:none}html.theme--documenter-dark .docstring>section>a.docs-sourcelink{transition:opacity 0.3s;opacity:0;position:absolute;right:.375rem;bottom:.375rem}html.theme--documenter-dark .docstring>section>a.docs-sourcelink:focus{opacity:1 !important}html.theme--documenter-dark .docstring:hover>section>a.docs-sourcelink{opacity:0.2}html.theme--documenter-dark .docstring:focus-within>section>a.docs-sourcelink{opacity:0.2}html.theme--documenter-dark .docstring>section:hover a.docs-sourcelink{opacity:1}html.theme--documenter-dark .documenter-example-output{background-color:#1f2424}html.theme--documenter-dark .outdated-warning-overlay{position:fixed;top:0;left:0;right:0;box-shadow:0 0 10px rgba(0,0,0,0.3);z-index:999;background-color:#282f2f;color:#fff;border-bottom:3px solid #9e1b0d;padding:10px 35px;text-align:center;font-size:15px}html.theme--documenter-dark .outdated-warning-overlay .outdated-warning-closer{position:absolute;top:calc(50% - 10px);right:18px;cursor:pointer;width:12px}html.theme--documenter-dark .outdated-warning-overlay a{color:#1abc9c}html.theme--documenter-dark .outdated-warning-overlay a:hover{color:#1dd2af}html.theme--documenter-dark .content pre{border:1px solid #5e6d6f}html.theme--documenter-dark .content code{font-weight:inherit}html.theme--documenter-dark .content a code{color:#1abc9c}html.theme--documenter-dark .content h1 code,html.theme--documenter-dark .content h2 code,html.theme--documenter-dark .content h3 code,html.theme--documenter-dark .content h4 code,html.theme--documenter-dark .content h5 code,html.theme--documenter-dark .content h6 code{color:#f2f2f2}html.theme--documenter-dark .content table{display:block;width:initial;max-width:100%;overflow-x:auto}html.theme--documenter-dark .content blockquote>ul:first-child,html.theme--documenter-dark .content blockquote>ol:first-child,html.theme--documenter-dark .content .admonition-body>ul:first-child,html.theme--documenter-dark .content .admonition-body>ol:first-child{margin-top:0}html.theme--documenter-dark pre,html.theme--documenter-dark code{font-variant-ligatures:no-contextual}html.theme--documenter-dark .breadcrumb a.is-disabled{cursor:default;pointer-events:none}html.theme--documenter-dark .breadcrumb a.is-disabled,html.theme--documenter-dark .breadcrumb a.is-disabled:hover{color:#f2f2f2}html.theme--documenter-dark .hljs{background:initial !important}html.theme--documenter-dark .katex .katex-mathml{top:0;right:0}html.theme--documenter-dark .katex-display,html.theme--documenter-dark mjx-container,html.theme--documenter-dark .MathJax_Display{margin:0.5em 0 !important}html.theme--documenter-dark html{-moz-osx-font-smoothing:auto;-webkit-font-smoothing:auto}html.theme--documenter-dark li.no-marker{list-style:none}html.theme--documenter-dark #documenter .docs-main>article{overflow-wrap:break-word}html.theme--documenter-dark #documenter .docs-main>article .math-container{overflow-x:auto;overflow-y:hidden}@media screen and (min-width: 1056px){html.theme--documenter-dark #documenter .docs-main{max-width:52rem;margin-left:20rem;padding-right:1rem}}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-main{width:100%}html.theme--documenter-dark #documenter .docs-main>article{max-width:52rem;margin-left:auto;margin-right:auto;margin-bottom:1rem;padding:0 1rem}html.theme--documenter-dark #documenter .docs-main>header,html.theme--documenter-dark #documenter .docs-main>nav{max-width:100%;width:100%;margin:0}}html.theme--documenter-dark #documenter .docs-main header.docs-navbar{background-color:#1f2424;border-bottom:1px solid #5e6d6f;z-index:2;min-height:4rem;margin-bottom:1rem;display:flex}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .breadcrumb{flex-grow:1}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-sidebar-button{display:block;font-size:1.5rem;padding-bottom:0.1rem;margin-right:1rem}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right{display:flex;white-space:nowrap;gap:1rem;align-items:center}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right .docs-icon,html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right .docs-label{display:inline-block}html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right .docs-label{padding:0;margin-left:0.3em}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-main header.docs-navbar .docs-right .docs-navbar-link{margin-left:0.4rem;margin-right:0.4rem}}html.theme--documenter-dark #documenter .docs-main header.docs-navbar>*{margin:auto 0}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-main header.docs-navbar{position:sticky;top:0;padding:0 1rem;transition-property:top, box-shadow;-webkit-transition-property:top, box-shadow;transition-duration:0.3s;-webkit-transition-duration:0.3s}html.theme--documenter-dark #documenter .docs-main header.docs-navbar.headroom--not-top{box-shadow:.2rem 0rem .4rem #171717;transition-duration:0.7s;-webkit-transition-duration:0.7s}html.theme--documenter-dark #documenter .docs-main header.docs-navbar.headroom--unpinned.headroom--not-top.headroom--not-bottom{top:-4.5rem;transition-duration:0.7s;-webkit-transition-duration:0.7s}}html.theme--documenter-dark #documenter .docs-main section.footnotes{border-top:1px solid #5e6d6f}html.theme--documenter-dark #documenter .docs-main section.footnotes li .tag:first-child,html.theme--documenter-dark #documenter .docs-main section.footnotes li .docstring>section>a.docs-sourcelink:first-child,html.theme--documenter-dark #documenter .docs-main section.footnotes li .content kbd:first-child,html.theme--documenter-dark .content #documenter .docs-main section.footnotes li kbd:first-child{margin-right:1em;margin-bottom:0.4em}html.theme--documenter-dark #documenter .docs-main .docs-footer{display:flex;flex-wrap:wrap;margin-left:0;margin-right:0;border-top:1px solid #5e6d6f;padding-top:1rem;padding-bottom:1rem}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-main .docs-footer{padding-left:1rem;padding-right:1rem}}html.theme--documenter-dark #documenter .docs-main .docs-footer .docs-footer-nextpage,html.theme--documenter-dark #documenter .docs-main .docs-footer .docs-footer-prevpage{flex-grow:1}html.theme--documenter-dark #documenter .docs-main .docs-footer .docs-footer-nextpage{text-align:right}html.theme--documenter-dark #documenter .docs-main .docs-footer .flexbox-break{flex-basis:100%;height:0}html.theme--documenter-dark #documenter .docs-main .docs-footer .footer-message{font-size:0.8em;margin:0.5em auto 0 auto;text-align:center}html.theme--documenter-dark #documenter .docs-sidebar{display:flex;flex-direction:column;color:#fff;background-color:#282f2f;border-right:1px solid #5e6d6f;padding:0;flex:0 0 18rem;z-index:5;font-size:1rem;position:fixed;left:-18rem;width:18rem;height:100%;transition:left 0.3s}html.theme--documenter-dark #documenter .docs-sidebar.visible{left:0;box-shadow:.4rem 0rem .8rem #171717}@media screen and (min-width: 1056px){html.theme--documenter-dark #documenter .docs-sidebar.visible{box-shadow:none}}@media screen and (min-width: 1056px){html.theme--documenter-dark #documenter .docs-sidebar{left:0;top:0}}html.theme--documenter-dark #documenter .docs-sidebar .docs-logo{margin-top:1rem;padding:0 1rem}html.theme--documenter-dark #documenter .docs-sidebar .docs-logo>img{max-height:6rem;margin:auto}html.theme--documenter-dark #documenter .docs-sidebar .docs-package-name{flex-shrink:0;font-size:1.5rem;font-weight:700;text-align:center;white-space:nowrap;overflow:hidden;padding:0.5rem 0}html.theme--documenter-dark #documenter .docs-sidebar .docs-package-name .docs-autofit{max-width:16.2rem}html.theme--documenter-dark #documenter .docs-sidebar .docs-package-name a,html.theme--documenter-dark #documenter .docs-sidebar .docs-package-name a:hover{color:#fff}html.theme--documenter-dark #documenter .docs-sidebar .docs-version-selector{border-top:1px solid #5e6d6f;display:none;padding:0.5rem}html.theme--documenter-dark #documenter .docs-sidebar .docs-version-selector.visible{display:flex}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu{flex-grow:1;user-select:none;border-top:1px solid #5e6d6f;padding-bottom:1.5rem}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu>li>.tocitem{font-weight:bold}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu>li li{font-size:.95rem;margin-left:1em;border-left:1px solid #5e6d6f}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu input.collapse-toggle{display:none}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.collapsed{display:none}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu input:checked~ul.collapsed{display:block}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem{display:flex}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem .docs-label{flex-grow:2}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem .docs-chevron{display:inline-block;font-style:normal;font-variant:normal;text-rendering:auto;line-height:1;font-size:.75rem;margin-left:1rem;margin-top:auto;margin-bottom:auto}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem .docs-chevron::before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f054"}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu input:checked~label.tocitem .docs-chevron::before{content:"\f078"}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu .tocitem{display:block;padding:0.5rem 0.5rem}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu .tocitem,html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu .tocitem:hover{color:#fff;background:#282f2f}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu a.tocitem:hover,html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu label.tocitem:hover{color:#fff;background-color:#32393a}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu li.is-active{border-top:1px solid #5e6d6f;border-bottom:1px solid #5e6d6f;background-color:#1f2424}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu li.is-active .tocitem,html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu li.is-active .tocitem:hover{background-color:#1f2424;color:#fff}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu li.is-active ul.internal .tocitem:hover{background-color:#32393a;color:#fff}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu>li.is-active:first-child{border-top:none}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.internal{margin:0 0.5rem 0.5rem;border-top:1px solid #5e6d6f}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.internal li{font-size:.85rem;border-left:none;margin-left:0;margin-top:0.5rem}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.internal .tocitem{width:100%;padding:0}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu ul.internal .tocitem::before{content:"⚬";margin-right:0.4em}html.theme--documenter-dark #documenter .docs-sidebar form.docs-search{margin:auto;margin-top:0.5rem;margin-bottom:0.5rem}html.theme--documenter-dark #documenter .docs-sidebar form.docs-search>input{width:14.4rem}html.theme--documenter-dark #documenter .docs-sidebar #documenter-search-query{color:#868c98;width:14.4rem;box-shadow:inset 0 1px 2px rgba(10,10,10,0.1)}@media screen and (min-width: 1056px){html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu{overflow-y:auto;-webkit-overflow-scroll:touch}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar{width:.3rem;background:none}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar-thumb{border-radius:5px 0px 0px 5px;background:#3b4445}html.theme--documenter-dark #documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar-thumb:hover{background:#4e5a5c}}@media screen and (max-width: 1055px){html.theme--documenter-dark #documenter .docs-sidebar{overflow-y:auto;-webkit-overflow-scroll:touch}html.theme--documenter-dark #documenter .docs-sidebar::-webkit-scrollbar{width:.3rem;background:none}html.theme--documenter-dark #documenter .docs-sidebar::-webkit-scrollbar-thumb{border-radius:5px 0px 0px 5px;background:#3b4445}html.theme--documenter-dark #documenter .docs-sidebar::-webkit-scrollbar-thumb:hover{background:#4e5a5c}}html.theme--documenter-dark kbd.search-modal-key-hints{border-radius:0.25rem;border:1px solid rgba(245,245,245,0.6);box-shadow:0 2px 0 1px rgba(245,245,245,0.6);cursor:default;font-size:0.9rem;line-height:1.5;min-width:0.75rem;text-align:center;padding:0.1rem 0.3rem;position:relative;top:-1px}html.theme--documenter-dark .search-min-width-50{min-width:50%}html.theme--documenter-dark .search-min-height-100{min-height:100%}html.theme--documenter-dark .search-modal-card-body{max-height:calc(100vh - 15rem)}html.theme--documenter-dark .search-result-link{border-radius:0.7em;transition:all 300ms}html.theme--documenter-dark .search-result-link:hover,html.theme--documenter-dark .search-result-link:focus{background-color:rgba(0,128,128,0.1)}html.theme--documenter-dark .search-result-link .property-search-result-badge,html.theme--documenter-dark .search-result-link .search-filter{transition:all 300ms}html.theme--documenter-dark .property-search-result-badge,html.theme--documenter-dark .search-filter{padding:0.15em 0.5em;font-size:0.8em;font-style:italic;text-transform:none !important;line-height:1.5;color:#f5f5f5;background-color:rgba(51,65,85,0.501961);border-radius:0.6rem}html.theme--documenter-dark .search-result-link:hover .property-search-result-badge,html.theme--documenter-dark .search-result-link:hover .search-filter,html.theme--documenter-dark .search-result-link:focus .property-search-result-badge,html.theme--documenter-dark .search-result-link:focus .search-filter{color:#333;background-color:#f1f5f9}html.theme--documenter-dark .search-filter{color:#333;background-color:#f5f5f5;transition:all 300ms}html.theme--documenter-dark .search-filter:hover,html.theme--documenter-dark .search-filter:focus{color:#333}html.theme--documenter-dark .search-filter-selected{color:#f5f5f5;background-color:rgba(139,0,139,0.5)}html.theme--documenter-dark .search-filter-selected:hover,html.theme--documenter-dark .search-filter-selected:focus{color:#f5f5f5}html.theme--documenter-dark .search-result-highlight{background-color:#ffdd57;color:black}html.theme--documenter-dark .search-divider{border-bottom:1px solid #5e6d6f}html.theme--documenter-dark .search-result-title{width:85%;color:#f5f5f5}html.theme--documenter-dark .search-result-code-title{font-size:0.875rem;font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace}html.theme--documenter-dark #search-modal .modal-card-body::-webkit-scrollbar,html.theme--documenter-dark #search-modal .filter-tabs::-webkit-scrollbar{height:10px;width:10px;background-color:transparent}html.theme--documenter-dark #search-modal .modal-card-body::-webkit-scrollbar-thumb,html.theme--documenter-dark #search-modal .filter-tabs::-webkit-scrollbar-thumb{background-color:gray;border-radius:1rem}html.theme--documenter-dark #search-modal .modal-card-body::-webkit-scrollbar-track,html.theme--documenter-dark #search-modal .filter-tabs::-webkit-scrollbar-track{-webkit-box-shadow:inset 0 0 6px rgba(0,0,0,0.6);background-color:transparent}html.theme--documenter-dark .w-100{width:100%}html.theme--documenter-dark .gap-2{gap:0.5rem}html.theme--documenter-dark .gap-4{gap:1rem}html.theme--documenter-dark .gap-8{gap:2rem}html.theme--documenter-dark{background-color:#1f2424;font-size:16px;min-width:300px;overflow-x:auto;overflow-y:scroll;text-rendering:optimizeLegibility;text-size-adjust:100%}html.theme--documenter-dark .ansi span.sgr1{font-weight:bolder}html.theme--documenter-dark .ansi span.sgr2{font-weight:lighter}html.theme--documenter-dark .ansi span.sgr3{font-style:italic}html.theme--documenter-dark .ansi span.sgr4{text-decoration:underline}html.theme--documenter-dark .ansi span.sgr7{color:#1f2424;background-color:#fff}html.theme--documenter-dark .ansi span.sgr8{color:transparent}html.theme--documenter-dark .ansi span.sgr8 span{color:transparent}html.theme--documenter-dark .ansi span.sgr9{text-decoration:line-through}html.theme--documenter-dark .ansi span.sgr30{color:#242424}html.theme--documenter-dark .ansi span.sgr31{color:#f6705f}html.theme--documenter-dark .ansi span.sgr32{color:#4fb43a}html.theme--documenter-dark .ansi span.sgr33{color:#f4c72f}html.theme--documenter-dark .ansi span.sgr34{color:#7587f0}html.theme--documenter-dark .ansi span.sgr35{color:#bc89d3}html.theme--documenter-dark .ansi span.sgr36{color:#49b6ca}html.theme--documenter-dark .ansi span.sgr37{color:#b3bdbe}html.theme--documenter-dark .ansi span.sgr40{background-color:#242424}html.theme--documenter-dark .ansi span.sgr41{background-color:#f6705f}html.theme--documenter-dark .ansi span.sgr42{background-color:#4fb43a}html.theme--documenter-dark .ansi span.sgr43{background-color:#f4c72f}html.theme--documenter-dark .ansi span.sgr44{background-color:#7587f0}html.theme--documenter-dark .ansi span.sgr45{background-color:#bc89d3}html.theme--documenter-dark .ansi span.sgr46{background-color:#49b6ca}html.theme--documenter-dark .ansi span.sgr47{background-color:#b3bdbe}html.theme--documenter-dark .ansi span.sgr90{color:#92a0a2}html.theme--documenter-dark .ansi span.sgr91{color:#ff8674}html.theme--documenter-dark .ansi span.sgr92{color:#79d462}html.theme--documenter-dark .ansi span.sgr93{color:#ffe76b}html.theme--documenter-dark .ansi span.sgr94{color:#8a98ff}html.theme--documenter-dark .ansi span.sgr95{color:#d2a4e6}html.theme--documenter-dark .ansi span.sgr96{color:#6bc8db}html.theme--documenter-dark .ansi span.sgr97{color:#ecf0f1}html.theme--documenter-dark .ansi span.sgr100{background-color:#92a0a2}html.theme--documenter-dark .ansi span.sgr101{background-color:#ff8674}html.theme--documenter-dark .ansi span.sgr102{background-color:#79d462}html.theme--documenter-dark .ansi span.sgr103{background-color:#ffe76b}html.theme--documenter-dark .ansi span.sgr104{background-color:#8a98ff}html.theme--documenter-dark .ansi span.sgr105{background-color:#d2a4e6}html.theme--documenter-dark .ansi span.sgr106{background-color:#6bc8db}html.theme--documenter-dark .ansi span.sgr107{background-color:#ecf0f1}html.theme--documenter-dark code.language-julia-repl>span.hljs-meta{color:#4fb43a;font-weight:bolder}html.theme--documenter-dark .hljs{background:#2b2b2b;color:#f8f8f2}html.theme--documenter-dark .hljs-comment,html.theme--documenter-dark .hljs-quote{color:#d4d0ab}html.theme--documenter-dark .hljs-variable,html.theme--documenter-dark .hljs-template-variable,html.theme--documenter-dark .hljs-tag,html.theme--documenter-dark .hljs-name,html.theme--documenter-dark .hljs-selector-id,html.theme--documenter-dark .hljs-selector-class,html.theme--documenter-dark .hljs-regexp,html.theme--documenter-dark .hljs-deletion{color:#ffa07a}html.theme--documenter-dark .hljs-number,html.theme--documenter-dark .hljs-built_in,html.theme--documenter-dark .hljs-literal,html.theme--documenter-dark .hljs-type,html.theme--documenter-dark .hljs-params,html.theme--documenter-dark .hljs-meta,html.theme--documenter-dark .hljs-link{color:#f5ab35}html.theme--documenter-dark .hljs-attribute{color:#ffd700}html.theme--documenter-dark .hljs-string,html.theme--documenter-dark .hljs-symbol,html.theme--documenter-dark .hljs-bullet,html.theme--documenter-dark .hljs-addition{color:#abe338}html.theme--documenter-dark .hljs-title,html.theme--documenter-dark .hljs-section{color:#00e0e0}html.theme--documenter-dark .hljs-keyword,html.theme--documenter-dark .hljs-selector-tag{color:#dcc6e0}html.theme--documenter-dark .hljs-emphasis{font-style:italic}html.theme--documenter-dark .hljs-strong{font-weight:bold}@media screen and (-ms-high-contrast: active){html.theme--documenter-dark .hljs-addition,html.theme--documenter-dark .hljs-attribute,html.theme--documenter-dark .hljs-built_in,html.theme--documenter-dark .hljs-bullet,html.theme--documenter-dark .hljs-comment,html.theme--documenter-dark .hljs-link,html.theme--documenter-dark .hljs-literal,html.theme--documenter-dark .hljs-meta,html.theme--documenter-dark .hljs-number,html.theme--documenter-dark .hljs-params,html.theme--documenter-dark .hljs-string,html.theme--documenter-dark .hljs-symbol,html.theme--documenter-dark .hljs-type,html.theme--documenter-dark .hljs-quote{color:highlight}html.theme--documenter-dark .hljs-keyword,html.theme--documenter-dark .hljs-selector-tag{font-weight:bold}}html.theme--documenter-dark .hljs-subst{color:#f8f8f2}
diff --git a/previews/PR176/assets/themes/documenter-light.css b/previews/PR176/assets/themes/documenter-light.css
new file mode 100644
index 000000000..60a317a4c
--- /dev/null
+++ b/previews/PR176/assets/themes/documenter-light.css
@@ -0,0 +1,9 @@
+.pagination-previous,.pagination-next,.pagination-link,.pagination-ellipsis,.file-cta,.file-name,.select select,.textarea,.input,#documenter .docs-sidebar form.docs-search>input,.button{-moz-appearance:none;-webkit-appearance:none;align-items:center;border:1px solid transparent;border-radius:4px;box-shadow:none;display:inline-flex;font-size:1rem;height:2.5em;justify-content:flex-start;line-height:1.5;padding-bottom:calc(0.5em - 1px);padding-left:calc(0.75em - 1px);padding-right:calc(0.75em - 1px);padding-top:calc(0.5em - 1px);position:relative;vertical-align:top}.pagination-previous:focus,.pagination-next:focus,.pagination-link:focus,.pagination-ellipsis:focus,.file-cta:focus,.file-name:focus,.select select:focus,.textarea:focus,.input:focus,#documenter .docs-sidebar form.docs-search>input:focus,.button:focus,.is-focused.pagination-previous,.is-focused.pagination-next,.is-focused.pagination-link,.is-focused.pagination-ellipsis,.is-focused.file-cta,.is-focused.file-name,.select select.is-focused,.is-focused.textarea,.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-focused.button,.pagination-previous:active,.pagination-next:active,.pagination-link:active,.pagination-ellipsis:active,.file-cta:active,.file-name:active,.select select:active,.textarea:active,.input:active,#documenter .docs-sidebar form.docs-search>input:active,.button:active,.is-active.pagination-previous,.is-active.pagination-next,.is-active.pagination-link,.is-active.pagination-ellipsis,.is-active.file-cta,.is-active.file-name,.select select.is-active,.is-active.textarea,.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active,.is-active.button{outline:none}.pagination-previous[disabled],.pagination-next[disabled],.pagination-link[disabled],.pagination-ellipsis[disabled],.file-cta[disabled],.file-name[disabled],.select select[disabled],.textarea[disabled],.input[disabled],#documenter .docs-sidebar form.docs-search>input[disabled],.button[disabled],fieldset[disabled] .pagination-previous,fieldset[disabled] .pagination-next,fieldset[disabled] .pagination-link,fieldset[disabled] .pagination-ellipsis,fieldset[disabled] .file-cta,fieldset[disabled] .file-name,fieldset[disabled] .select select,.select fieldset[disabled] select,fieldset[disabled] .textarea,fieldset[disabled] .input,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input,fieldset[disabled] .button{cursor:not-allowed}.tabs,.pagination-previous,.pagination-next,.pagination-link,.pagination-ellipsis,.breadcrumb,.file,.button,.is-unselectable{-webkit-touch-callout:none;-webkit-user-select:none;-moz-user-select:none;-ms-user-select:none;user-select:none}.navbar-link:not(.is-arrowless)::after,.select:not(.is-multiple):not(.is-loading)::after{border:3px solid rgba(0,0,0,0);border-radius:2px;border-right:0;border-top:0;content:" ";display:block;height:0.625em;margin-top:-0.4375em;pointer-events:none;position:absolute;top:50%;transform:rotate(-45deg);transform-origin:center;width:0.625em}.admonition:not(:last-child),.tabs:not(:last-child),.pagination:not(:last-child),.message:not(:last-child),.level:not(:last-child),.breadcrumb:not(:last-child),.block:not(:last-child),.title:not(:last-child),.subtitle:not(:last-child),.table-container:not(:last-child),.table:not(:last-child),.progress:not(:last-child),.notification:not(:last-child),.content:not(:last-child),.box:not(:last-child){margin-bottom:1.5rem}.modal-close,.delete{-webkit-touch-callout:none;-webkit-user-select:none;-moz-user-select:none;-ms-user-select:none;user-select:none;-moz-appearance:none;-webkit-appearance:none;background-color:rgba(10,10,10,0.2);border:none;border-radius:9999px;cursor:pointer;pointer-events:auto;display:inline-block;flex-grow:0;flex-shrink:0;font-size:0;height:20px;max-height:20px;max-width:20px;min-height:20px;min-width:20px;outline:none;position:relative;vertical-align:top;width:20px}.modal-close::before,.delete::before,.modal-close::after,.delete::after{background-color:#fff;content:"";display:block;left:50%;position:absolute;top:50%;transform:translateX(-50%) translateY(-50%) rotate(45deg);transform-origin:center center}.modal-close::before,.delete::before{height:2px;width:50%}.modal-close::after,.delete::after{height:50%;width:2px}.modal-close:hover,.delete:hover,.modal-close:focus,.delete:focus{background-color:rgba(10,10,10,0.3)}.modal-close:active,.delete:active{background-color:rgba(10,10,10,0.4)}.is-small.modal-close,#documenter .docs-sidebar form.docs-search>input.modal-close,.is-small.delete,#documenter .docs-sidebar form.docs-search>input.delete{height:16px;max-height:16px;max-width:16px;min-height:16px;min-width:16px;width:16px}.is-medium.modal-close,.is-medium.delete{height:24px;max-height:24px;max-width:24px;min-height:24px;min-width:24px;width:24px}.is-large.modal-close,.is-large.delete{height:32px;max-height:32px;max-width:32px;min-height:32px;min-width:32px;width:32px}.control.is-loading::after,.select.is-loading::after,.loader,.button.is-loading::after{animation:spinAround 500ms infinite linear;border:2px solid #dbdbdb;border-radius:9999px;border-right-color:transparent;border-top-color:transparent;content:"";display:block;height:1em;position:relative;width:1em}.hero-video,.modal-background,.modal,.image.is-square img,#documenter .docs-sidebar .docs-logo>img.is-square img,.image.is-square .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-square .has-ratio,.image.is-1by1 img,#documenter .docs-sidebar .docs-logo>img.is-1by1 img,.image.is-1by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by1 .has-ratio,.image.is-5by4 img,#documenter .docs-sidebar .docs-logo>img.is-5by4 img,.image.is-5by4 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-5by4 .has-ratio,.image.is-4by3 img,#documenter .docs-sidebar .docs-logo>img.is-4by3 img,.image.is-4by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-4by3 .has-ratio,.image.is-3by2 img,#documenter .docs-sidebar .docs-logo>img.is-3by2 img,.image.is-3by2 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by2 .has-ratio,.image.is-5by3 img,#documenter .docs-sidebar .docs-logo>img.is-5by3 img,.image.is-5by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-5by3 .has-ratio,.image.is-16by9 img,#documenter .docs-sidebar .docs-logo>img.is-16by9 img,.image.is-16by9 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-16by9 .has-ratio,.image.is-2by1 img,#documenter .docs-sidebar .docs-logo>img.is-2by1 img,.image.is-2by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-2by1 .has-ratio,.image.is-3by1 img,#documenter .docs-sidebar .docs-logo>img.is-3by1 img,.image.is-3by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by1 .has-ratio,.image.is-4by5 img,#documenter .docs-sidebar .docs-logo>img.is-4by5 img,.image.is-4by5 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-4by5 .has-ratio,.image.is-3by4 img,#documenter .docs-sidebar .docs-logo>img.is-3by4 img,.image.is-3by4 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by4 .has-ratio,.image.is-2by3 img,#documenter .docs-sidebar .docs-logo>img.is-2by3 img,.image.is-2by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-2by3 .has-ratio,.image.is-3by5 img,#documenter .docs-sidebar .docs-logo>img.is-3by5 img,.image.is-3by5 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by5 .has-ratio,.image.is-9by16 img,#documenter .docs-sidebar .docs-logo>img.is-9by16 img,.image.is-9by16 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-9by16 .has-ratio,.image.is-1by2 img,#documenter .docs-sidebar .docs-logo>img.is-1by2 img,.image.is-1by2 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by2 .has-ratio,.image.is-1by3 img,#documenter .docs-sidebar .docs-logo>img.is-1by3 img,.image.is-1by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by3 .has-ratio,.is-overlay{bottom:0;left:0;position:absolute;right:0;top:0}.navbar-burger{-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;color:currentColor;font-family:inherit;font-size:1em;margin:0;padding:0}.has-text-white{color:#fff !important}a.has-text-white:hover,a.has-text-white:focus{color:#e6e6e6 !important}.has-background-white{background-color:#fff !important}.has-text-black{color:#0a0a0a !important}a.has-text-black:hover,a.has-text-black:focus{color:#000 !important}.has-background-black{background-color:#0a0a0a !important}.has-text-light{color:#f5f5f5 !important}a.has-text-light:hover,a.has-text-light:focus{color:#dbdbdb !important}.has-background-light{background-color:#f5f5f5 !important}.has-text-dark{color:#363636 !important}a.has-text-dark:hover,a.has-text-dark:focus{color:#1c1c1c !important}.has-background-dark{background-color:#363636 !important}.has-text-primary{color:#4eb5de !important}a.has-text-primary:hover,a.has-text-primary:focus{color:#27a1d2 !important}.has-background-primary{background-color:#4eb5de !important}.has-text-primary-light{color:#eef8fc !important}a.has-text-primary-light:hover,a.has-text-primary-light:focus{color:#c3e6f4 !important}.has-background-primary-light{background-color:#eef8fc !important}.has-text-primary-dark{color:#1a6d8e !important}a.has-text-primary-dark:hover,a.has-text-primary-dark:focus{color:#228eb9 !important}.has-background-primary-dark{background-color:#1a6d8e !important}.has-text-link{color:#2e63b8 !important}a.has-text-link:hover,a.has-text-link:focus{color:#244d8f !important}.has-background-link{background-color:#2e63b8 !important}.has-text-link-light{color:#eff3fb !important}a.has-text-link-light:hover,a.has-text-link-light:focus{color:#c6d6f1 !important}.has-background-link-light{background-color:#eff3fb !important}.has-text-link-dark{color:#3169c4 !important}a.has-text-link-dark:hover,a.has-text-link-dark:focus{color:#5485d4 !important}.has-background-link-dark{background-color:#3169c4 !important}.has-text-info{color:#209cee !important}a.has-text-info:hover,a.has-text-info:focus{color:#1081cb !important}.has-background-info{background-color:#209cee !important}.has-text-info-light{color:#ecf7fe !important}a.has-text-info-light:hover,a.has-text-info-light:focus{color:#bde2fa !important}.has-background-info-light{background-color:#ecf7fe !important}.has-text-info-dark{color:#0e72b4 !important}a.has-text-info-dark:hover,a.has-text-info-dark:focus{color:#1190e3 !important}.has-background-info-dark{background-color:#0e72b4 !important}.has-text-success{color:#22c35b !important}a.has-text-success:hover,a.has-text-success:focus{color:#1a9847 !important}.has-background-success{background-color:#22c35b !important}.has-text-success-light{color:#eefcf3 !important}a.has-text-success-light:hover,a.has-text-success-light:focus{color:#c2f4d4 !important}.has-background-success-light{background-color:#eefcf3 !important}.has-text-success-dark{color:#198f43 !important}a.has-text-success-dark:hover,a.has-text-success-dark:focus{color:#21bb57 !important}.has-background-success-dark{background-color:#198f43 !important}.has-text-warning{color:#ffdd57 !important}a.has-text-warning:hover,a.has-text-warning:focus{color:#ffd324 !important}.has-background-warning{background-color:#ffdd57 !important}.has-text-warning-light{color:#fffbeb !important}a.has-text-warning-light:hover,a.has-text-warning-light:focus{color:#fff1b8 !important}.has-background-warning-light{background-color:#fffbeb !important}.has-text-warning-dark{color:#947600 !important}a.has-text-warning-dark:hover,a.has-text-warning-dark:focus{color:#c79f00 !important}.has-background-warning-dark{background-color:#947600 !important}.has-text-danger{color:#da0b00 !important}a.has-text-danger:hover,a.has-text-danger:focus{color:#a70800 !important}.has-background-danger{background-color:#da0b00 !important}.has-text-danger-light{color:#ffeceb !important}a.has-text-danger-light:hover,a.has-text-danger-light:focus{color:#ffbbb8 !important}.has-background-danger-light{background-color:#ffeceb !important}.has-text-danger-dark{color:#f50c00 !important}a.has-text-danger-dark:hover,a.has-text-danger-dark:focus{color:#ff3429 !important}.has-background-danger-dark{background-color:#f50c00 !important}.has-text-black-bis{color:#121212 !important}.has-background-black-bis{background-color:#121212 !important}.has-text-black-ter{color:#242424 !important}.has-background-black-ter{background-color:#242424 !important}.has-text-grey-darker{color:#363636 !important}.has-background-grey-darker{background-color:#363636 !important}.has-text-grey-dark{color:#4a4a4a !important}.has-background-grey-dark{background-color:#4a4a4a !important}.has-text-grey{color:#6b6b6b !important}.has-background-grey{background-color:#6b6b6b !important}.has-text-grey-light{color:#b5b5b5 !important}.has-background-grey-light{background-color:#b5b5b5 !important}.has-text-grey-lighter{color:#dbdbdb !important}.has-background-grey-lighter{background-color:#dbdbdb !important}.has-text-white-ter{color:#f5f5f5 !important}.has-background-white-ter{background-color:#f5f5f5 !important}.has-text-white-bis{color:#fafafa !important}.has-background-white-bis{background-color:#fafafa !important}.is-flex-direction-row{flex-direction:row !important}.is-flex-direction-row-reverse{flex-direction:row-reverse !important}.is-flex-direction-column{flex-direction:column !important}.is-flex-direction-column-reverse{flex-direction:column-reverse !important}.is-flex-wrap-nowrap{flex-wrap:nowrap !important}.is-flex-wrap-wrap{flex-wrap:wrap !important}.is-flex-wrap-wrap-reverse{flex-wrap:wrap-reverse !important}.is-justify-content-flex-start{justify-content:flex-start !important}.is-justify-content-flex-end{justify-content:flex-end !important}.is-justify-content-center{justify-content:center !important}.is-justify-content-space-between{justify-content:space-between !important}.is-justify-content-space-around{justify-content:space-around !important}.is-justify-content-space-evenly{justify-content:space-evenly !important}.is-justify-content-start{justify-content:start !important}.is-justify-content-end{justify-content:end !important}.is-justify-content-left{justify-content:left !important}.is-justify-content-right{justify-content:right !important}.is-align-content-flex-start{align-content:flex-start !important}.is-align-content-flex-end{align-content:flex-end !important}.is-align-content-center{align-content:center !important}.is-align-content-space-between{align-content:space-between !important}.is-align-content-space-around{align-content:space-around !important}.is-align-content-space-evenly{align-content:space-evenly !important}.is-align-content-stretch{align-content:stretch !important}.is-align-content-start{align-content:start !important}.is-align-content-end{align-content:end !important}.is-align-content-baseline{align-content:baseline !important}.is-align-items-stretch{align-items:stretch !important}.is-align-items-flex-start{align-items:flex-start !important}.is-align-items-flex-end{align-items:flex-end !important}.is-align-items-center{align-items:center !important}.is-align-items-baseline{align-items:baseline !important}.is-align-items-start{align-items:start !important}.is-align-items-end{align-items:end !important}.is-align-items-self-start{align-items:self-start !important}.is-align-items-self-end{align-items:self-end !important}.is-align-self-auto{align-self:auto !important}.is-align-self-flex-start{align-self:flex-start !important}.is-align-self-flex-end{align-self:flex-end !important}.is-align-self-center{align-self:center !important}.is-align-self-baseline{align-self:baseline !important}.is-align-self-stretch{align-self:stretch !important}.is-flex-grow-0{flex-grow:0 !important}.is-flex-grow-1{flex-grow:1 !important}.is-flex-grow-2{flex-grow:2 !important}.is-flex-grow-3{flex-grow:3 !important}.is-flex-grow-4{flex-grow:4 !important}.is-flex-grow-5{flex-grow:5 !important}.is-flex-shrink-0{flex-shrink:0 !important}.is-flex-shrink-1{flex-shrink:1 !important}.is-flex-shrink-2{flex-shrink:2 !important}.is-flex-shrink-3{flex-shrink:3 !important}.is-flex-shrink-4{flex-shrink:4 !important}.is-flex-shrink-5{flex-shrink:5 !important}.is-clearfix::after{clear:both;content:" ";display:table}.is-pulled-left{float:left !important}.is-pulled-right{float:right !important}.is-radiusless{border-radius:0 !important}.is-shadowless{box-shadow:none !important}.is-clickable{cursor:pointer !important;pointer-events:all !important}.is-clipped{overflow:hidden !important}.is-relative{position:relative !important}.is-marginless{margin:0 !important}.is-paddingless{padding:0 !important}.m-0{margin:0 !important}.mt-0{margin-top:0 !important}.mr-0{margin-right:0 !important}.mb-0{margin-bottom:0 !important}.ml-0{margin-left:0 !important}.mx-0{margin-left:0 !important;margin-right:0 !important}.my-0{margin-top:0 !important;margin-bottom:0 !important}.m-1{margin:.25rem !important}.mt-1{margin-top:.25rem !important}.mr-1{margin-right:.25rem !important}.mb-1{margin-bottom:.25rem !important}.ml-1{margin-left:.25rem !important}.mx-1{margin-left:.25rem !important;margin-right:.25rem !important}.my-1{margin-top:.25rem !important;margin-bottom:.25rem !important}.m-2{margin:.5rem !important}.mt-2{margin-top:.5rem !important}.mr-2{margin-right:.5rem !important}.mb-2{margin-bottom:.5rem !important}.ml-2{margin-left:.5rem !important}.mx-2{margin-left:.5rem !important;margin-right:.5rem !important}.my-2{margin-top:.5rem !important;margin-bottom:.5rem !important}.m-3{margin:.75rem !important}.mt-3{margin-top:.75rem !important}.mr-3{margin-right:.75rem !important}.mb-3{margin-bottom:.75rem !important}.ml-3{margin-left:.75rem !important}.mx-3{margin-left:.75rem !important;margin-right:.75rem !important}.my-3{margin-top:.75rem !important;margin-bottom:.75rem !important}.m-4{margin:1rem !important}.mt-4{margin-top:1rem !important}.mr-4{margin-right:1rem !important}.mb-4{margin-bottom:1rem !important}.ml-4{margin-left:1rem !important}.mx-4{margin-left:1rem !important;margin-right:1rem !important}.my-4{margin-top:1rem !important;margin-bottom:1rem !important}.m-5{margin:1.5rem !important}.mt-5{margin-top:1.5rem !important}.mr-5{margin-right:1.5rem !important}.mb-5{margin-bottom:1.5rem !important}.ml-5{margin-left:1.5rem !important}.mx-5{margin-left:1.5rem !important;margin-right:1.5rem !important}.my-5{margin-top:1.5rem !important;margin-bottom:1.5rem !important}.m-6{margin:3rem !important}.mt-6{margin-top:3rem !important}.mr-6{margin-right:3rem !important}.mb-6{margin-bottom:3rem !important}.ml-6{margin-left:3rem !important}.mx-6{margin-left:3rem !important;margin-right:3rem !important}.my-6{margin-top:3rem !important;margin-bottom:3rem !important}.m-auto{margin:auto !important}.mt-auto{margin-top:auto !important}.mr-auto{margin-right:auto !important}.mb-auto{margin-bottom:auto !important}.ml-auto{margin-left:auto !important}.mx-auto{margin-left:auto !important;margin-right:auto !important}.my-auto{margin-top:auto !important;margin-bottom:auto !important}.p-0{padding:0 !important}.pt-0{padding-top:0 !important}.pr-0{padding-right:0 !important}.pb-0{padding-bottom:0 !important}.pl-0{padding-left:0 !important}.px-0{padding-left:0 !important;padding-right:0 !important}.py-0{padding-top:0 !important;padding-bottom:0 !important}.p-1{padding:.25rem !important}.pt-1{padding-top:.25rem !important}.pr-1{padding-right:.25rem !important}.pb-1{padding-bottom:.25rem !important}.pl-1{padding-left:.25rem !important}.px-1{padding-left:.25rem !important;padding-right:.25rem !important}.py-1{padding-top:.25rem !important;padding-bottom:.25rem !important}.p-2{padding:.5rem !important}.pt-2{padding-top:.5rem !important}.pr-2{padding-right:.5rem !important}.pb-2{padding-bottom:.5rem !important}.pl-2{padding-left:.5rem !important}.px-2{padding-left:.5rem !important;padding-right:.5rem !important}.py-2{padding-top:.5rem !important;padding-bottom:.5rem !important}.p-3{padding:.75rem !important}.pt-3{padding-top:.75rem !important}.pr-3{padding-right:.75rem !important}.pb-3{padding-bottom:.75rem !important}.pl-3{padding-left:.75rem !important}.px-3{padding-left:.75rem !important;padding-right:.75rem !important}.py-3{padding-top:.75rem !important;padding-bottom:.75rem !important}.p-4{padding:1rem !important}.pt-4{padding-top:1rem !important}.pr-4{padding-right:1rem !important}.pb-4{padding-bottom:1rem !important}.pl-4{padding-left:1rem !important}.px-4{padding-left:1rem !important;padding-right:1rem !important}.py-4{padding-top:1rem !important;padding-bottom:1rem !important}.p-5{padding:1.5rem !important}.pt-5{padding-top:1.5rem !important}.pr-5{padding-right:1.5rem !important}.pb-5{padding-bottom:1.5rem !important}.pl-5{padding-left:1.5rem !important}.px-5{padding-left:1.5rem !important;padding-right:1.5rem !important}.py-5{padding-top:1.5rem !important;padding-bottom:1.5rem !important}.p-6{padding:3rem !important}.pt-6{padding-top:3rem !important}.pr-6{padding-right:3rem !important}.pb-6{padding-bottom:3rem !important}.pl-6{padding-left:3rem !important}.px-6{padding-left:3rem !important;padding-right:3rem !important}.py-6{padding-top:3rem !important;padding-bottom:3rem !important}.p-auto{padding:auto !important}.pt-auto{padding-top:auto !important}.pr-auto{padding-right:auto !important}.pb-auto{padding-bottom:auto !important}.pl-auto{padding-left:auto !important}.px-auto{padding-left:auto !important;padding-right:auto !important}.py-auto{padding-top:auto !important;padding-bottom:auto !important}.is-size-1{font-size:3rem !important}.is-size-2{font-size:2.5rem !important}.is-size-3{font-size:2rem !important}.is-size-4{font-size:1.5rem !important}.is-size-5{font-size:1.25rem !important}.is-size-6{font-size:1rem !important}.is-size-7,.docstring>section>a.docs-sourcelink{font-size:.75rem !important}@media screen and (max-width: 768px){.is-size-1-mobile{font-size:3rem !important}.is-size-2-mobile{font-size:2.5rem !important}.is-size-3-mobile{font-size:2rem !important}.is-size-4-mobile{font-size:1.5rem !important}.is-size-5-mobile{font-size:1.25rem !important}.is-size-6-mobile{font-size:1rem !important}.is-size-7-mobile{font-size:.75rem !important}}@media screen and (min-width: 769px),print{.is-size-1-tablet{font-size:3rem !important}.is-size-2-tablet{font-size:2.5rem !important}.is-size-3-tablet{font-size:2rem !important}.is-size-4-tablet{font-size:1.5rem !important}.is-size-5-tablet{font-size:1.25rem !important}.is-size-6-tablet{font-size:1rem !important}.is-size-7-tablet{font-size:.75rem !important}}@media screen and (max-width: 1055px){.is-size-1-touch{font-size:3rem !important}.is-size-2-touch{font-size:2.5rem !important}.is-size-3-touch{font-size:2rem !important}.is-size-4-touch{font-size:1.5rem !important}.is-size-5-touch{font-size:1.25rem !important}.is-size-6-touch{font-size:1rem !important}.is-size-7-touch{font-size:.75rem !important}}@media screen and (min-width: 1056px){.is-size-1-desktop{font-size:3rem !important}.is-size-2-desktop{font-size:2.5rem !important}.is-size-3-desktop{font-size:2rem !important}.is-size-4-desktop{font-size:1.5rem !important}.is-size-5-desktop{font-size:1.25rem !important}.is-size-6-desktop{font-size:1rem !important}.is-size-7-desktop{font-size:.75rem !important}}@media screen and (min-width: 1216px){.is-size-1-widescreen{font-size:3rem !important}.is-size-2-widescreen{font-size:2.5rem !important}.is-size-3-widescreen{font-size:2rem !important}.is-size-4-widescreen{font-size:1.5rem !important}.is-size-5-widescreen{font-size:1.25rem !important}.is-size-6-widescreen{font-size:1rem !important}.is-size-7-widescreen{font-size:.75rem !important}}@media screen and (min-width: 1408px){.is-size-1-fullhd{font-size:3rem !important}.is-size-2-fullhd{font-size:2.5rem !important}.is-size-3-fullhd{font-size:2rem !important}.is-size-4-fullhd{font-size:1.5rem !important}.is-size-5-fullhd{font-size:1.25rem !important}.is-size-6-fullhd{font-size:1rem !important}.is-size-7-fullhd{font-size:.75rem !important}}.has-text-centered{text-align:center !important}.has-text-justified{text-align:justify !important}.has-text-left{text-align:left !important}.has-text-right{text-align:right !important}@media screen and (max-width: 768px){.has-text-centered-mobile{text-align:center !important}}@media screen and (min-width: 769px),print{.has-text-centered-tablet{text-align:center !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-centered-tablet-only{text-align:center !important}}@media screen and (max-width: 1055px){.has-text-centered-touch{text-align:center !important}}@media screen and (min-width: 1056px){.has-text-centered-desktop{text-align:center !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-centered-desktop-only{text-align:center !important}}@media screen and (min-width: 1216px){.has-text-centered-widescreen{text-align:center !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-centered-widescreen-only{text-align:center !important}}@media screen and (min-width: 1408px){.has-text-centered-fullhd{text-align:center !important}}@media screen and (max-width: 768px){.has-text-justified-mobile{text-align:justify !important}}@media screen and (min-width: 769px),print{.has-text-justified-tablet{text-align:justify !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-justified-tablet-only{text-align:justify !important}}@media screen and (max-width: 1055px){.has-text-justified-touch{text-align:justify !important}}@media screen and (min-width: 1056px){.has-text-justified-desktop{text-align:justify !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-justified-desktop-only{text-align:justify !important}}@media screen and (min-width: 1216px){.has-text-justified-widescreen{text-align:justify !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-justified-widescreen-only{text-align:justify !important}}@media screen and (min-width: 1408px){.has-text-justified-fullhd{text-align:justify !important}}@media screen and (max-width: 768px){.has-text-left-mobile{text-align:left !important}}@media screen and (min-width: 769px),print{.has-text-left-tablet{text-align:left !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-left-tablet-only{text-align:left !important}}@media screen and (max-width: 1055px){.has-text-left-touch{text-align:left !important}}@media screen and (min-width: 1056px){.has-text-left-desktop{text-align:left !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-left-desktop-only{text-align:left !important}}@media screen and (min-width: 1216px){.has-text-left-widescreen{text-align:left !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-left-widescreen-only{text-align:left !important}}@media screen and (min-width: 1408px){.has-text-left-fullhd{text-align:left !important}}@media screen and (max-width: 768px){.has-text-right-mobile{text-align:right !important}}@media screen and (min-width: 769px),print{.has-text-right-tablet{text-align:right !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.has-text-right-tablet-only{text-align:right !important}}@media screen and (max-width: 1055px){.has-text-right-touch{text-align:right !important}}@media screen and (min-width: 1056px){.has-text-right-desktop{text-align:right !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.has-text-right-desktop-only{text-align:right !important}}@media screen and (min-width: 1216px){.has-text-right-widescreen{text-align:right !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.has-text-right-widescreen-only{text-align:right !important}}@media screen and (min-width: 1408px){.has-text-right-fullhd{text-align:right !important}}.is-capitalized{text-transform:capitalize !important}.is-lowercase{text-transform:lowercase !important}.is-uppercase{text-transform:uppercase !important}.is-italic{font-style:italic !important}.is-underlined{text-decoration:underline !important}.has-text-weight-light{font-weight:300 !important}.has-text-weight-normal{font-weight:400 !important}.has-text-weight-medium{font-weight:500 !important}.has-text-weight-semibold{font-weight:600 !important}.has-text-weight-bold{font-weight:700 !important}.is-family-primary{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-secondary{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-sans-serif{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif !important}.is-family-monospace{font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace !important}.is-family-code{font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace !important}.is-block{display:block !important}@media screen and (max-width: 768px){.is-block-mobile{display:block !important}}@media screen and (min-width: 769px),print{.is-block-tablet{display:block !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-block-tablet-only{display:block !important}}@media screen and (max-width: 1055px){.is-block-touch{display:block !important}}@media screen and (min-width: 1056px){.is-block-desktop{display:block !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-block-desktop-only{display:block !important}}@media screen and (min-width: 1216px){.is-block-widescreen{display:block !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-block-widescreen-only{display:block !important}}@media screen and (min-width: 1408px){.is-block-fullhd{display:block !important}}.is-flex{display:flex !important}@media screen and (max-width: 768px){.is-flex-mobile{display:flex !important}}@media screen and (min-width: 769px),print{.is-flex-tablet{display:flex !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-flex-tablet-only{display:flex !important}}@media screen and (max-width: 1055px){.is-flex-touch{display:flex !important}}@media screen and (min-width: 1056px){.is-flex-desktop{display:flex !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-flex-desktop-only{display:flex !important}}@media screen and (min-width: 1216px){.is-flex-widescreen{display:flex !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-flex-widescreen-only{display:flex !important}}@media screen and (min-width: 1408px){.is-flex-fullhd{display:flex !important}}.is-inline{display:inline !important}@media screen and (max-width: 768px){.is-inline-mobile{display:inline !important}}@media screen and (min-width: 769px),print{.is-inline-tablet{display:inline !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-tablet-only{display:inline !important}}@media screen and (max-width: 1055px){.is-inline-touch{display:inline !important}}@media screen and (min-width: 1056px){.is-inline-desktop{display:inline !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-desktop-only{display:inline !important}}@media screen and (min-width: 1216px){.is-inline-widescreen{display:inline !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-widescreen-only{display:inline !important}}@media screen and (min-width: 1408px){.is-inline-fullhd{display:inline !important}}.is-inline-block{display:inline-block !important}@media screen and (max-width: 768px){.is-inline-block-mobile{display:inline-block !important}}@media screen and (min-width: 769px),print{.is-inline-block-tablet{display:inline-block !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-block-tablet-only{display:inline-block !important}}@media screen and (max-width: 1055px){.is-inline-block-touch{display:inline-block !important}}@media screen and (min-width: 1056px){.is-inline-block-desktop{display:inline-block !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-block-desktop-only{display:inline-block !important}}@media screen and (min-width: 1216px){.is-inline-block-widescreen{display:inline-block !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-block-widescreen-only{display:inline-block !important}}@media screen and (min-width: 1408px){.is-inline-block-fullhd{display:inline-block !important}}.is-inline-flex{display:inline-flex !important}@media screen and (max-width: 768px){.is-inline-flex-mobile{display:inline-flex !important}}@media screen and (min-width: 769px),print{.is-inline-flex-tablet{display:inline-flex !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-inline-flex-tablet-only{display:inline-flex !important}}@media screen and (max-width: 1055px){.is-inline-flex-touch{display:inline-flex !important}}@media screen and (min-width: 1056px){.is-inline-flex-desktop{display:inline-flex !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-inline-flex-desktop-only{display:inline-flex !important}}@media screen and (min-width: 1216px){.is-inline-flex-widescreen{display:inline-flex !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-inline-flex-widescreen-only{display:inline-flex !important}}@media screen and (min-width: 1408px){.is-inline-flex-fullhd{display:inline-flex !important}}.is-hidden{display:none !important}.is-sr-only{border:none !important;clip:rect(0, 0, 0, 0) !important;height:0.01em !important;overflow:hidden !important;padding:0 !important;position:absolute !important;white-space:nowrap !important;width:0.01em !important}@media screen and (max-width: 768px){.is-hidden-mobile{display:none !important}}@media screen and (min-width: 769px),print{.is-hidden-tablet{display:none !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-hidden-tablet-only{display:none !important}}@media screen and (max-width: 1055px){.is-hidden-touch{display:none !important}}@media screen and (min-width: 1056px){.is-hidden-desktop{display:none !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-hidden-desktop-only{display:none !important}}@media screen and (min-width: 1216px){.is-hidden-widescreen{display:none !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-hidden-widescreen-only{display:none !important}}@media screen and (min-width: 1408px){.is-hidden-fullhd{display:none !important}}.is-invisible{visibility:hidden !important}@media screen and (max-width: 768px){.is-invisible-mobile{visibility:hidden !important}}@media screen and (min-width: 769px),print{.is-invisible-tablet{visibility:hidden !important}}@media screen and (min-width: 769px) and (max-width: 1055px){.is-invisible-tablet-only{visibility:hidden !important}}@media screen and (max-width: 1055px){.is-invisible-touch{visibility:hidden !important}}@media screen and (min-width: 1056px){.is-invisible-desktop{visibility:hidden !important}}@media screen and (min-width: 1056px) and (max-width: 1215px){.is-invisible-desktop-only{visibility:hidden !important}}@media screen and (min-width: 1216px){.is-invisible-widescreen{visibility:hidden !important}}@media screen and (min-width: 1216px) and (max-width: 1407px){.is-invisible-widescreen-only{visibility:hidden !important}}@media screen and (min-width: 1408px){.is-invisible-fullhd{visibility:hidden !important}}/*! minireset.css v0.0.6 | MIT License | github.com/jgthms/minireset.css */html,body,p,ol,ul,li,dl,dt,dd,blockquote,figure,fieldset,legend,textarea,pre,iframe,hr,h1,h2,h3,h4,h5,h6{margin:0;padding:0}h1,h2,h3,h4,h5,h6{font-size:100%;font-weight:normal}ul{list-style:none}button,input,select,textarea{margin:0}html{box-sizing:border-box}*,*::before,*::after{box-sizing:inherit}img,video{height:auto;max-width:100%}iframe{border:0}table{border-collapse:collapse;border-spacing:0}td,th{padding:0}td:not([align]),th:not([align]){text-align:inherit}html{background-color:#fff;font-size:16px;-moz-osx-font-smoothing:grayscale;-webkit-font-smoothing:antialiased;min-width:300px;overflow-x:auto;overflow-y:scroll;text-rendering:optimizeLegibility;text-size-adjust:100%}article,aside,figure,footer,header,hgroup,section{display:block}body,button,input,optgroup,select,textarea{font-family:"Lato Medium",-apple-system,BlinkMacSystemFont,"Segoe UI","Helvetica Neue","Helvetica","Arial",sans-serif}code,pre{-moz-osx-font-smoothing:auto;-webkit-font-smoothing:auto;font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace}body{color:#222;font-size:1em;font-weight:400;line-height:1.5}a{color:#2e63b8;cursor:pointer;text-decoration:none}a strong{color:currentColor}a:hover{color:#363636}code{background-color:rgba(0,0,0,0.05);color:#000;font-size:.875em;font-weight:normal;padding:.1em}hr{background-color:#f5f5f5;border:none;display:block;height:2px;margin:1.5rem 0}img{height:auto;max-width:100%}input[type="checkbox"],input[type="radio"]{vertical-align:baseline}small{font-size:.875em}span{font-style:inherit;font-weight:inherit}strong{color:#222;font-weight:700}fieldset{border:none}pre{-webkit-overflow-scrolling:touch;background-color:#f5f5f5;color:#222;font-size:.875em;overflow-x:auto;padding:1.25rem 1.5rem;white-space:pre;word-wrap:normal}pre code{background-color:transparent;color:currentColor;font-size:1em;padding:0}table td,table th{vertical-align:top}table td:not([align]),table th:not([align]){text-align:inherit}table th{color:#222}@keyframes spinAround{from{transform:rotate(0deg)}to{transform:rotate(359deg)}}.box{background-color:#fff;border-radius:6px;box-shadow:#bbb;color:#222;display:block;padding:1.25rem}a.box:hover,a.box:focus{box-shadow:0 0.5em 1em -0.125em rgba(10,10,10,0.1),0 0 0 1px #2e63b8}a.box:active{box-shadow:inset 0 1px 2px rgba(10,10,10,0.2),0 0 0 1px #2e63b8}.button{background-color:#fff;border-color:#dbdbdb;border-width:1px;color:#222;cursor:pointer;justify-content:center;padding-bottom:calc(0.5em - 1px);padding-left:1em;padding-right:1em;padding-top:calc(0.5em - 1px);text-align:center;white-space:nowrap}.button strong{color:inherit}.button .icon,.button .icon.is-small,.button #documenter .docs-sidebar form.docs-search>input.icon,#documenter .docs-sidebar .button form.docs-search>input.icon,.button .icon.is-medium,.button .icon.is-large{height:1.5em;width:1.5em}.button .icon:first-child:not(:last-child){margin-left:calc(-0.5em - 1px);margin-right:.25em}.button .icon:last-child:not(:first-child){margin-left:.25em;margin-right:calc(-0.5em - 1px)}.button .icon:first-child:last-child{margin-left:calc(-0.5em - 1px);margin-right:calc(-0.5em - 1px)}.button:hover,.button.is-hovered{border-color:#b5b5b5;color:#363636}.button:focus,.button.is-focused{border-color:#3c5dcd;color:#363636}.button:focus:not(:active),.button.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.button:active,.button.is-active{border-color:#4a4a4a;color:#363636}.button.is-text{background-color:transparent;border-color:transparent;color:#222;text-decoration:underline}.button.is-text:hover,.button.is-text.is-hovered,.button.is-text:focus,.button.is-text.is-focused{background-color:#f5f5f5;color:#222}.button.is-text:active,.button.is-text.is-active{background-color:#e8e8e8;color:#222}.button.is-text[disabled],fieldset[disabled] .button.is-text{background-color:transparent;border-color:transparent;box-shadow:none}.button.is-ghost{background:none;border-color:rgba(0,0,0,0);color:#2e63b8;text-decoration:none}.button.is-ghost:hover,.button.is-ghost.is-hovered{color:#2e63b8;text-decoration:underline}.button.is-white{background-color:#fff;border-color:transparent;color:#0a0a0a}.button.is-white:hover,.button.is-white.is-hovered{background-color:#f9f9f9;border-color:transparent;color:#0a0a0a}.button.is-white:focus,.button.is-white.is-focused{border-color:transparent;color:#0a0a0a}.button.is-white:focus:not(:active),.button.is-white.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}.button.is-white:active,.button.is-white.is-active{background-color:#f2f2f2;border-color:transparent;color:#0a0a0a}.button.is-white[disabled],fieldset[disabled] .button.is-white{background-color:#fff;border-color:#fff;box-shadow:none}.button.is-white.is-inverted{background-color:#0a0a0a;color:#fff}.button.is-white.is-inverted:hover,.button.is-white.is-inverted.is-hovered{background-color:#000}.button.is-white.is-inverted[disabled],fieldset[disabled] .button.is-white.is-inverted{background-color:#0a0a0a;border-color:transparent;box-shadow:none;color:#fff}.button.is-white.is-loading::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}.button.is-white.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-white.is-outlined:hover,.button.is-white.is-outlined.is-hovered,.button.is-white.is-outlined:focus,.button.is-white.is-outlined.is-focused{background-color:#fff;border-color:#fff;color:#0a0a0a}.button.is-white.is-outlined.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-white.is-outlined.is-loading:hover::after,.button.is-white.is-outlined.is-loading.is-hovered::after,.button.is-white.is-outlined.is-loading:focus::after,.button.is-white.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}.button.is-white.is-outlined[disabled],fieldset[disabled] .button.is-white.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-white.is-inverted.is-outlined{background-color:transparent;border-color:#0a0a0a;color:#0a0a0a}.button.is-white.is-inverted.is-outlined:hover,.button.is-white.is-inverted.is-outlined.is-hovered,.button.is-white.is-inverted.is-outlined:focus,.button.is-white.is-inverted.is-outlined.is-focused{background-color:#0a0a0a;color:#fff}.button.is-white.is-inverted.is-outlined.is-loading:hover::after,.button.is-white.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-white.is-inverted.is-outlined.is-loading:focus::after,.button.is-white.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-white.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-white.is-inverted.is-outlined{background-color:transparent;border-color:#0a0a0a;box-shadow:none;color:#0a0a0a}.button.is-black{background-color:#0a0a0a;border-color:transparent;color:#fff}.button.is-black:hover,.button.is-black.is-hovered{background-color:#040404;border-color:transparent;color:#fff}.button.is-black:focus,.button.is-black.is-focused{border-color:transparent;color:#fff}.button.is-black:focus:not(:active),.button.is-black.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}.button.is-black:active,.button.is-black.is-active{background-color:#000;border-color:transparent;color:#fff}.button.is-black[disabled],fieldset[disabled] .button.is-black{background-color:#0a0a0a;border-color:#0a0a0a;box-shadow:none}.button.is-black.is-inverted{background-color:#fff;color:#0a0a0a}.button.is-black.is-inverted:hover,.button.is-black.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-black.is-inverted[disabled],fieldset[disabled] .button.is-black.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#0a0a0a}.button.is-black.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-black.is-outlined{background-color:transparent;border-color:#0a0a0a;color:#0a0a0a}.button.is-black.is-outlined:hover,.button.is-black.is-outlined.is-hovered,.button.is-black.is-outlined:focus,.button.is-black.is-outlined.is-focused{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}.button.is-black.is-outlined.is-loading::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}.button.is-black.is-outlined.is-loading:hover::after,.button.is-black.is-outlined.is-loading.is-hovered::after,.button.is-black.is-outlined.is-loading:focus::after,.button.is-black.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-black.is-outlined[disabled],fieldset[disabled] .button.is-black.is-outlined{background-color:transparent;border-color:#0a0a0a;box-shadow:none;color:#0a0a0a}.button.is-black.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-black.is-inverted.is-outlined:hover,.button.is-black.is-inverted.is-outlined.is-hovered,.button.is-black.is-inverted.is-outlined:focus,.button.is-black.is-inverted.is-outlined.is-focused{background-color:#fff;color:#0a0a0a}.button.is-black.is-inverted.is-outlined.is-loading:hover::after,.button.is-black.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-black.is-inverted.is-outlined.is-loading:focus::after,.button.is-black.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #0a0a0a #0a0a0a !important}.button.is-black.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-black.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-light{background-color:#f5f5f5;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-light:hover,.button.is-light.is-hovered{background-color:#eee;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-light:focus,.button.is-light.is-focused{border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-light:focus:not(:active),.button.is-light.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(245,245,245,0.25)}.button.is-light:active,.button.is-light.is-active{background-color:#e8e8e8;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-light[disabled],fieldset[disabled] .button.is-light{background-color:#f5f5f5;border-color:#f5f5f5;box-shadow:none}.button.is-light.is-inverted{background-color:rgba(0,0,0,0.7);color:#f5f5f5}.button.is-light.is-inverted:hover,.button.is-light.is-inverted.is-hovered{background-color:rgba(0,0,0,0.7)}.button.is-light.is-inverted[disabled],fieldset[disabled] .button.is-light.is-inverted{background-color:rgba(0,0,0,0.7);border-color:transparent;box-shadow:none;color:#f5f5f5}.button.is-light.is-loading::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}.button.is-light.is-outlined{background-color:transparent;border-color:#f5f5f5;color:#f5f5f5}.button.is-light.is-outlined:hover,.button.is-light.is-outlined.is-hovered,.button.is-light.is-outlined:focus,.button.is-light.is-outlined.is-focused{background-color:#f5f5f5;border-color:#f5f5f5;color:rgba(0,0,0,0.7)}.button.is-light.is-outlined.is-loading::after{border-color:transparent transparent #f5f5f5 #f5f5f5 !important}.button.is-light.is-outlined.is-loading:hover::after,.button.is-light.is-outlined.is-loading.is-hovered::after,.button.is-light.is-outlined.is-loading:focus::after,.button.is-light.is-outlined.is-loading.is-focused::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}.button.is-light.is-outlined[disabled],fieldset[disabled] .button.is-light.is-outlined{background-color:transparent;border-color:#f5f5f5;box-shadow:none;color:#f5f5f5}.button.is-light.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);color:rgba(0,0,0,0.7)}.button.is-light.is-inverted.is-outlined:hover,.button.is-light.is-inverted.is-outlined.is-hovered,.button.is-light.is-inverted.is-outlined:focus,.button.is-light.is-inverted.is-outlined.is-focused{background-color:rgba(0,0,0,0.7);color:#f5f5f5}.button.is-light.is-inverted.is-outlined.is-loading:hover::after,.button.is-light.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-light.is-inverted.is-outlined.is-loading:focus::after,.button.is-light.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #f5f5f5 #f5f5f5 !important}.button.is-light.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-light.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);box-shadow:none;color:rgba(0,0,0,0.7)}.button.is-dark,.content kbd.button{background-color:#363636;border-color:transparent;color:#fff}.button.is-dark:hover,.content kbd.button:hover,.button.is-dark.is-hovered,.content kbd.button.is-hovered{background-color:#2f2f2f;border-color:transparent;color:#fff}.button.is-dark:focus,.content kbd.button:focus,.button.is-dark.is-focused,.content kbd.button.is-focused{border-color:transparent;color:#fff}.button.is-dark:focus:not(:active),.content kbd.button:focus:not(:active),.button.is-dark.is-focused:not(:active),.content kbd.button.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(54,54,54,0.25)}.button.is-dark:active,.content kbd.button:active,.button.is-dark.is-active,.content kbd.button.is-active{background-color:#292929;border-color:transparent;color:#fff}.button.is-dark[disabled],.content kbd.button[disabled],fieldset[disabled] .button.is-dark,fieldset[disabled] .content kbd.button,.content fieldset[disabled] kbd.button{background-color:#363636;border-color:#363636;box-shadow:none}.button.is-dark.is-inverted,.content kbd.button.is-inverted{background-color:#fff;color:#363636}.button.is-dark.is-inverted:hover,.content kbd.button.is-inverted:hover,.button.is-dark.is-inverted.is-hovered,.content kbd.button.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-dark.is-inverted[disabled],.content kbd.button.is-inverted[disabled],fieldset[disabled] .button.is-dark.is-inverted,fieldset[disabled] .content kbd.button.is-inverted,.content fieldset[disabled] kbd.button.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#363636}.button.is-dark.is-loading::after,.content kbd.button.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-dark.is-outlined,.content kbd.button.is-outlined{background-color:transparent;border-color:#363636;color:#363636}.button.is-dark.is-outlined:hover,.content kbd.button.is-outlined:hover,.button.is-dark.is-outlined.is-hovered,.content kbd.button.is-outlined.is-hovered,.button.is-dark.is-outlined:focus,.content kbd.button.is-outlined:focus,.button.is-dark.is-outlined.is-focused,.content kbd.button.is-outlined.is-focused{background-color:#363636;border-color:#363636;color:#fff}.button.is-dark.is-outlined.is-loading::after,.content kbd.button.is-outlined.is-loading::after{border-color:transparent transparent #363636 #363636 !important}.button.is-dark.is-outlined.is-loading:hover::after,.content kbd.button.is-outlined.is-loading:hover::after,.button.is-dark.is-outlined.is-loading.is-hovered::after,.content kbd.button.is-outlined.is-loading.is-hovered::after,.button.is-dark.is-outlined.is-loading:focus::after,.content kbd.button.is-outlined.is-loading:focus::after,.button.is-dark.is-outlined.is-loading.is-focused::after,.content kbd.button.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-dark.is-outlined[disabled],.content kbd.button.is-outlined[disabled],fieldset[disabled] .button.is-dark.is-outlined,fieldset[disabled] .content kbd.button.is-outlined,.content fieldset[disabled] kbd.button.is-outlined{background-color:transparent;border-color:#363636;box-shadow:none;color:#363636}.button.is-dark.is-inverted.is-outlined,.content kbd.button.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-dark.is-inverted.is-outlined:hover,.content kbd.button.is-inverted.is-outlined:hover,.button.is-dark.is-inverted.is-outlined.is-hovered,.content kbd.button.is-inverted.is-outlined.is-hovered,.button.is-dark.is-inverted.is-outlined:focus,.content kbd.button.is-inverted.is-outlined:focus,.button.is-dark.is-inverted.is-outlined.is-focused,.content kbd.button.is-inverted.is-outlined.is-focused{background-color:#fff;color:#363636}.button.is-dark.is-inverted.is-outlined.is-loading:hover::after,.content kbd.button.is-inverted.is-outlined.is-loading:hover::after,.button.is-dark.is-inverted.is-outlined.is-loading.is-hovered::after,.content kbd.button.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-dark.is-inverted.is-outlined.is-loading:focus::after,.content kbd.button.is-inverted.is-outlined.is-loading:focus::after,.button.is-dark.is-inverted.is-outlined.is-loading.is-focused::after,.content kbd.button.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #363636 #363636 !important}.button.is-dark.is-inverted.is-outlined[disabled],.content kbd.button.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-dark.is-inverted.is-outlined,fieldset[disabled] .content kbd.button.is-inverted.is-outlined,.content fieldset[disabled] kbd.button.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-primary,.docstring>section>a.button.docs-sourcelink{background-color:#4eb5de;border-color:transparent;color:#fff}.button.is-primary:hover,.docstring>section>a.button.docs-sourcelink:hover,.button.is-primary.is-hovered,.docstring>section>a.button.is-hovered.docs-sourcelink{background-color:#43b1dc;border-color:transparent;color:#fff}.button.is-primary:focus,.docstring>section>a.button.docs-sourcelink:focus,.button.is-primary.is-focused,.docstring>section>a.button.is-focused.docs-sourcelink{border-color:transparent;color:#fff}.button.is-primary:focus:not(:active),.docstring>section>a.button.docs-sourcelink:focus:not(:active),.button.is-primary.is-focused:not(:active),.docstring>section>a.button.is-focused.docs-sourcelink:not(:active){box-shadow:0 0 0 0.125em rgba(78,181,222,0.25)}.button.is-primary:active,.docstring>section>a.button.docs-sourcelink:active,.button.is-primary.is-active,.docstring>section>a.button.is-active.docs-sourcelink{background-color:#39acda;border-color:transparent;color:#fff}.button.is-primary[disabled],.docstring>section>a.button.docs-sourcelink[disabled],fieldset[disabled] .button.is-primary,fieldset[disabled] .docstring>section>a.button.docs-sourcelink{background-color:#4eb5de;border-color:#4eb5de;box-shadow:none}.button.is-primary.is-inverted,.docstring>section>a.button.is-inverted.docs-sourcelink{background-color:#fff;color:#4eb5de}.button.is-primary.is-inverted:hover,.docstring>section>a.button.is-inverted.docs-sourcelink:hover,.button.is-primary.is-inverted.is-hovered,.docstring>section>a.button.is-inverted.is-hovered.docs-sourcelink{background-color:#f2f2f2}.button.is-primary.is-inverted[disabled],.docstring>section>a.button.is-inverted.docs-sourcelink[disabled],fieldset[disabled] .button.is-primary.is-inverted,fieldset[disabled] .docstring>section>a.button.is-inverted.docs-sourcelink{background-color:#fff;border-color:transparent;box-shadow:none;color:#4eb5de}.button.is-primary.is-loading::after,.docstring>section>a.button.is-loading.docs-sourcelink::after{border-color:transparent transparent #fff #fff !important}.button.is-primary.is-outlined,.docstring>section>a.button.is-outlined.docs-sourcelink{background-color:transparent;border-color:#4eb5de;color:#4eb5de}.button.is-primary.is-outlined:hover,.docstring>section>a.button.is-outlined.docs-sourcelink:hover,.button.is-primary.is-outlined.is-hovered,.docstring>section>a.button.is-outlined.is-hovered.docs-sourcelink,.button.is-primary.is-outlined:focus,.docstring>section>a.button.is-outlined.docs-sourcelink:focus,.button.is-primary.is-outlined.is-focused,.docstring>section>a.button.is-outlined.is-focused.docs-sourcelink{background-color:#4eb5de;border-color:#4eb5de;color:#fff}.button.is-primary.is-outlined.is-loading::after,.docstring>section>a.button.is-outlined.is-loading.docs-sourcelink::after{border-color:transparent transparent #4eb5de #4eb5de !important}.button.is-primary.is-outlined.is-loading:hover::after,.docstring>section>a.button.is-outlined.is-loading.docs-sourcelink:hover::after,.button.is-primary.is-outlined.is-loading.is-hovered::after,.docstring>section>a.button.is-outlined.is-loading.is-hovered.docs-sourcelink::after,.button.is-primary.is-outlined.is-loading:focus::after,.docstring>section>a.button.is-outlined.is-loading.docs-sourcelink:focus::after,.button.is-primary.is-outlined.is-loading.is-focused::after,.docstring>section>a.button.is-outlined.is-loading.is-focused.docs-sourcelink::after{border-color:transparent transparent #fff #fff !important}.button.is-primary.is-outlined[disabled],.docstring>section>a.button.is-outlined.docs-sourcelink[disabled],fieldset[disabled] .button.is-primary.is-outlined,fieldset[disabled] .docstring>section>a.button.is-outlined.docs-sourcelink{background-color:transparent;border-color:#4eb5de;box-shadow:none;color:#4eb5de}.button.is-primary.is-inverted.is-outlined,.docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink{background-color:transparent;border-color:#fff;color:#fff}.button.is-primary.is-inverted.is-outlined:hover,.docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink:hover,.button.is-primary.is-inverted.is-outlined.is-hovered,.docstring>section>a.button.is-inverted.is-outlined.is-hovered.docs-sourcelink,.button.is-primary.is-inverted.is-outlined:focus,.docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink:focus,.button.is-primary.is-inverted.is-outlined.is-focused,.docstring>section>a.button.is-inverted.is-outlined.is-focused.docs-sourcelink{background-color:#fff;color:#4eb5de}.button.is-primary.is-inverted.is-outlined.is-loading:hover::after,.docstring>section>a.button.is-inverted.is-outlined.is-loading.docs-sourcelink:hover::after,.button.is-primary.is-inverted.is-outlined.is-loading.is-hovered::after,.docstring>section>a.button.is-inverted.is-outlined.is-loading.is-hovered.docs-sourcelink::after,.button.is-primary.is-inverted.is-outlined.is-loading:focus::after,.docstring>section>a.button.is-inverted.is-outlined.is-loading.docs-sourcelink:focus::after,.button.is-primary.is-inverted.is-outlined.is-loading.is-focused::after,.docstring>section>a.button.is-inverted.is-outlined.is-loading.is-focused.docs-sourcelink::after{border-color:transparent transparent #4eb5de #4eb5de !important}.button.is-primary.is-inverted.is-outlined[disabled],.docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink[disabled],fieldset[disabled] .button.is-primary.is-inverted.is-outlined,fieldset[disabled] .docstring>section>a.button.is-inverted.is-outlined.docs-sourcelink{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-primary.is-light,.docstring>section>a.button.is-light.docs-sourcelink{background-color:#eef8fc;color:#1a6d8e}.button.is-primary.is-light:hover,.docstring>section>a.button.is-light.docs-sourcelink:hover,.button.is-primary.is-light.is-hovered,.docstring>section>a.button.is-light.is-hovered.docs-sourcelink{background-color:#e3f3fa;border-color:transparent;color:#1a6d8e}.button.is-primary.is-light:active,.docstring>section>a.button.is-light.docs-sourcelink:active,.button.is-primary.is-light.is-active,.docstring>section>a.button.is-light.is-active.docs-sourcelink{background-color:#d8eff8;border-color:transparent;color:#1a6d8e}.button.is-link{background-color:#2e63b8;border-color:transparent;color:#fff}.button.is-link:hover,.button.is-link.is-hovered{background-color:#2b5eae;border-color:transparent;color:#fff}.button.is-link:focus,.button.is-link.is-focused{border-color:transparent;color:#fff}.button.is-link:focus:not(:active),.button.is-link.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.button.is-link:active,.button.is-link.is-active{background-color:#2958a4;border-color:transparent;color:#fff}.button.is-link[disabled],fieldset[disabled] .button.is-link{background-color:#2e63b8;border-color:#2e63b8;box-shadow:none}.button.is-link.is-inverted{background-color:#fff;color:#2e63b8}.button.is-link.is-inverted:hover,.button.is-link.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-link.is-inverted[disabled],fieldset[disabled] .button.is-link.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#2e63b8}.button.is-link.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-link.is-outlined{background-color:transparent;border-color:#2e63b8;color:#2e63b8}.button.is-link.is-outlined:hover,.button.is-link.is-outlined.is-hovered,.button.is-link.is-outlined:focus,.button.is-link.is-outlined.is-focused{background-color:#2e63b8;border-color:#2e63b8;color:#fff}.button.is-link.is-outlined.is-loading::after{border-color:transparent transparent #2e63b8 #2e63b8 !important}.button.is-link.is-outlined.is-loading:hover::after,.button.is-link.is-outlined.is-loading.is-hovered::after,.button.is-link.is-outlined.is-loading:focus::after,.button.is-link.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-link.is-outlined[disabled],fieldset[disabled] .button.is-link.is-outlined{background-color:transparent;border-color:#2e63b8;box-shadow:none;color:#2e63b8}.button.is-link.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-link.is-inverted.is-outlined:hover,.button.is-link.is-inverted.is-outlined.is-hovered,.button.is-link.is-inverted.is-outlined:focus,.button.is-link.is-inverted.is-outlined.is-focused{background-color:#fff;color:#2e63b8}.button.is-link.is-inverted.is-outlined.is-loading:hover::after,.button.is-link.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-link.is-inverted.is-outlined.is-loading:focus::after,.button.is-link.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #2e63b8 #2e63b8 !important}.button.is-link.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-link.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-link.is-light{background-color:#eff3fb;color:#3169c4}.button.is-link.is-light:hover,.button.is-link.is-light.is-hovered{background-color:#e4ecf8;border-color:transparent;color:#3169c4}.button.is-link.is-light:active,.button.is-link.is-light.is-active{background-color:#dae5f6;border-color:transparent;color:#3169c4}.button.is-info{background-color:#209cee;border-color:transparent;color:#fff}.button.is-info:hover,.button.is-info.is-hovered{background-color:#1497ed;border-color:transparent;color:#fff}.button.is-info:focus,.button.is-info.is-focused{border-color:transparent;color:#fff}.button.is-info:focus:not(:active),.button.is-info.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(32,156,238,0.25)}.button.is-info:active,.button.is-info.is-active{background-color:#1190e3;border-color:transparent;color:#fff}.button.is-info[disabled],fieldset[disabled] .button.is-info{background-color:#209cee;border-color:#209cee;box-shadow:none}.button.is-info.is-inverted{background-color:#fff;color:#209cee}.button.is-info.is-inverted:hover,.button.is-info.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-info.is-inverted[disabled],fieldset[disabled] .button.is-info.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#209cee}.button.is-info.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-info.is-outlined{background-color:transparent;border-color:#209cee;color:#209cee}.button.is-info.is-outlined:hover,.button.is-info.is-outlined.is-hovered,.button.is-info.is-outlined:focus,.button.is-info.is-outlined.is-focused{background-color:#209cee;border-color:#209cee;color:#fff}.button.is-info.is-outlined.is-loading::after{border-color:transparent transparent #209cee #209cee !important}.button.is-info.is-outlined.is-loading:hover::after,.button.is-info.is-outlined.is-loading.is-hovered::after,.button.is-info.is-outlined.is-loading:focus::after,.button.is-info.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-info.is-outlined[disabled],fieldset[disabled] .button.is-info.is-outlined{background-color:transparent;border-color:#209cee;box-shadow:none;color:#209cee}.button.is-info.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-info.is-inverted.is-outlined:hover,.button.is-info.is-inverted.is-outlined.is-hovered,.button.is-info.is-inverted.is-outlined:focus,.button.is-info.is-inverted.is-outlined.is-focused{background-color:#fff;color:#209cee}.button.is-info.is-inverted.is-outlined.is-loading:hover::after,.button.is-info.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-info.is-inverted.is-outlined.is-loading:focus::after,.button.is-info.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #209cee #209cee !important}.button.is-info.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-info.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-info.is-light{background-color:#ecf7fe;color:#0e72b4}.button.is-info.is-light:hover,.button.is-info.is-light.is-hovered{background-color:#e0f1fd;border-color:transparent;color:#0e72b4}.button.is-info.is-light:active,.button.is-info.is-light.is-active{background-color:#d4ecfc;border-color:transparent;color:#0e72b4}.button.is-success{background-color:#22c35b;border-color:transparent;color:#fff}.button.is-success:hover,.button.is-success.is-hovered{background-color:#20b856;border-color:transparent;color:#fff}.button.is-success:focus,.button.is-success.is-focused{border-color:transparent;color:#fff}.button.is-success:focus:not(:active),.button.is-success.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(34,195,91,0.25)}.button.is-success:active,.button.is-success.is-active{background-color:#1ead51;border-color:transparent;color:#fff}.button.is-success[disabled],fieldset[disabled] .button.is-success{background-color:#22c35b;border-color:#22c35b;box-shadow:none}.button.is-success.is-inverted{background-color:#fff;color:#22c35b}.button.is-success.is-inverted:hover,.button.is-success.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-success.is-inverted[disabled],fieldset[disabled] .button.is-success.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#22c35b}.button.is-success.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-success.is-outlined{background-color:transparent;border-color:#22c35b;color:#22c35b}.button.is-success.is-outlined:hover,.button.is-success.is-outlined.is-hovered,.button.is-success.is-outlined:focus,.button.is-success.is-outlined.is-focused{background-color:#22c35b;border-color:#22c35b;color:#fff}.button.is-success.is-outlined.is-loading::after{border-color:transparent transparent #22c35b #22c35b !important}.button.is-success.is-outlined.is-loading:hover::after,.button.is-success.is-outlined.is-loading.is-hovered::after,.button.is-success.is-outlined.is-loading:focus::after,.button.is-success.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-success.is-outlined[disabled],fieldset[disabled] .button.is-success.is-outlined{background-color:transparent;border-color:#22c35b;box-shadow:none;color:#22c35b}.button.is-success.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-success.is-inverted.is-outlined:hover,.button.is-success.is-inverted.is-outlined.is-hovered,.button.is-success.is-inverted.is-outlined:focus,.button.is-success.is-inverted.is-outlined.is-focused{background-color:#fff;color:#22c35b}.button.is-success.is-inverted.is-outlined.is-loading:hover::after,.button.is-success.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-success.is-inverted.is-outlined.is-loading:focus::after,.button.is-success.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #22c35b #22c35b !important}.button.is-success.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-success.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-success.is-light{background-color:#eefcf3;color:#198f43}.button.is-success.is-light:hover,.button.is-success.is-light.is-hovered{background-color:#e3faeb;border-color:transparent;color:#198f43}.button.is-success.is-light:active,.button.is-success.is-light.is-active{background-color:#d8f8e3;border-color:transparent;color:#198f43}.button.is-warning{background-color:#ffdd57;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-warning:hover,.button.is-warning.is-hovered{background-color:#ffda4a;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-warning:focus,.button.is-warning.is-focused{border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-warning:focus:not(:active),.button.is-warning.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(255,221,87,0.25)}.button.is-warning:active,.button.is-warning.is-active{background-color:#ffd83e;border-color:transparent;color:rgba(0,0,0,0.7)}.button.is-warning[disabled],fieldset[disabled] .button.is-warning{background-color:#ffdd57;border-color:#ffdd57;box-shadow:none}.button.is-warning.is-inverted{background-color:rgba(0,0,0,0.7);color:#ffdd57}.button.is-warning.is-inverted:hover,.button.is-warning.is-inverted.is-hovered{background-color:rgba(0,0,0,0.7)}.button.is-warning.is-inverted[disabled],fieldset[disabled] .button.is-warning.is-inverted{background-color:rgba(0,0,0,0.7);border-color:transparent;box-shadow:none;color:#ffdd57}.button.is-warning.is-loading::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}.button.is-warning.is-outlined{background-color:transparent;border-color:#ffdd57;color:#ffdd57}.button.is-warning.is-outlined:hover,.button.is-warning.is-outlined.is-hovered,.button.is-warning.is-outlined:focus,.button.is-warning.is-outlined.is-focused{background-color:#ffdd57;border-color:#ffdd57;color:rgba(0,0,0,0.7)}.button.is-warning.is-outlined.is-loading::after{border-color:transparent transparent #ffdd57 #ffdd57 !important}.button.is-warning.is-outlined.is-loading:hover::after,.button.is-warning.is-outlined.is-loading.is-hovered::after,.button.is-warning.is-outlined.is-loading:focus::after,.button.is-warning.is-outlined.is-loading.is-focused::after{border-color:transparent transparent rgba(0,0,0,0.7) rgba(0,0,0,0.7) !important}.button.is-warning.is-outlined[disabled],fieldset[disabled] .button.is-warning.is-outlined{background-color:transparent;border-color:#ffdd57;box-shadow:none;color:#ffdd57}.button.is-warning.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);color:rgba(0,0,0,0.7)}.button.is-warning.is-inverted.is-outlined:hover,.button.is-warning.is-inverted.is-outlined.is-hovered,.button.is-warning.is-inverted.is-outlined:focus,.button.is-warning.is-inverted.is-outlined.is-focused{background-color:rgba(0,0,0,0.7);color:#ffdd57}.button.is-warning.is-inverted.is-outlined.is-loading:hover::after,.button.is-warning.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-warning.is-inverted.is-outlined.is-loading:focus::after,.button.is-warning.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #ffdd57 #ffdd57 !important}.button.is-warning.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-warning.is-inverted.is-outlined{background-color:transparent;border-color:rgba(0,0,0,0.7);box-shadow:none;color:rgba(0,0,0,0.7)}.button.is-warning.is-light{background-color:#fffbeb;color:#947600}.button.is-warning.is-light:hover,.button.is-warning.is-light.is-hovered{background-color:#fff8de;border-color:transparent;color:#947600}.button.is-warning.is-light:active,.button.is-warning.is-light.is-active{background-color:#fff6d1;border-color:transparent;color:#947600}.button.is-danger{background-color:#da0b00;border-color:transparent;color:#fff}.button.is-danger:hover,.button.is-danger.is-hovered{background-color:#cd0a00;border-color:transparent;color:#fff}.button.is-danger:focus,.button.is-danger.is-focused{border-color:transparent;color:#fff}.button.is-danger:focus:not(:active),.button.is-danger.is-focused:not(:active){box-shadow:0 0 0 0.125em rgba(218,11,0,0.25)}.button.is-danger:active,.button.is-danger.is-active{background-color:#c10a00;border-color:transparent;color:#fff}.button.is-danger[disabled],fieldset[disabled] .button.is-danger{background-color:#da0b00;border-color:#da0b00;box-shadow:none}.button.is-danger.is-inverted{background-color:#fff;color:#da0b00}.button.is-danger.is-inverted:hover,.button.is-danger.is-inverted.is-hovered{background-color:#f2f2f2}.button.is-danger.is-inverted[disabled],fieldset[disabled] .button.is-danger.is-inverted{background-color:#fff;border-color:transparent;box-shadow:none;color:#da0b00}.button.is-danger.is-loading::after{border-color:transparent transparent #fff #fff !important}.button.is-danger.is-outlined{background-color:transparent;border-color:#da0b00;color:#da0b00}.button.is-danger.is-outlined:hover,.button.is-danger.is-outlined.is-hovered,.button.is-danger.is-outlined:focus,.button.is-danger.is-outlined.is-focused{background-color:#da0b00;border-color:#da0b00;color:#fff}.button.is-danger.is-outlined.is-loading::after{border-color:transparent transparent #da0b00 #da0b00 !important}.button.is-danger.is-outlined.is-loading:hover::after,.button.is-danger.is-outlined.is-loading.is-hovered::after,.button.is-danger.is-outlined.is-loading:focus::after,.button.is-danger.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #fff #fff !important}.button.is-danger.is-outlined[disabled],fieldset[disabled] .button.is-danger.is-outlined{background-color:transparent;border-color:#da0b00;box-shadow:none;color:#da0b00}.button.is-danger.is-inverted.is-outlined{background-color:transparent;border-color:#fff;color:#fff}.button.is-danger.is-inverted.is-outlined:hover,.button.is-danger.is-inverted.is-outlined.is-hovered,.button.is-danger.is-inverted.is-outlined:focus,.button.is-danger.is-inverted.is-outlined.is-focused{background-color:#fff;color:#da0b00}.button.is-danger.is-inverted.is-outlined.is-loading:hover::after,.button.is-danger.is-inverted.is-outlined.is-loading.is-hovered::after,.button.is-danger.is-inverted.is-outlined.is-loading:focus::after,.button.is-danger.is-inverted.is-outlined.is-loading.is-focused::after{border-color:transparent transparent #da0b00 #da0b00 !important}.button.is-danger.is-inverted.is-outlined[disabled],fieldset[disabled] .button.is-danger.is-inverted.is-outlined{background-color:transparent;border-color:#fff;box-shadow:none;color:#fff}.button.is-danger.is-light{background-color:#ffeceb;color:#f50c00}.button.is-danger.is-light:hover,.button.is-danger.is-light.is-hovered{background-color:#ffe0de;border-color:transparent;color:#f50c00}.button.is-danger.is-light:active,.button.is-danger.is-light.is-active{background-color:#ffd3d1;border-color:transparent;color:#f50c00}.button.is-small,#documenter .docs-sidebar form.docs-search>input.button{font-size:.75rem}.button.is-small:not(.is-rounded),#documenter .docs-sidebar form.docs-search>input.button:not(.is-rounded){border-radius:2px}.button.is-normal{font-size:1rem}.button.is-medium{font-size:1.25rem}.button.is-large{font-size:1.5rem}.button[disabled],fieldset[disabled] .button{background-color:#fff;border-color:#dbdbdb;box-shadow:none;opacity:.5}.button.is-fullwidth{display:flex;width:100%}.button.is-loading{color:transparent !important;pointer-events:none}.button.is-loading::after{position:absolute;left:calc(50% - (1em * 0.5));top:calc(50% - (1em * 0.5));position:absolute !important}.button.is-static{background-color:#f5f5f5;border-color:#dbdbdb;color:#6b6b6b;box-shadow:none;pointer-events:none}.button.is-rounded,#documenter .docs-sidebar form.docs-search>input.button{border-radius:9999px;padding-left:calc(1em + 0.25em);padding-right:calc(1em + 0.25em)}.buttons{align-items:center;display:flex;flex-wrap:wrap;justify-content:flex-start}.buttons .button{margin-bottom:0.5rem}.buttons .button:not(:last-child):not(.is-fullwidth){margin-right:.5rem}.buttons:last-child{margin-bottom:-0.5rem}.buttons:not(:last-child){margin-bottom:1rem}.buttons.are-small .button:not(.is-normal):not(.is-medium):not(.is-large){font-size:.75rem}.buttons.are-small .button:not(.is-normal):not(.is-medium):not(.is-large):not(.is-rounded){border-radius:2px}.buttons.are-medium .button:not(.is-small):not(.is-normal):not(.is-large){font-size:1.25rem}.buttons.are-large .button:not(.is-small):not(.is-normal):not(.is-medium){font-size:1.5rem}.buttons.has-addons .button:not(:first-child){border-bottom-left-radius:0;border-top-left-radius:0}.buttons.has-addons .button:not(:last-child){border-bottom-right-radius:0;border-top-right-radius:0;margin-right:-1px}.buttons.has-addons .button:last-child{margin-right:0}.buttons.has-addons .button:hover,.buttons.has-addons .button.is-hovered{z-index:2}.buttons.has-addons .button:focus,.buttons.has-addons .button.is-focused,.buttons.has-addons .button:active,.buttons.has-addons .button.is-active,.buttons.has-addons .button.is-selected{z-index:3}.buttons.has-addons .button:focus:hover,.buttons.has-addons .button.is-focused:hover,.buttons.has-addons .button:active:hover,.buttons.has-addons .button.is-active:hover,.buttons.has-addons .button.is-selected:hover{z-index:4}.buttons.has-addons .button.is-expanded{flex-grow:1;flex-shrink:1}.buttons.is-centered{justify-content:center}.buttons.is-centered:not(.has-addons) .button:not(.is-fullwidth){margin-left:0.25rem;margin-right:0.25rem}.buttons.is-right{justify-content:flex-end}.buttons.is-right:not(.has-addons) .button:not(.is-fullwidth){margin-left:0.25rem;margin-right:0.25rem}@media screen and (max-width: 768px){.button.is-responsive.is-small,#documenter .docs-sidebar form.docs-search>input.is-responsive{font-size:.5625rem}.button.is-responsive,.button.is-responsive.is-normal{font-size:.65625rem}.button.is-responsive.is-medium{font-size:.75rem}.button.is-responsive.is-large{font-size:1rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.button.is-responsive.is-small,#documenter .docs-sidebar form.docs-search>input.is-responsive{font-size:.65625rem}.button.is-responsive,.button.is-responsive.is-normal{font-size:.75rem}.button.is-responsive.is-medium{font-size:1rem}.button.is-responsive.is-large{font-size:1.25rem}}.container{flex-grow:1;margin:0 auto;position:relative;width:auto}.container.is-fluid{max-width:none !important;padding-left:32px;padding-right:32px;width:100%}@media screen and (min-width: 1056px){.container{max-width:992px}}@media screen and (max-width: 1215px){.container.is-widescreen:not(.is-max-desktop){max-width:1152px}}@media screen and (max-width: 1407px){.container.is-fullhd:not(.is-max-desktop):not(.is-max-widescreen){max-width:1344px}}@media screen and (min-width: 1216px){.container:not(.is-max-desktop){max-width:1152px}}@media screen and (min-width: 1408px){.container:not(.is-max-desktop):not(.is-max-widescreen){max-width:1344px}}.content li+li{margin-top:0.25em}.content p:not(:last-child),.content dl:not(:last-child),.content ol:not(:last-child),.content ul:not(:last-child),.content blockquote:not(:last-child),.content pre:not(:last-child),.content table:not(:last-child){margin-bottom:1em}.content h1,.content h2,.content h3,.content h4,.content h5,.content h6{color:#222;font-weight:600;line-height:1.125}.content h1{font-size:2em;margin-bottom:0.5em}.content h1:not(:first-child){margin-top:1em}.content h2{font-size:1.75em;margin-bottom:0.5714em}.content h2:not(:first-child){margin-top:1.1428em}.content h3{font-size:1.5em;margin-bottom:0.6666em}.content h3:not(:first-child){margin-top:1.3333em}.content h4{font-size:1.25em;margin-bottom:0.8em}.content h5{font-size:1.125em;margin-bottom:0.8888em}.content h6{font-size:1em;margin-bottom:1em}.content blockquote{background-color:#f5f5f5;border-left:5px solid #dbdbdb;padding:1.25em 1.5em}.content ol{list-style-position:outside;margin-left:2em;margin-top:1em}.content ol:not([type]){list-style-type:decimal}.content ol.is-lower-alpha:not([type]){list-style-type:lower-alpha}.content ol.is-lower-roman:not([type]){list-style-type:lower-roman}.content ol.is-upper-alpha:not([type]){list-style-type:upper-alpha}.content ol.is-upper-roman:not([type]){list-style-type:upper-roman}.content ul{list-style:disc outside;margin-left:2em;margin-top:1em}.content ul ul{list-style-type:circle;margin-top:0.5em}.content ul ul ul{list-style-type:square}.content dd{margin-left:2em}.content figure{margin-left:2em;margin-right:2em;text-align:center}.content figure:not(:first-child){margin-top:2em}.content figure:not(:last-child){margin-bottom:2em}.content figure img{display:inline-block}.content figure figcaption{font-style:italic}.content pre{-webkit-overflow-scrolling:touch;overflow-x:auto;padding:0;white-space:pre;word-wrap:normal}.content sup,.content sub{font-size:75%}.content table{width:100%}.content table td,.content table th{border:1px solid #dbdbdb;border-width:0 0 1px;padding:0.5em 0.75em;vertical-align:top}.content table th{color:#222}.content table th:not([align]){text-align:inherit}.content table thead td,.content table thead th{border-width:0 0 2px;color:#222}.content table tfoot td,.content table tfoot th{border-width:2px 0 0;color:#222}.content table tbody tr:last-child td,.content table tbody tr:last-child th{border-bottom-width:0}.content .tabs li+li{margin-top:0}.content.is-small,#documenter .docs-sidebar form.docs-search>input.content{font-size:.75rem}.content.is-normal{font-size:1rem}.content.is-medium{font-size:1.25rem}.content.is-large{font-size:1.5rem}.icon{align-items:center;display:inline-flex;justify-content:center;height:1.5rem;width:1.5rem}.icon.is-small,#documenter .docs-sidebar form.docs-search>input.icon{height:1rem;width:1rem}.icon.is-medium{height:2rem;width:2rem}.icon.is-large{height:3rem;width:3rem}.icon-text{align-items:flex-start;color:inherit;display:inline-flex;flex-wrap:wrap;line-height:1.5rem;vertical-align:top}.icon-text .icon{flex-grow:0;flex-shrink:0}.icon-text .icon:not(:last-child){margin-right:.25em}.icon-text .icon:not(:first-child){margin-left:.25em}div.icon-text{display:flex}.image,#documenter .docs-sidebar .docs-logo>img{display:block;position:relative}.image img,#documenter .docs-sidebar .docs-logo>img img{display:block;height:auto;width:100%}.image img.is-rounded,#documenter .docs-sidebar .docs-logo>img img.is-rounded{border-radius:9999px}.image.is-fullwidth,#documenter .docs-sidebar .docs-logo>img.is-fullwidth{width:100%}.image.is-square img,#documenter .docs-sidebar .docs-logo>img.is-square img,.image.is-square .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-square .has-ratio,.image.is-1by1 img,#documenter .docs-sidebar .docs-logo>img.is-1by1 img,.image.is-1by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by1 .has-ratio,.image.is-5by4 img,#documenter .docs-sidebar .docs-logo>img.is-5by4 img,.image.is-5by4 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-5by4 .has-ratio,.image.is-4by3 img,#documenter .docs-sidebar .docs-logo>img.is-4by3 img,.image.is-4by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-4by3 .has-ratio,.image.is-3by2 img,#documenter .docs-sidebar .docs-logo>img.is-3by2 img,.image.is-3by2 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by2 .has-ratio,.image.is-5by3 img,#documenter .docs-sidebar .docs-logo>img.is-5by3 img,.image.is-5by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-5by3 .has-ratio,.image.is-16by9 img,#documenter .docs-sidebar .docs-logo>img.is-16by9 img,.image.is-16by9 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-16by9 .has-ratio,.image.is-2by1 img,#documenter .docs-sidebar .docs-logo>img.is-2by1 img,.image.is-2by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-2by1 .has-ratio,.image.is-3by1 img,#documenter .docs-sidebar .docs-logo>img.is-3by1 img,.image.is-3by1 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by1 .has-ratio,.image.is-4by5 img,#documenter .docs-sidebar .docs-logo>img.is-4by5 img,.image.is-4by5 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-4by5 .has-ratio,.image.is-3by4 img,#documenter .docs-sidebar .docs-logo>img.is-3by4 img,.image.is-3by4 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by4 .has-ratio,.image.is-2by3 img,#documenter .docs-sidebar .docs-logo>img.is-2by3 img,.image.is-2by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-2by3 .has-ratio,.image.is-3by5 img,#documenter .docs-sidebar .docs-logo>img.is-3by5 img,.image.is-3by5 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-3by5 .has-ratio,.image.is-9by16 img,#documenter .docs-sidebar .docs-logo>img.is-9by16 img,.image.is-9by16 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-9by16 .has-ratio,.image.is-1by2 img,#documenter .docs-sidebar .docs-logo>img.is-1by2 img,.image.is-1by2 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by2 .has-ratio,.image.is-1by3 img,#documenter .docs-sidebar .docs-logo>img.is-1by3 img,.image.is-1by3 .has-ratio,#documenter .docs-sidebar .docs-logo>img.is-1by3 .has-ratio{height:100%;width:100%}.image.is-square,#documenter .docs-sidebar .docs-logo>img.is-square,.image.is-1by1,#documenter .docs-sidebar .docs-logo>img.is-1by1{padding-top:100%}.image.is-5by4,#documenter .docs-sidebar .docs-logo>img.is-5by4{padding-top:80%}.image.is-4by3,#documenter .docs-sidebar .docs-logo>img.is-4by3{padding-top:75%}.image.is-3by2,#documenter .docs-sidebar .docs-logo>img.is-3by2{padding-top:66.6666%}.image.is-5by3,#documenter .docs-sidebar .docs-logo>img.is-5by3{padding-top:60%}.image.is-16by9,#documenter .docs-sidebar .docs-logo>img.is-16by9{padding-top:56.25%}.image.is-2by1,#documenter .docs-sidebar .docs-logo>img.is-2by1{padding-top:50%}.image.is-3by1,#documenter .docs-sidebar .docs-logo>img.is-3by1{padding-top:33.3333%}.image.is-4by5,#documenter .docs-sidebar .docs-logo>img.is-4by5{padding-top:125%}.image.is-3by4,#documenter .docs-sidebar .docs-logo>img.is-3by4{padding-top:133.3333%}.image.is-2by3,#documenter .docs-sidebar .docs-logo>img.is-2by3{padding-top:150%}.image.is-3by5,#documenter .docs-sidebar .docs-logo>img.is-3by5{padding-top:166.6666%}.image.is-9by16,#documenter .docs-sidebar .docs-logo>img.is-9by16{padding-top:177.7777%}.image.is-1by2,#documenter .docs-sidebar .docs-logo>img.is-1by2{padding-top:200%}.image.is-1by3,#documenter .docs-sidebar .docs-logo>img.is-1by3{padding-top:300%}.image.is-16x16,#documenter .docs-sidebar .docs-logo>img.is-16x16{height:16px;width:16px}.image.is-24x24,#documenter .docs-sidebar .docs-logo>img.is-24x24{height:24px;width:24px}.image.is-32x32,#documenter .docs-sidebar .docs-logo>img.is-32x32{height:32px;width:32px}.image.is-48x48,#documenter .docs-sidebar .docs-logo>img.is-48x48{height:48px;width:48px}.image.is-64x64,#documenter .docs-sidebar .docs-logo>img.is-64x64{height:64px;width:64px}.image.is-96x96,#documenter .docs-sidebar .docs-logo>img.is-96x96{height:96px;width:96px}.image.is-128x128,#documenter .docs-sidebar .docs-logo>img.is-128x128{height:128px;width:128px}.notification{background-color:#f5f5f5;border-radius:4px;position:relative;padding:1.25rem 2.5rem 1.25rem 1.5rem}.notification a:not(.button):not(.dropdown-item){color:currentColor;text-decoration:underline}.notification strong{color:currentColor}.notification code,.notification pre{background:#fff}.notification pre code{background:transparent}.notification>.delete{right:.5rem;position:absolute;top:0.5rem}.notification .title,.notification .subtitle,.notification .content{color:currentColor}.notification.is-white{background-color:#fff;color:#0a0a0a}.notification.is-black{background-color:#0a0a0a;color:#fff}.notification.is-light{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.notification.is-dark,.content kbd.notification{background-color:#363636;color:#fff}.notification.is-primary,.docstring>section>a.notification.docs-sourcelink{background-color:#4eb5de;color:#fff}.notification.is-primary.is-light,.docstring>section>a.notification.is-light.docs-sourcelink{background-color:#eef8fc;color:#1a6d8e}.notification.is-link{background-color:#2e63b8;color:#fff}.notification.is-link.is-light{background-color:#eff3fb;color:#3169c4}.notification.is-info{background-color:#209cee;color:#fff}.notification.is-info.is-light{background-color:#ecf7fe;color:#0e72b4}.notification.is-success{background-color:#22c35b;color:#fff}.notification.is-success.is-light{background-color:#eefcf3;color:#198f43}.notification.is-warning{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.notification.is-warning.is-light{background-color:#fffbeb;color:#947600}.notification.is-danger{background-color:#da0b00;color:#fff}.notification.is-danger.is-light{background-color:#ffeceb;color:#f50c00}.progress{-moz-appearance:none;-webkit-appearance:none;border:none;border-radius:9999px;display:block;height:1rem;overflow:hidden;padding:0;width:100%}.progress::-webkit-progress-bar{background-color:#ededed}.progress::-webkit-progress-value{background-color:#222}.progress::-moz-progress-bar{background-color:#222}.progress::-ms-fill{background-color:#222;border:none}.progress.is-white::-webkit-progress-value{background-color:#fff}.progress.is-white::-moz-progress-bar{background-color:#fff}.progress.is-white::-ms-fill{background-color:#fff}.progress.is-white:indeterminate{background-image:linear-gradient(to right, #fff 30%, #ededed 30%)}.progress.is-black::-webkit-progress-value{background-color:#0a0a0a}.progress.is-black::-moz-progress-bar{background-color:#0a0a0a}.progress.is-black::-ms-fill{background-color:#0a0a0a}.progress.is-black:indeterminate{background-image:linear-gradient(to right, #0a0a0a 30%, #ededed 30%)}.progress.is-light::-webkit-progress-value{background-color:#f5f5f5}.progress.is-light::-moz-progress-bar{background-color:#f5f5f5}.progress.is-light::-ms-fill{background-color:#f5f5f5}.progress.is-light:indeterminate{background-image:linear-gradient(to right, #f5f5f5 30%, #ededed 30%)}.progress.is-dark::-webkit-progress-value,.content kbd.progress::-webkit-progress-value{background-color:#363636}.progress.is-dark::-moz-progress-bar,.content kbd.progress::-moz-progress-bar{background-color:#363636}.progress.is-dark::-ms-fill,.content kbd.progress::-ms-fill{background-color:#363636}.progress.is-dark:indeterminate,.content kbd.progress:indeterminate{background-image:linear-gradient(to right, #363636 30%, #ededed 30%)}.progress.is-primary::-webkit-progress-value,.docstring>section>a.progress.docs-sourcelink::-webkit-progress-value{background-color:#4eb5de}.progress.is-primary::-moz-progress-bar,.docstring>section>a.progress.docs-sourcelink::-moz-progress-bar{background-color:#4eb5de}.progress.is-primary::-ms-fill,.docstring>section>a.progress.docs-sourcelink::-ms-fill{background-color:#4eb5de}.progress.is-primary:indeterminate,.docstring>section>a.progress.docs-sourcelink:indeterminate{background-image:linear-gradient(to right, #4eb5de 30%, #ededed 30%)}.progress.is-link::-webkit-progress-value{background-color:#2e63b8}.progress.is-link::-moz-progress-bar{background-color:#2e63b8}.progress.is-link::-ms-fill{background-color:#2e63b8}.progress.is-link:indeterminate{background-image:linear-gradient(to right, #2e63b8 30%, #ededed 30%)}.progress.is-info::-webkit-progress-value{background-color:#209cee}.progress.is-info::-moz-progress-bar{background-color:#209cee}.progress.is-info::-ms-fill{background-color:#209cee}.progress.is-info:indeterminate{background-image:linear-gradient(to right, #209cee 30%, #ededed 30%)}.progress.is-success::-webkit-progress-value{background-color:#22c35b}.progress.is-success::-moz-progress-bar{background-color:#22c35b}.progress.is-success::-ms-fill{background-color:#22c35b}.progress.is-success:indeterminate{background-image:linear-gradient(to right, #22c35b 30%, #ededed 30%)}.progress.is-warning::-webkit-progress-value{background-color:#ffdd57}.progress.is-warning::-moz-progress-bar{background-color:#ffdd57}.progress.is-warning::-ms-fill{background-color:#ffdd57}.progress.is-warning:indeterminate{background-image:linear-gradient(to right, #ffdd57 30%, #ededed 30%)}.progress.is-danger::-webkit-progress-value{background-color:#da0b00}.progress.is-danger::-moz-progress-bar{background-color:#da0b00}.progress.is-danger::-ms-fill{background-color:#da0b00}.progress.is-danger:indeterminate{background-image:linear-gradient(to right, #da0b00 30%, #ededed 30%)}.progress:indeterminate{animation-duration:1.5s;animation-iteration-count:infinite;animation-name:moveIndeterminate;animation-timing-function:linear;background-color:#ededed;background-image:linear-gradient(to right, #222 30%, #ededed 30%);background-position:top left;background-repeat:no-repeat;background-size:150% 150%}.progress:indeterminate::-webkit-progress-bar{background-color:transparent}.progress:indeterminate::-moz-progress-bar{background-color:transparent}.progress:indeterminate::-ms-fill{animation-name:none}.progress.is-small,#documenter .docs-sidebar form.docs-search>input.progress{height:.75rem}.progress.is-medium{height:1.25rem}.progress.is-large{height:1.5rem}@keyframes moveIndeterminate{from{background-position:200% 0}to{background-position:-200% 0}}.table{background-color:#fff;color:#222}.table td,.table th{border:1px solid #dbdbdb;border-width:0 0 1px;padding:0.5em 0.75em;vertical-align:top}.table td.is-white,.table th.is-white{background-color:#fff;border-color:#fff;color:#0a0a0a}.table td.is-black,.table th.is-black{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}.table td.is-light,.table th.is-light{background-color:#f5f5f5;border-color:#f5f5f5;color:rgba(0,0,0,0.7)}.table td.is-dark,.table th.is-dark{background-color:#363636;border-color:#363636;color:#fff}.table td.is-primary,.table th.is-primary{background-color:#4eb5de;border-color:#4eb5de;color:#fff}.table td.is-link,.table th.is-link{background-color:#2e63b8;border-color:#2e63b8;color:#fff}.table td.is-info,.table th.is-info{background-color:#209cee;border-color:#209cee;color:#fff}.table td.is-success,.table th.is-success{background-color:#22c35b;border-color:#22c35b;color:#fff}.table td.is-warning,.table th.is-warning{background-color:#ffdd57;border-color:#ffdd57;color:rgba(0,0,0,0.7)}.table td.is-danger,.table th.is-danger{background-color:#da0b00;border-color:#da0b00;color:#fff}.table td.is-narrow,.table th.is-narrow{white-space:nowrap;width:1%}.table td.is-selected,.table th.is-selected{background-color:#4eb5de;color:#fff}.table td.is-selected a,.table td.is-selected strong,.table th.is-selected a,.table th.is-selected strong{color:currentColor}.table td.is-vcentered,.table th.is-vcentered{vertical-align:middle}.table th{color:#222}.table th:not([align]){text-align:left}.table tr.is-selected{background-color:#4eb5de;color:#fff}.table tr.is-selected a,.table tr.is-selected strong{color:currentColor}.table tr.is-selected td,.table tr.is-selected th{border-color:#fff;color:currentColor}.table thead{background-color:rgba(0,0,0,0)}.table thead td,.table thead th{border-width:0 0 2px;color:#222}.table tfoot{background-color:rgba(0,0,0,0)}.table tfoot td,.table tfoot th{border-width:2px 0 0;color:#222}.table tbody{background-color:rgba(0,0,0,0)}.table tbody tr:last-child td,.table tbody tr:last-child th{border-bottom-width:0}.table.is-bordered td,.table.is-bordered th{border-width:1px}.table.is-bordered tr:last-child td,.table.is-bordered tr:last-child th{border-bottom-width:1px}.table.is-fullwidth{width:100%}.table.is-hoverable tbody tr:not(.is-selected):hover{background-color:#fafafa}.table.is-hoverable.is-striped tbody tr:not(.is-selected):hover{background-color:#fafafa}.table.is-hoverable.is-striped tbody tr:not(.is-selected):hover:nth-child(even){background-color:#f5f5f5}.table.is-narrow td,.table.is-narrow th{padding:0.25em 0.5em}.table.is-striped tbody tr:not(.is-selected):nth-child(even){background-color:#fafafa}.table-container{-webkit-overflow-scrolling:touch;overflow:auto;overflow-y:hidden;max-width:100%}.tags{align-items:center;display:flex;flex-wrap:wrap;justify-content:flex-start}.tags .tag,.tags .content kbd,.content .tags kbd,.tags .docstring>section>a.docs-sourcelink{margin-bottom:0.5rem}.tags .tag:not(:last-child),.tags .content kbd:not(:last-child),.content .tags kbd:not(:last-child),.tags .docstring>section>a.docs-sourcelink:not(:last-child){margin-right:.5rem}.tags:last-child{margin-bottom:-0.5rem}.tags:not(:last-child){margin-bottom:1rem}.tags.are-medium .tag:not(.is-normal):not(.is-large),.tags.are-medium .content kbd:not(.is-normal):not(.is-large),.content .tags.are-medium kbd:not(.is-normal):not(.is-large),.tags.are-medium .docstring>section>a.docs-sourcelink:not(.is-normal):not(.is-large){font-size:1rem}.tags.are-large .tag:not(.is-normal):not(.is-medium),.tags.are-large .content kbd:not(.is-normal):not(.is-medium),.content .tags.are-large kbd:not(.is-normal):not(.is-medium),.tags.are-large .docstring>section>a.docs-sourcelink:not(.is-normal):not(.is-medium){font-size:1.25rem}.tags.is-centered{justify-content:center}.tags.is-centered .tag,.tags.is-centered .content kbd,.content .tags.is-centered kbd,.tags.is-centered .docstring>section>a.docs-sourcelink{margin-right:0.25rem;margin-left:0.25rem}.tags.is-right{justify-content:flex-end}.tags.is-right .tag:not(:first-child),.tags.is-right .content kbd:not(:first-child),.content .tags.is-right kbd:not(:first-child),.tags.is-right .docstring>section>a.docs-sourcelink:not(:first-child){margin-left:0.5rem}.tags.is-right .tag:not(:last-child),.tags.is-right .content kbd:not(:last-child),.content .tags.is-right kbd:not(:last-child),.tags.is-right .docstring>section>a.docs-sourcelink:not(:last-child){margin-right:0}.tags.has-addons .tag,.tags.has-addons .content kbd,.content .tags.has-addons kbd,.tags.has-addons .docstring>section>a.docs-sourcelink{margin-right:0}.tags.has-addons .tag:not(:first-child),.tags.has-addons .content kbd:not(:first-child),.content .tags.has-addons kbd:not(:first-child),.tags.has-addons .docstring>section>a.docs-sourcelink:not(:first-child){margin-left:0;border-top-left-radius:0;border-bottom-left-radius:0}.tags.has-addons .tag:not(:last-child),.tags.has-addons .content kbd:not(:last-child),.content .tags.has-addons kbd:not(:last-child),.tags.has-addons .docstring>section>a.docs-sourcelink:not(:last-child){border-top-right-radius:0;border-bottom-right-radius:0}.tag:not(body),.content kbd:not(body),.docstring>section>a.docs-sourcelink:not(body){align-items:center;background-color:#f5f5f5;border-radius:4px;color:#222;display:inline-flex;font-size:.75rem;height:2em;justify-content:center;line-height:1.5;padding-left:0.75em;padding-right:0.75em;white-space:nowrap}.tag:not(body) .delete,.content kbd:not(body) .delete,.docstring>section>a.docs-sourcelink:not(body) .delete{margin-left:.25rem;margin-right:-.375rem}.tag.is-white:not(body),.content kbd.is-white:not(body),.docstring>section>a.docs-sourcelink.is-white:not(body){background-color:#fff;color:#0a0a0a}.tag.is-black:not(body),.content kbd.is-black:not(body),.docstring>section>a.docs-sourcelink.is-black:not(body){background-color:#0a0a0a;color:#fff}.tag.is-light:not(body),.content kbd.is-light:not(body),.docstring>section>a.docs-sourcelink.is-light:not(body){background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.tag.is-dark:not(body),.content kbd:not(body),.docstring>section>a.docs-sourcelink.is-dark:not(body),.content .docstring>section>kbd:not(body){background-color:#363636;color:#fff}.tag.is-primary:not(body),.content kbd.is-primary:not(body),.docstring>section>a.docs-sourcelink:not(body){background-color:#4eb5de;color:#fff}.tag.is-primary.is-light:not(body),.content kbd.is-primary.is-light:not(body),.docstring>section>a.docs-sourcelink.is-light:not(body){background-color:#eef8fc;color:#1a6d8e}.tag.is-link:not(body),.content kbd.is-link:not(body),.docstring>section>a.docs-sourcelink.is-link:not(body){background-color:#2e63b8;color:#fff}.tag.is-link.is-light:not(body),.content kbd.is-link.is-light:not(body),.docstring>section>a.docs-sourcelink.is-link.is-light:not(body){background-color:#eff3fb;color:#3169c4}.tag.is-info:not(body),.content kbd.is-info:not(body),.docstring>section>a.docs-sourcelink.is-info:not(body){background-color:#209cee;color:#fff}.tag.is-info.is-light:not(body),.content kbd.is-info.is-light:not(body),.docstring>section>a.docs-sourcelink.is-info.is-light:not(body){background-color:#ecf7fe;color:#0e72b4}.tag.is-success:not(body),.content kbd.is-success:not(body),.docstring>section>a.docs-sourcelink.is-success:not(body){background-color:#22c35b;color:#fff}.tag.is-success.is-light:not(body),.content kbd.is-success.is-light:not(body),.docstring>section>a.docs-sourcelink.is-success.is-light:not(body){background-color:#eefcf3;color:#198f43}.tag.is-warning:not(body),.content kbd.is-warning:not(body),.docstring>section>a.docs-sourcelink.is-warning:not(body){background-color:#ffdd57;color:rgba(0,0,0,0.7)}.tag.is-warning.is-light:not(body),.content kbd.is-warning.is-light:not(body),.docstring>section>a.docs-sourcelink.is-warning.is-light:not(body){background-color:#fffbeb;color:#947600}.tag.is-danger:not(body),.content kbd.is-danger:not(body),.docstring>section>a.docs-sourcelink.is-danger:not(body){background-color:#da0b00;color:#fff}.tag.is-danger.is-light:not(body),.content kbd.is-danger.is-light:not(body),.docstring>section>a.docs-sourcelink.is-danger.is-light:not(body){background-color:#ffeceb;color:#f50c00}.tag.is-normal:not(body),.content kbd.is-normal:not(body),.docstring>section>a.docs-sourcelink.is-normal:not(body){font-size:.75rem}.tag.is-medium:not(body),.content kbd.is-medium:not(body),.docstring>section>a.docs-sourcelink.is-medium:not(body){font-size:1rem}.tag.is-large:not(body),.content kbd.is-large:not(body),.docstring>section>a.docs-sourcelink.is-large:not(body){font-size:1.25rem}.tag:not(body) .icon:first-child:not(:last-child),.content kbd:not(body) .icon:first-child:not(:last-child),.docstring>section>a.docs-sourcelink:not(body) .icon:first-child:not(:last-child){margin-left:-.375em;margin-right:.1875em}.tag:not(body) .icon:last-child:not(:first-child),.content kbd:not(body) .icon:last-child:not(:first-child),.docstring>section>a.docs-sourcelink:not(body) .icon:last-child:not(:first-child){margin-left:.1875em;margin-right:-.375em}.tag:not(body) .icon:first-child:last-child,.content kbd:not(body) .icon:first-child:last-child,.docstring>section>a.docs-sourcelink:not(body) .icon:first-child:last-child{margin-left:-.375em;margin-right:-.375em}.tag.is-delete:not(body),.content kbd.is-delete:not(body),.docstring>section>a.docs-sourcelink.is-delete:not(body){margin-left:1px;padding:0;position:relative;width:2em}.tag.is-delete:not(body)::before,.content kbd.is-delete:not(body)::before,.docstring>section>a.docs-sourcelink.is-delete:not(body)::before,.tag.is-delete:not(body)::after,.content kbd.is-delete:not(body)::after,.docstring>section>a.docs-sourcelink.is-delete:not(body)::after{background-color:currentColor;content:"";display:block;left:50%;position:absolute;top:50%;transform:translateX(-50%) translateY(-50%) rotate(45deg);transform-origin:center center}.tag.is-delete:not(body)::before,.content kbd.is-delete:not(body)::before,.docstring>section>a.docs-sourcelink.is-delete:not(body)::before{height:1px;width:50%}.tag.is-delete:not(body)::after,.content kbd.is-delete:not(body)::after,.docstring>section>a.docs-sourcelink.is-delete:not(body)::after{height:50%;width:1px}.tag.is-delete:not(body):hover,.content kbd.is-delete:not(body):hover,.docstring>section>a.docs-sourcelink.is-delete:not(body):hover,.tag.is-delete:not(body):focus,.content kbd.is-delete:not(body):focus,.docstring>section>a.docs-sourcelink.is-delete:not(body):focus{background-color:#e8e8e8}.tag.is-delete:not(body):active,.content kbd.is-delete:not(body):active,.docstring>section>a.docs-sourcelink.is-delete:not(body):active{background-color:#dbdbdb}.tag.is-rounded:not(body),#documenter .docs-sidebar form.docs-search>input:not(body),.content kbd.is-rounded:not(body),#documenter .docs-sidebar .content form.docs-search>input:not(body),.docstring>section>a.docs-sourcelink.is-rounded:not(body){border-radius:9999px}a.tag:hover,.docstring>section>a.docs-sourcelink:hover{text-decoration:underline}.title,.subtitle{word-break:break-word}.title em,.title span,.subtitle em,.subtitle span{font-weight:inherit}.title sub,.subtitle sub{font-size:.75em}.title sup,.subtitle sup{font-size:.75em}.title .tag,.title .content kbd,.content .title kbd,.title .docstring>section>a.docs-sourcelink,.subtitle .tag,.subtitle .content kbd,.content .subtitle kbd,.subtitle .docstring>section>a.docs-sourcelink{vertical-align:middle}.title{color:#222;font-size:2rem;font-weight:600;line-height:1.125}.title strong{color:inherit;font-weight:inherit}.title:not(.is-spaced)+.subtitle{margin-top:-1.25rem}.title.is-1{font-size:3rem}.title.is-2{font-size:2.5rem}.title.is-3{font-size:2rem}.title.is-4{font-size:1.5rem}.title.is-5{font-size:1.25rem}.title.is-6{font-size:1rem}.title.is-7{font-size:.75rem}.subtitle{color:#222;font-size:1.25rem;font-weight:400;line-height:1.25}.subtitle strong{color:#222;font-weight:600}.subtitle:not(.is-spaced)+.title{margin-top:-1.25rem}.subtitle.is-1{font-size:3rem}.subtitle.is-2{font-size:2.5rem}.subtitle.is-3{font-size:2rem}.subtitle.is-4{font-size:1.5rem}.subtitle.is-5{font-size:1.25rem}.subtitle.is-6{font-size:1rem}.subtitle.is-7{font-size:.75rem}.heading{display:block;font-size:11px;letter-spacing:1px;margin-bottom:5px;text-transform:uppercase}.number{align-items:center;background-color:#f5f5f5;border-radius:9999px;display:inline-flex;font-size:1.25rem;height:2em;justify-content:center;margin-right:1.5rem;min-width:2.5em;padding:0.25rem 0.5rem;text-align:center;vertical-align:top}.select select,.textarea,.input,#documenter .docs-sidebar form.docs-search>input{background-color:#fff;border-color:#dbdbdb;border-radius:4px;color:#222}.select select::-moz-placeholder,.textarea::-moz-placeholder,.input::-moz-placeholder,#documenter .docs-sidebar form.docs-search>input::-moz-placeholder{color:#707070}.select select::-webkit-input-placeholder,.textarea::-webkit-input-placeholder,.input::-webkit-input-placeholder,#documenter .docs-sidebar form.docs-search>input::-webkit-input-placeholder{color:#707070}.select select:-moz-placeholder,.textarea:-moz-placeholder,.input:-moz-placeholder,#documenter .docs-sidebar form.docs-search>input:-moz-placeholder{color:#707070}.select select:-ms-input-placeholder,.textarea:-ms-input-placeholder,.input:-ms-input-placeholder,#documenter .docs-sidebar form.docs-search>input:-ms-input-placeholder{color:#707070}.select select:hover,.textarea:hover,.input:hover,#documenter .docs-sidebar form.docs-search>input:hover,.select select.is-hovered,.is-hovered.textarea,.is-hovered.input,#documenter .docs-sidebar form.docs-search>input.is-hovered{border-color:#b5b5b5}.select select:focus,.textarea:focus,.input:focus,#documenter .docs-sidebar form.docs-search>input:focus,.select select.is-focused,.is-focused.textarea,.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.select select:active,.textarea:active,.input:active,#documenter .docs-sidebar form.docs-search>input:active,.select select.is-active,.is-active.textarea,.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{border-color:#2e63b8;box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.select select[disabled],.textarea[disabled],.input[disabled],#documenter .docs-sidebar form.docs-search>input[disabled],fieldset[disabled] .select select,.select fieldset[disabled] select,fieldset[disabled] .textarea,fieldset[disabled] .input,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input{background-color:#f5f5f5;border-color:#f5f5f5;box-shadow:none;color:#6b6b6b}.select select[disabled]::-moz-placeholder,.textarea[disabled]::-moz-placeholder,.input[disabled]::-moz-placeholder,#documenter .docs-sidebar form.docs-search>input[disabled]::-moz-placeholder,fieldset[disabled] .select select::-moz-placeholder,.select fieldset[disabled] select::-moz-placeholder,fieldset[disabled] .textarea::-moz-placeholder,fieldset[disabled] .input::-moz-placeholder,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input::-moz-placeholder,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input::-moz-placeholder{color:rgba(107,107,107,0.3)}.select select[disabled]::-webkit-input-placeholder,.textarea[disabled]::-webkit-input-placeholder,.input[disabled]::-webkit-input-placeholder,#documenter .docs-sidebar form.docs-search>input[disabled]::-webkit-input-placeholder,fieldset[disabled] .select select::-webkit-input-placeholder,.select fieldset[disabled] select::-webkit-input-placeholder,fieldset[disabled] .textarea::-webkit-input-placeholder,fieldset[disabled] .input::-webkit-input-placeholder,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input::-webkit-input-placeholder,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input::-webkit-input-placeholder{color:rgba(107,107,107,0.3)}.select select[disabled]:-moz-placeholder,.textarea[disabled]:-moz-placeholder,.input[disabled]:-moz-placeholder,#documenter .docs-sidebar form.docs-search>input[disabled]:-moz-placeholder,fieldset[disabled] .select select:-moz-placeholder,.select fieldset[disabled] select:-moz-placeholder,fieldset[disabled] .textarea:-moz-placeholder,fieldset[disabled] .input:-moz-placeholder,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input:-moz-placeholder,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input:-moz-placeholder{color:rgba(107,107,107,0.3)}.select select[disabled]:-ms-input-placeholder,.textarea[disabled]:-ms-input-placeholder,.input[disabled]:-ms-input-placeholder,#documenter .docs-sidebar form.docs-search>input[disabled]:-ms-input-placeholder,fieldset[disabled] .select select:-ms-input-placeholder,.select fieldset[disabled] select:-ms-input-placeholder,fieldset[disabled] .textarea:-ms-input-placeholder,fieldset[disabled] .input:-ms-input-placeholder,fieldset[disabled] #documenter .docs-sidebar form.docs-search>input:-ms-input-placeholder,#documenter .docs-sidebar fieldset[disabled] form.docs-search>input:-ms-input-placeholder{color:rgba(107,107,107,0.3)}.textarea,.input,#documenter .docs-sidebar form.docs-search>input{box-shadow:inset 0 0.0625em 0.125em rgba(10,10,10,0.05);max-width:100%;width:100%}.textarea[readonly],.input[readonly],#documenter .docs-sidebar form.docs-search>input[readonly]{box-shadow:none}.is-white.textarea,.is-white.input,#documenter .docs-sidebar form.docs-search>input.is-white{border-color:#fff}.is-white.textarea:focus,.is-white.input:focus,#documenter .docs-sidebar form.docs-search>input.is-white:focus,.is-white.is-focused.textarea,.is-white.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-white.textarea:active,.is-white.input:active,#documenter .docs-sidebar form.docs-search>input.is-white:active,.is-white.is-active.textarea,.is-white.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}.is-black.textarea,.is-black.input,#documenter .docs-sidebar form.docs-search>input.is-black{border-color:#0a0a0a}.is-black.textarea:focus,.is-black.input:focus,#documenter .docs-sidebar form.docs-search>input.is-black:focus,.is-black.is-focused.textarea,.is-black.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-black.textarea:active,.is-black.input:active,#documenter .docs-sidebar form.docs-search>input.is-black:active,.is-black.is-active.textarea,.is-black.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}.is-light.textarea,.is-light.input,#documenter .docs-sidebar form.docs-search>input.is-light{border-color:#f5f5f5}.is-light.textarea:focus,.is-light.input:focus,#documenter .docs-sidebar form.docs-search>input.is-light:focus,.is-light.is-focused.textarea,.is-light.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-light.textarea:active,.is-light.input:active,#documenter .docs-sidebar form.docs-search>input.is-light:active,.is-light.is-active.textarea,.is-light.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(245,245,245,0.25)}.is-dark.textarea,.content kbd.textarea,.is-dark.input,#documenter .docs-sidebar form.docs-search>input.is-dark,.content kbd.input{border-color:#363636}.is-dark.textarea:focus,.content kbd.textarea:focus,.is-dark.input:focus,#documenter .docs-sidebar form.docs-search>input.is-dark:focus,.content kbd.input:focus,.is-dark.is-focused.textarea,.content kbd.is-focused.textarea,.is-dark.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.content kbd.is-focused.input,#documenter .docs-sidebar .content form.docs-search>input.is-focused,.is-dark.textarea:active,.content kbd.textarea:active,.is-dark.input:active,#documenter .docs-sidebar form.docs-search>input.is-dark:active,.content kbd.input:active,.is-dark.is-active.textarea,.content kbd.is-active.textarea,.is-dark.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active,.content kbd.is-active.input,#documenter .docs-sidebar .content form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(54,54,54,0.25)}.is-primary.textarea,.docstring>section>a.textarea.docs-sourcelink,.is-primary.input,#documenter .docs-sidebar form.docs-search>input.is-primary,.docstring>section>a.input.docs-sourcelink{border-color:#4eb5de}.is-primary.textarea:focus,.docstring>section>a.textarea.docs-sourcelink:focus,.is-primary.input:focus,#documenter .docs-sidebar form.docs-search>input.is-primary:focus,.docstring>section>a.input.docs-sourcelink:focus,.is-primary.is-focused.textarea,.docstring>section>a.is-focused.textarea.docs-sourcelink,.is-primary.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.docstring>section>a.is-focused.input.docs-sourcelink,.is-primary.textarea:active,.docstring>section>a.textarea.docs-sourcelink:active,.is-primary.input:active,#documenter .docs-sidebar form.docs-search>input.is-primary:active,.docstring>section>a.input.docs-sourcelink:active,.is-primary.is-active.textarea,.docstring>section>a.is-active.textarea.docs-sourcelink,.is-primary.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active,.docstring>section>a.is-active.input.docs-sourcelink{box-shadow:0 0 0 0.125em rgba(78,181,222,0.25)}.is-link.textarea,.is-link.input,#documenter .docs-sidebar form.docs-search>input.is-link{border-color:#2e63b8}.is-link.textarea:focus,.is-link.input:focus,#documenter .docs-sidebar form.docs-search>input.is-link:focus,.is-link.is-focused.textarea,.is-link.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-link.textarea:active,.is-link.input:active,#documenter .docs-sidebar form.docs-search>input.is-link:active,.is-link.is-active.textarea,.is-link.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.is-info.textarea,.is-info.input,#documenter .docs-sidebar form.docs-search>input.is-info{border-color:#209cee}.is-info.textarea:focus,.is-info.input:focus,#documenter .docs-sidebar form.docs-search>input.is-info:focus,.is-info.is-focused.textarea,.is-info.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-info.textarea:active,.is-info.input:active,#documenter .docs-sidebar form.docs-search>input.is-info:active,.is-info.is-active.textarea,.is-info.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(32,156,238,0.25)}.is-success.textarea,.is-success.input,#documenter .docs-sidebar form.docs-search>input.is-success{border-color:#22c35b}.is-success.textarea:focus,.is-success.input:focus,#documenter .docs-sidebar form.docs-search>input.is-success:focus,.is-success.is-focused.textarea,.is-success.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-success.textarea:active,.is-success.input:active,#documenter .docs-sidebar form.docs-search>input.is-success:active,.is-success.is-active.textarea,.is-success.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(34,195,91,0.25)}.is-warning.textarea,.is-warning.input,#documenter .docs-sidebar form.docs-search>input.is-warning{border-color:#ffdd57}.is-warning.textarea:focus,.is-warning.input:focus,#documenter .docs-sidebar form.docs-search>input.is-warning:focus,.is-warning.is-focused.textarea,.is-warning.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-warning.textarea:active,.is-warning.input:active,#documenter .docs-sidebar form.docs-search>input.is-warning:active,.is-warning.is-active.textarea,.is-warning.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(255,221,87,0.25)}.is-danger.textarea,.is-danger.input,#documenter .docs-sidebar form.docs-search>input.is-danger{border-color:#da0b00}.is-danger.textarea:focus,.is-danger.input:focus,#documenter .docs-sidebar form.docs-search>input.is-danger:focus,.is-danger.is-focused.textarea,.is-danger.is-focused.input,#documenter .docs-sidebar form.docs-search>input.is-focused,.is-danger.textarea:active,.is-danger.input:active,#documenter .docs-sidebar form.docs-search>input.is-danger:active,.is-danger.is-active.textarea,.is-danger.is-active.input,#documenter .docs-sidebar form.docs-search>input.is-active{box-shadow:0 0 0 0.125em rgba(218,11,0,0.25)}.is-small.textarea,.is-small.input,#documenter .docs-sidebar form.docs-search>input{border-radius:2px;font-size:.75rem}.is-medium.textarea,.is-medium.input,#documenter .docs-sidebar form.docs-search>input.is-medium{font-size:1.25rem}.is-large.textarea,.is-large.input,#documenter .docs-sidebar form.docs-search>input.is-large{font-size:1.5rem}.is-fullwidth.textarea,.is-fullwidth.input,#documenter .docs-sidebar form.docs-search>input.is-fullwidth{display:block;width:100%}.is-inline.textarea,.is-inline.input,#documenter .docs-sidebar form.docs-search>input.is-inline{display:inline;width:auto}.input.is-rounded,#documenter .docs-sidebar form.docs-search>input{border-radius:9999px;padding-left:calc(calc(0.75em - 1px) + 0.375em);padding-right:calc(calc(0.75em - 1px) + 0.375em)}.input.is-static,#documenter .docs-sidebar form.docs-search>input.is-static{background-color:transparent;border-color:transparent;box-shadow:none;padding-left:0;padding-right:0}.textarea{display:block;max-width:100%;min-width:100%;padding:calc(0.75em - 1px);resize:vertical}.textarea:not([rows]){max-height:40em;min-height:8em}.textarea[rows]{height:initial}.textarea.has-fixed-size{resize:none}.radio,.checkbox{cursor:pointer;display:inline-block;line-height:1.25;position:relative}.radio input,.checkbox input{cursor:pointer}.radio:hover,.checkbox:hover{color:#222}.radio[disabled],.checkbox[disabled],fieldset[disabled] .radio,fieldset[disabled] .checkbox,.radio input[disabled],.checkbox input[disabled]{color:#6b6b6b;cursor:not-allowed}.radio+.radio{margin-left:.5em}.select{display:inline-block;max-width:100%;position:relative;vertical-align:top}.select:not(.is-multiple){height:2.5em}.select:not(.is-multiple):not(.is-loading)::after{border-color:#2e63b8;right:1.125em;z-index:4}.select.is-rounded select,#documenter .docs-sidebar form.docs-search>input.select select{border-radius:9999px;padding-left:1em}.select select{cursor:pointer;display:block;font-size:1em;max-width:100%;outline:none}.select select::-ms-expand{display:none}.select select[disabled]:hover,fieldset[disabled] .select select:hover{border-color:#f5f5f5}.select select:not([multiple]){padding-right:2.5em}.select select[multiple]{height:auto;padding:0}.select select[multiple] option{padding:0.5em 1em}.select:not(.is-multiple):not(.is-loading):hover::after{border-color:#222}.select.is-white:not(:hover)::after{border-color:#fff}.select.is-white select{border-color:#fff}.select.is-white select:hover,.select.is-white select.is-hovered{border-color:#f2f2f2}.select.is-white select:focus,.select.is-white select.is-focused,.select.is-white select:active,.select.is-white select.is-active{box-shadow:0 0 0 0.125em rgba(255,255,255,0.25)}.select.is-black:not(:hover)::after{border-color:#0a0a0a}.select.is-black select{border-color:#0a0a0a}.select.is-black select:hover,.select.is-black select.is-hovered{border-color:#000}.select.is-black select:focus,.select.is-black select.is-focused,.select.is-black select:active,.select.is-black select.is-active{box-shadow:0 0 0 0.125em rgba(10,10,10,0.25)}.select.is-light:not(:hover)::after{border-color:#f5f5f5}.select.is-light select{border-color:#f5f5f5}.select.is-light select:hover,.select.is-light select.is-hovered{border-color:#e8e8e8}.select.is-light select:focus,.select.is-light select.is-focused,.select.is-light select:active,.select.is-light select.is-active{box-shadow:0 0 0 0.125em rgba(245,245,245,0.25)}.select.is-dark:not(:hover)::after,.content kbd.select:not(:hover)::after{border-color:#363636}.select.is-dark select,.content kbd.select select{border-color:#363636}.select.is-dark select:hover,.content kbd.select select:hover,.select.is-dark select.is-hovered,.content kbd.select select.is-hovered{border-color:#292929}.select.is-dark select:focus,.content kbd.select select:focus,.select.is-dark select.is-focused,.content kbd.select select.is-focused,.select.is-dark select:active,.content kbd.select select:active,.select.is-dark select.is-active,.content kbd.select select.is-active{box-shadow:0 0 0 0.125em rgba(54,54,54,0.25)}.select.is-primary:not(:hover)::after,.docstring>section>a.select.docs-sourcelink:not(:hover)::after{border-color:#4eb5de}.select.is-primary select,.docstring>section>a.select.docs-sourcelink select{border-color:#4eb5de}.select.is-primary select:hover,.docstring>section>a.select.docs-sourcelink select:hover,.select.is-primary select.is-hovered,.docstring>section>a.select.docs-sourcelink select.is-hovered{border-color:#39acda}.select.is-primary select:focus,.docstring>section>a.select.docs-sourcelink select:focus,.select.is-primary select.is-focused,.docstring>section>a.select.docs-sourcelink select.is-focused,.select.is-primary select:active,.docstring>section>a.select.docs-sourcelink select:active,.select.is-primary select.is-active,.docstring>section>a.select.docs-sourcelink select.is-active{box-shadow:0 0 0 0.125em rgba(78,181,222,0.25)}.select.is-link:not(:hover)::after{border-color:#2e63b8}.select.is-link select{border-color:#2e63b8}.select.is-link select:hover,.select.is-link select.is-hovered{border-color:#2958a4}.select.is-link select:focus,.select.is-link select.is-focused,.select.is-link select:active,.select.is-link select.is-active{box-shadow:0 0 0 0.125em rgba(46,99,184,0.25)}.select.is-info:not(:hover)::after{border-color:#209cee}.select.is-info select{border-color:#209cee}.select.is-info select:hover,.select.is-info select.is-hovered{border-color:#1190e3}.select.is-info select:focus,.select.is-info select.is-focused,.select.is-info select:active,.select.is-info select.is-active{box-shadow:0 0 0 0.125em rgba(32,156,238,0.25)}.select.is-success:not(:hover)::after{border-color:#22c35b}.select.is-success select{border-color:#22c35b}.select.is-success select:hover,.select.is-success select.is-hovered{border-color:#1ead51}.select.is-success select:focus,.select.is-success select.is-focused,.select.is-success select:active,.select.is-success select.is-active{box-shadow:0 0 0 0.125em rgba(34,195,91,0.25)}.select.is-warning:not(:hover)::after{border-color:#ffdd57}.select.is-warning select{border-color:#ffdd57}.select.is-warning select:hover,.select.is-warning select.is-hovered{border-color:#ffd83e}.select.is-warning select:focus,.select.is-warning select.is-focused,.select.is-warning select:active,.select.is-warning select.is-active{box-shadow:0 0 0 0.125em rgba(255,221,87,0.25)}.select.is-danger:not(:hover)::after{border-color:#da0b00}.select.is-danger select{border-color:#da0b00}.select.is-danger select:hover,.select.is-danger select.is-hovered{border-color:#c10a00}.select.is-danger select:focus,.select.is-danger select.is-focused,.select.is-danger select:active,.select.is-danger select.is-active{box-shadow:0 0 0 0.125em rgba(218,11,0,0.25)}.select.is-small,#documenter .docs-sidebar form.docs-search>input.select{border-radius:2px;font-size:.75rem}.select.is-medium{font-size:1.25rem}.select.is-large{font-size:1.5rem}.select.is-disabled::after{border-color:#6b6b6b !important;opacity:0.5}.select.is-fullwidth{width:100%}.select.is-fullwidth select{width:100%}.select.is-loading::after{margin-top:0;position:absolute;right:.625em;top:0.625em;transform:none}.select.is-loading.is-small:after,#documenter .docs-sidebar form.docs-search>input.is-loading:after{font-size:.75rem}.select.is-loading.is-medium:after{font-size:1.25rem}.select.is-loading.is-large:after{font-size:1.5rem}.file{align-items:stretch;display:flex;justify-content:flex-start;position:relative}.file.is-white .file-cta{background-color:#fff;border-color:transparent;color:#0a0a0a}.file.is-white:hover .file-cta,.file.is-white.is-hovered .file-cta{background-color:#f9f9f9;border-color:transparent;color:#0a0a0a}.file.is-white:focus .file-cta,.file.is-white.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(255,255,255,0.25);color:#0a0a0a}.file.is-white:active .file-cta,.file.is-white.is-active .file-cta{background-color:#f2f2f2;border-color:transparent;color:#0a0a0a}.file.is-black .file-cta{background-color:#0a0a0a;border-color:transparent;color:#fff}.file.is-black:hover .file-cta,.file.is-black.is-hovered .file-cta{background-color:#040404;border-color:transparent;color:#fff}.file.is-black:focus .file-cta,.file.is-black.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(10,10,10,0.25);color:#fff}.file.is-black:active .file-cta,.file.is-black.is-active .file-cta{background-color:#000;border-color:transparent;color:#fff}.file.is-light .file-cta{background-color:#f5f5f5;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-light:hover .file-cta,.file.is-light.is-hovered .file-cta{background-color:#eee;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-light:focus .file-cta,.file.is-light.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(245,245,245,0.25);color:rgba(0,0,0,0.7)}.file.is-light:active .file-cta,.file.is-light.is-active .file-cta{background-color:#e8e8e8;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-dark .file-cta,.content kbd.file .file-cta{background-color:#363636;border-color:transparent;color:#fff}.file.is-dark:hover .file-cta,.content kbd.file:hover .file-cta,.file.is-dark.is-hovered .file-cta,.content kbd.file.is-hovered .file-cta{background-color:#2f2f2f;border-color:transparent;color:#fff}.file.is-dark:focus .file-cta,.content kbd.file:focus .file-cta,.file.is-dark.is-focused .file-cta,.content kbd.file.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(54,54,54,0.25);color:#fff}.file.is-dark:active .file-cta,.content kbd.file:active .file-cta,.file.is-dark.is-active .file-cta,.content kbd.file.is-active .file-cta{background-color:#292929;border-color:transparent;color:#fff}.file.is-primary .file-cta,.docstring>section>a.file.docs-sourcelink .file-cta{background-color:#4eb5de;border-color:transparent;color:#fff}.file.is-primary:hover .file-cta,.docstring>section>a.file.docs-sourcelink:hover .file-cta,.file.is-primary.is-hovered .file-cta,.docstring>section>a.file.is-hovered.docs-sourcelink .file-cta{background-color:#43b1dc;border-color:transparent;color:#fff}.file.is-primary:focus .file-cta,.docstring>section>a.file.docs-sourcelink:focus .file-cta,.file.is-primary.is-focused .file-cta,.docstring>section>a.file.is-focused.docs-sourcelink .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(78,181,222,0.25);color:#fff}.file.is-primary:active .file-cta,.docstring>section>a.file.docs-sourcelink:active .file-cta,.file.is-primary.is-active .file-cta,.docstring>section>a.file.is-active.docs-sourcelink .file-cta{background-color:#39acda;border-color:transparent;color:#fff}.file.is-link .file-cta{background-color:#2e63b8;border-color:transparent;color:#fff}.file.is-link:hover .file-cta,.file.is-link.is-hovered .file-cta{background-color:#2b5eae;border-color:transparent;color:#fff}.file.is-link:focus .file-cta,.file.is-link.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(46,99,184,0.25);color:#fff}.file.is-link:active .file-cta,.file.is-link.is-active .file-cta{background-color:#2958a4;border-color:transparent;color:#fff}.file.is-info .file-cta{background-color:#209cee;border-color:transparent;color:#fff}.file.is-info:hover .file-cta,.file.is-info.is-hovered .file-cta{background-color:#1497ed;border-color:transparent;color:#fff}.file.is-info:focus .file-cta,.file.is-info.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(32,156,238,0.25);color:#fff}.file.is-info:active .file-cta,.file.is-info.is-active .file-cta{background-color:#1190e3;border-color:transparent;color:#fff}.file.is-success .file-cta{background-color:#22c35b;border-color:transparent;color:#fff}.file.is-success:hover .file-cta,.file.is-success.is-hovered .file-cta{background-color:#20b856;border-color:transparent;color:#fff}.file.is-success:focus .file-cta,.file.is-success.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(34,195,91,0.25);color:#fff}.file.is-success:active .file-cta,.file.is-success.is-active .file-cta{background-color:#1ead51;border-color:transparent;color:#fff}.file.is-warning .file-cta{background-color:#ffdd57;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-warning:hover .file-cta,.file.is-warning.is-hovered .file-cta{background-color:#ffda4a;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-warning:focus .file-cta,.file.is-warning.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(255,221,87,0.25);color:rgba(0,0,0,0.7)}.file.is-warning:active .file-cta,.file.is-warning.is-active .file-cta{background-color:#ffd83e;border-color:transparent;color:rgba(0,0,0,0.7)}.file.is-danger .file-cta{background-color:#da0b00;border-color:transparent;color:#fff}.file.is-danger:hover .file-cta,.file.is-danger.is-hovered .file-cta{background-color:#cd0a00;border-color:transparent;color:#fff}.file.is-danger:focus .file-cta,.file.is-danger.is-focused .file-cta{border-color:transparent;box-shadow:0 0 0.5em rgba(218,11,0,0.25);color:#fff}.file.is-danger:active .file-cta,.file.is-danger.is-active .file-cta{background-color:#c10a00;border-color:transparent;color:#fff}.file.is-small,#documenter .docs-sidebar form.docs-search>input.file{font-size:.75rem}.file.is-normal{font-size:1rem}.file.is-medium{font-size:1.25rem}.file.is-medium .file-icon .fa{font-size:21px}.file.is-large{font-size:1.5rem}.file.is-large .file-icon .fa{font-size:28px}.file.has-name .file-cta{border-bottom-right-radius:0;border-top-right-radius:0}.file.has-name .file-name{border-bottom-left-radius:0;border-top-left-radius:0}.file.has-name.is-empty .file-cta{border-radius:4px}.file.has-name.is-empty .file-name{display:none}.file.is-boxed .file-label{flex-direction:column}.file.is-boxed .file-cta{flex-direction:column;height:auto;padding:1em 3em}.file.is-boxed .file-name{border-width:0 1px 1px}.file.is-boxed .file-icon{height:1.5em;width:1.5em}.file.is-boxed .file-icon .fa{font-size:21px}.file.is-boxed.is-small .file-icon .fa,#documenter .docs-sidebar form.docs-search>input.is-boxed .file-icon .fa{font-size:14px}.file.is-boxed.is-medium .file-icon .fa{font-size:28px}.file.is-boxed.is-large .file-icon .fa{font-size:35px}.file.is-boxed.has-name .file-cta{border-radius:4px 4px 0 0}.file.is-boxed.has-name .file-name{border-radius:0 0 4px 4px;border-width:0 1px 1px}.file.is-centered{justify-content:center}.file.is-fullwidth .file-label{width:100%}.file.is-fullwidth .file-name{flex-grow:1;max-width:none}.file.is-right{justify-content:flex-end}.file.is-right .file-cta{border-radius:0 4px 4px 0}.file.is-right .file-name{border-radius:4px 0 0 4px;border-width:1px 0 1px 1px;order:-1}.file-label{align-items:stretch;display:flex;cursor:pointer;justify-content:flex-start;overflow:hidden;position:relative}.file-label:hover .file-cta{background-color:#eee;color:#222}.file-label:hover .file-name{border-color:#d5d5d5}.file-label:active .file-cta{background-color:#e8e8e8;color:#222}.file-label:active .file-name{border-color:#cfcfcf}.file-input{height:100%;left:0;opacity:0;outline:none;position:absolute;top:0;width:100%}.file-cta,.file-name{border-color:#dbdbdb;border-radius:4px;font-size:1em;padding-left:1em;padding-right:1em;white-space:nowrap}.file-cta{background-color:#f5f5f5;color:#222}.file-name{border-color:#dbdbdb;border-style:solid;border-width:1px 1px 1px 0;display:block;max-width:16em;overflow:hidden;text-align:inherit;text-overflow:ellipsis}.file-icon{align-items:center;display:flex;height:1em;justify-content:center;margin-right:.5em;width:1em}.file-icon .fa{font-size:14px}.label{color:#222;display:block;font-size:1rem;font-weight:700}.label:not(:last-child){margin-bottom:0.5em}.label.is-small,#documenter .docs-sidebar form.docs-search>input.label{font-size:.75rem}.label.is-medium{font-size:1.25rem}.label.is-large{font-size:1.5rem}.help{display:block;font-size:.75rem;margin-top:0.25rem}.help.is-white{color:#fff}.help.is-black{color:#0a0a0a}.help.is-light{color:#f5f5f5}.help.is-dark,.content kbd.help{color:#363636}.help.is-primary,.docstring>section>a.help.docs-sourcelink{color:#4eb5de}.help.is-link{color:#2e63b8}.help.is-info{color:#209cee}.help.is-success{color:#22c35b}.help.is-warning{color:#ffdd57}.help.is-danger{color:#da0b00}.field:not(:last-child){margin-bottom:0.75rem}.field.has-addons{display:flex;justify-content:flex-start}.field.has-addons .control:not(:last-child){margin-right:-1px}.field.has-addons .control:not(:first-child):not(:last-child) .button,.field.has-addons .control:not(:first-child):not(:last-child) .input,.field.has-addons .control:not(:first-child):not(:last-child) #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .field.has-addons .control:not(:first-child):not(:last-child) form.docs-search>input,.field.has-addons .control:not(:first-child):not(:last-child) .select select{border-radius:0}.field.has-addons .control:first-child:not(:only-child) .button,.field.has-addons .control:first-child:not(:only-child) .input,.field.has-addons .control:first-child:not(:only-child) #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .field.has-addons .control:first-child:not(:only-child) form.docs-search>input,.field.has-addons .control:first-child:not(:only-child) .select select{border-bottom-right-radius:0;border-top-right-radius:0}.field.has-addons .control:last-child:not(:only-child) .button,.field.has-addons .control:last-child:not(:only-child) .input,.field.has-addons .control:last-child:not(:only-child) #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .field.has-addons .control:last-child:not(:only-child) form.docs-search>input,.field.has-addons .control:last-child:not(:only-child) .select select{border-bottom-left-radius:0;border-top-left-radius:0}.field.has-addons .control .button:not([disabled]):hover,.field.has-addons .control .button.is-hovered:not([disabled]),.field.has-addons .control .input:not([disabled]):hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):hover,.field.has-addons .control .input.is-hovered:not([disabled]),.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-hovered:not([disabled]),#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-hovered:not([disabled]),.field.has-addons .control .select select:not([disabled]):hover,.field.has-addons .control .select select.is-hovered:not([disabled]){z-index:2}.field.has-addons .control .button:not([disabled]):focus,.field.has-addons .control .button.is-focused:not([disabled]),.field.has-addons .control .button:not([disabled]):active,.field.has-addons .control .button.is-active:not([disabled]),.field.has-addons .control .input:not([disabled]):focus,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):focus,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):focus,.field.has-addons .control .input.is-focused:not([disabled]),.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-focused:not([disabled]),#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-focused:not([disabled]),.field.has-addons .control .input:not([disabled]):active,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):active,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):active,.field.has-addons .control .input.is-active:not([disabled]),.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-active:not([disabled]),#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-active:not([disabled]),.field.has-addons .control .select select:not([disabled]):focus,.field.has-addons .control .select select.is-focused:not([disabled]),.field.has-addons .control .select select:not([disabled]):active,.field.has-addons .control .select select.is-active:not([disabled]){z-index:3}.field.has-addons .control .button:not([disabled]):focus:hover,.field.has-addons .control .button.is-focused:not([disabled]):hover,.field.has-addons .control .button:not([disabled]):active:hover,.field.has-addons .control .button.is-active:not([disabled]):hover,.field.has-addons .control .input:not([disabled]):focus:hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):focus:hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):focus:hover,.field.has-addons .control .input.is-focused:not([disabled]):hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-focused:not([disabled]):hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-focused:not([disabled]):hover,.field.has-addons .control .input:not([disabled]):active:hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input:not([disabled]):active:hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input:not([disabled]):active:hover,.field.has-addons .control .input.is-active:not([disabled]):hover,.field.has-addons .control #documenter .docs-sidebar form.docs-search>input.is-active:not([disabled]):hover,#documenter .docs-sidebar .field.has-addons .control form.docs-search>input.is-active:not([disabled]):hover,.field.has-addons .control .select select:not([disabled]):focus:hover,.field.has-addons .control .select select.is-focused:not([disabled]):hover,.field.has-addons .control .select select:not([disabled]):active:hover,.field.has-addons .control .select select.is-active:not([disabled]):hover{z-index:4}.field.has-addons .control.is-expanded{flex-grow:1;flex-shrink:1}.field.has-addons.has-addons-centered{justify-content:center}.field.has-addons.has-addons-right{justify-content:flex-end}.field.has-addons.has-addons-fullwidth .control{flex-grow:1;flex-shrink:0}.field.is-grouped{display:flex;justify-content:flex-start}.field.is-grouped>.control{flex-shrink:0}.field.is-grouped>.control:not(:last-child){margin-bottom:0;margin-right:.75rem}.field.is-grouped>.control.is-expanded{flex-grow:1;flex-shrink:1}.field.is-grouped.is-grouped-centered{justify-content:center}.field.is-grouped.is-grouped-right{justify-content:flex-end}.field.is-grouped.is-grouped-multiline{flex-wrap:wrap}.field.is-grouped.is-grouped-multiline>.control:last-child,.field.is-grouped.is-grouped-multiline>.control:not(:last-child){margin-bottom:0.75rem}.field.is-grouped.is-grouped-multiline:last-child{margin-bottom:-0.75rem}.field.is-grouped.is-grouped-multiline:not(:last-child){margin-bottom:0}@media screen and (min-width: 769px),print{.field.is-horizontal{display:flex}}.field-label .label{font-size:inherit}@media screen and (max-width: 768px){.field-label{margin-bottom:0.5rem}}@media screen and (min-width: 769px),print{.field-label{flex-basis:0;flex-grow:1;flex-shrink:0;margin-right:1.5rem;text-align:right}.field-label.is-small,#documenter .docs-sidebar form.docs-search>input.field-label{font-size:.75rem;padding-top:0.375em}.field-label.is-normal{padding-top:0.375em}.field-label.is-medium{font-size:1.25rem;padding-top:0.375em}.field-label.is-large{font-size:1.5rem;padding-top:0.375em}}.field-body .field .field{margin-bottom:0}@media screen and (min-width: 769px),print{.field-body{display:flex;flex-basis:0;flex-grow:5;flex-shrink:1}.field-body .field{margin-bottom:0}.field-body>.field{flex-shrink:1}.field-body>.field:not(.is-narrow){flex-grow:1}.field-body>.field:not(:last-child){margin-right:.75rem}}.control{box-sizing:border-box;clear:both;font-size:1rem;position:relative;text-align:inherit}.control.has-icons-left .input:focus~.icon,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input:focus~.icon,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input:focus~.icon,.control.has-icons-left .select:focus~.icon,.control.has-icons-right .input:focus~.icon,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input:focus~.icon,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input:focus~.icon,.control.has-icons-right .select:focus~.icon{color:#222}.control.has-icons-left .input.is-small~.icon,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input~.icon,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input~.icon,.control.has-icons-left .select.is-small~.icon,.control.has-icons-right .input.is-small~.icon,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input~.icon,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input~.icon,.control.has-icons-right .select.is-small~.icon{font-size:.75rem}.control.has-icons-left .input.is-medium~.icon,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input.is-medium~.icon,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input.is-medium~.icon,.control.has-icons-left .select.is-medium~.icon,.control.has-icons-right .input.is-medium~.icon,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input.is-medium~.icon,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input.is-medium~.icon,.control.has-icons-right .select.is-medium~.icon{font-size:1.25rem}.control.has-icons-left .input.is-large~.icon,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input.is-large~.icon,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input.is-large~.icon,.control.has-icons-left .select.is-large~.icon,.control.has-icons-right .input.is-large~.icon,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input.is-large~.icon,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input.is-large~.icon,.control.has-icons-right .select.is-large~.icon{font-size:1.5rem}.control.has-icons-left .icon,.control.has-icons-right .icon{color:#dbdbdb;height:2.5em;pointer-events:none;position:absolute;top:0;width:2.5em;z-index:4}.control.has-icons-left .input,.control.has-icons-left #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .control.has-icons-left form.docs-search>input,.control.has-icons-left .select select{padding-left:2.5em}.control.has-icons-left .icon.is-left{left:0}.control.has-icons-right .input,.control.has-icons-right #documenter .docs-sidebar form.docs-search>input,#documenter .docs-sidebar .control.has-icons-right form.docs-search>input,.control.has-icons-right .select select{padding-right:2.5em}.control.has-icons-right .icon.is-right{right:0}.control.is-loading::after{position:absolute !important;right:.625em;top:0.625em;z-index:4}.control.is-loading.is-small:after,#documenter .docs-sidebar form.docs-search>input.is-loading:after{font-size:.75rem}.control.is-loading.is-medium:after{font-size:1.25rem}.control.is-loading.is-large:after{font-size:1.5rem}.breadcrumb{font-size:1rem;white-space:nowrap}.breadcrumb a{align-items:center;color:#2e63b8;display:flex;justify-content:center;padding:0 .75em}.breadcrumb a:hover{color:#363636}.breadcrumb li{align-items:center;display:flex}.breadcrumb li:first-child a{padding-left:0}.breadcrumb li.is-active a{color:#222;cursor:default;pointer-events:none}.breadcrumb li+li::before{color:#b5b5b5;content:"\0002f"}.breadcrumb ul,.breadcrumb ol{align-items:flex-start;display:flex;flex-wrap:wrap;justify-content:flex-start}.breadcrumb .icon:first-child{margin-right:.5em}.breadcrumb .icon:last-child{margin-left:.5em}.breadcrumb.is-centered ol,.breadcrumb.is-centered ul{justify-content:center}.breadcrumb.is-right ol,.breadcrumb.is-right ul{justify-content:flex-end}.breadcrumb.is-small,#documenter .docs-sidebar form.docs-search>input.breadcrumb{font-size:.75rem}.breadcrumb.is-medium{font-size:1.25rem}.breadcrumb.is-large{font-size:1.5rem}.breadcrumb.has-arrow-separator li+li::before{content:"\02192"}.breadcrumb.has-bullet-separator li+li::before{content:"\02022"}.breadcrumb.has-dot-separator li+li::before{content:"\000b7"}.breadcrumb.has-succeeds-separator li+li::before{content:"\0227B"}.card{background-color:#fff;border-radius:.25rem;box-shadow:#bbb;color:#222;max-width:100%;position:relative}.card-footer:first-child,.card-content:first-child,.card-header:first-child{border-top-left-radius:.25rem;border-top-right-radius:.25rem}.card-footer:last-child,.card-content:last-child,.card-header:last-child{border-bottom-left-radius:.25rem;border-bottom-right-radius:.25rem}.card-header{background-color:rgba(0,0,0,0);align-items:stretch;box-shadow:0 0.125em 0.25em rgba(10,10,10,0.1);display:flex}.card-header-title{align-items:center;color:#222;display:flex;flex-grow:1;font-weight:700;padding:0.75rem 1rem}.card-header-title.is-centered{justify-content:center}.card-header-icon{-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;color:currentColor;font-family:inherit;font-size:1em;margin:0;padding:0;align-items:center;cursor:pointer;display:flex;justify-content:center;padding:0.75rem 1rem}.card-image{display:block;position:relative}.card-image:first-child img{border-top-left-radius:.25rem;border-top-right-radius:.25rem}.card-image:last-child img{border-bottom-left-radius:.25rem;border-bottom-right-radius:.25rem}.card-content{background-color:rgba(0,0,0,0);padding:1.5rem}.card-footer{background-color:rgba(0,0,0,0);border-top:1px solid #ededed;align-items:stretch;display:flex}.card-footer-item{align-items:center;display:flex;flex-basis:0;flex-grow:1;flex-shrink:0;justify-content:center;padding:.75rem}.card-footer-item:not(:last-child){border-right:1px solid #ededed}.card .media:not(:last-child){margin-bottom:1.5rem}.dropdown{display:inline-flex;position:relative;vertical-align:top}.dropdown.is-active .dropdown-menu,.dropdown.is-hoverable:hover .dropdown-menu{display:block}.dropdown.is-right .dropdown-menu{left:auto;right:0}.dropdown.is-up .dropdown-menu{bottom:100%;padding-bottom:4px;padding-top:initial;top:auto}.dropdown-menu{display:none;left:0;min-width:12rem;padding-top:4px;position:absolute;top:100%;z-index:20}.dropdown-content{background-color:#fff;border-radius:4px;box-shadow:#bbb;padding-bottom:.5rem;padding-top:.5rem}.dropdown-item{color:#222;display:block;font-size:0.875rem;line-height:1.5;padding:0.375rem 1rem;position:relative}a.dropdown-item,button.dropdown-item{padding-right:3rem;text-align:inherit;white-space:nowrap;width:100%}a.dropdown-item:hover,button.dropdown-item:hover{background-color:#f5f5f5;color:#0a0a0a}a.dropdown-item.is-active,button.dropdown-item.is-active{background-color:#2e63b8;color:#fff}.dropdown-divider{background-color:#ededed;border:none;display:block;height:1px;margin:0.5rem 0}.level{align-items:center;justify-content:space-between}.level code{border-radius:4px}.level img{display:inline-block;vertical-align:top}.level.is-mobile{display:flex}.level.is-mobile .level-left,.level.is-mobile .level-right{display:flex}.level.is-mobile .level-left+.level-right{margin-top:0}.level.is-mobile .level-item:not(:last-child){margin-bottom:0;margin-right:.75rem}.level.is-mobile .level-item:not(.is-narrow){flex-grow:1}@media screen and (min-width: 769px),print{.level{display:flex}.level>.level-item:not(.is-narrow){flex-grow:1}}.level-item{align-items:center;display:flex;flex-basis:auto;flex-grow:0;flex-shrink:0;justify-content:center}.level-item .title,.level-item .subtitle{margin-bottom:0}@media screen and (max-width: 768px){.level-item:not(:last-child){margin-bottom:.75rem}}.level-left,.level-right{flex-basis:auto;flex-grow:0;flex-shrink:0}.level-left .level-item.is-flexible,.level-right .level-item.is-flexible{flex-grow:1}@media screen and (min-width: 769px),print{.level-left .level-item:not(:last-child),.level-right .level-item:not(:last-child){margin-right:.75rem}}.level-left{align-items:center;justify-content:flex-start}@media screen and (max-width: 768px){.level-left+.level-right{margin-top:1.5rem}}@media screen and (min-width: 769px),print{.level-left{display:flex}}.level-right{align-items:center;justify-content:flex-end}@media screen and (min-width: 769px),print{.level-right{display:flex}}.media{align-items:flex-start;display:flex;text-align:inherit}.media .content:not(:last-child){margin-bottom:.75rem}.media .media{border-top:1px solid rgba(219,219,219,0.5);display:flex;padding-top:.75rem}.media .media .content:not(:last-child),.media .media .control:not(:last-child){margin-bottom:.5rem}.media .media .media{padding-top:.5rem}.media .media .media+.media{margin-top:.5rem}.media+.media{border-top:1px solid rgba(219,219,219,0.5);margin-top:1rem;padding-top:1rem}.media.is-large+.media{margin-top:1.5rem;padding-top:1.5rem}.media-left,.media-right{flex-basis:auto;flex-grow:0;flex-shrink:0}.media-left{margin-right:1rem}.media-right{margin-left:1rem}.media-content{flex-basis:auto;flex-grow:1;flex-shrink:1;text-align:inherit}@media screen and (max-width: 768px){.media-content{overflow-x:auto}}.menu{font-size:1rem}.menu.is-small,#documenter .docs-sidebar form.docs-search>input.menu{font-size:.75rem}.menu.is-medium{font-size:1.25rem}.menu.is-large{font-size:1.5rem}.menu-list{line-height:1.25}.menu-list a{border-radius:2px;color:#222;display:block;padding:0.5em 0.75em}.menu-list a:hover{background-color:#f5f5f5;color:#222}.menu-list a.is-active{background-color:#2e63b8;color:#fff}.menu-list li ul{border-left:1px solid #dbdbdb;margin:.75em;padding-left:.75em}.menu-label{color:#6b6b6b;font-size:.75em;letter-spacing:.1em;text-transform:uppercase}.menu-label:not(:first-child){margin-top:1em}.menu-label:not(:last-child){margin-bottom:1em}.message{background-color:#f5f5f5;border-radius:4px;font-size:1rem}.message strong{color:currentColor}.message a:not(.button):not(.tag):not(.dropdown-item){color:currentColor;text-decoration:underline}.message.is-small,#documenter .docs-sidebar form.docs-search>input.message{font-size:.75rem}.message.is-medium{font-size:1.25rem}.message.is-large{font-size:1.5rem}.message.is-white{background-color:#fff}.message.is-white .message-header{background-color:#fff;color:#0a0a0a}.message.is-white .message-body{border-color:#fff}.message.is-black{background-color:#fafafa}.message.is-black .message-header{background-color:#0a0a0a;color:#fff}.message.is-black .message-body{border-color:#0a0a0a}.message.is-light{background-color:#fafafa}.message.is-light .message-header{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.message.is-light .message-body{border-color:#f5f5f5}.message.is-dark,.content kbd.message{background-color:#fafafa}.message.is-dark .message-header,.content kbd.message .message-header{background-color:#363636;color:#fff}.message.is-dark .message-body,.content kbd.message .message-body{border-color:#363636}.message.is-primary,.docstring>section>a.message.docs-sourcelink{background-color:#eef8fc}.message.is-primary .message-header,.docstring>section>a.message.docs-sourcelink .message-header{background-color:#4eb5de;color:#fff}.message.is-primary .message-body,.docstring>section>a.message.docs-sourcelink .message-body{border-color:#4eb5de;color:#1a6d8e}.message.is-link{background-color:#eff3fb}.message.is-link .message-header{background-color:#2e63b8;color:#fff}.message.is-link .message-body{border-color:#2e63b8;color:#3169c4}.message.is-info{background-color:#ecf7fe}.message.is-info .message-header{background-color:#209cee;color:#fff}.message.is-info .message-body{border-color:#209cee;color:#0e72b4}.message.is-success{background-color:#eefcf3}.message.is-success .message-header{background-color:#22c35b;color:#fff}.message.is-success .message-body{border-color:#22c35b;color:#198f43}.message.is-warning{background-color:#fffbeb}.message.is-warning .message-header{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.message.is-warning .message-body{border-color:#ffdd57;color:#947600}.message.is-danger{background-color:#ffeceb}.message.is-danger .message-header{background-color:#da0b00;color:#fff}.message.is-danger .message-body{border-color:#da0b00;color:#f50c00}.message-header{align-items:center;background-color:#222;border-radius:4px 4px 0 0;color:#fff;display:flex;font-weight:700;justify-content:space-between;line-height:1.25;padding:0.75em 1em;position:relative}.message-header .delete{flex-grow:0;flex-shrink:0;margin-left:.75em}.message-header+.message-body{border-width:0;border-top-left-radius:0;border-top-right-radius:0}.message-body{border-color:#dbdbdb;border-radius:4px;border-style:solid;border-width:0 0 0 4px;color:#222;padding:1.25em 1.5em}.message-body code,.message-body pre{background-color:#fff}.message-body pre code{background-color:rgba(0,0,0,0)}.modal{align-items:center;display:none;flex-direction:column;justify-content:center;overflow:hidden;position:fixed;z-index:40}.modal.is-active{display:flex}.modal-background{background-color:rgba(10,10,10,0.86)}.modal-content,.modal-card{margin:0 20px;max-height:calc(100vh - 160px);overflow:auto;position:relative;width:100%}@media screen and (min-width: 769px){.modal-content,.modal-card{margin:0 auto;max-height:calc(100vh - 40px);width:640px}}.modal-close{background:none;height:40px;position:fixed;right:20px;top:20px;width:40px}.modal-card{display:flex;flex-direction:column;max-height:calc(100vh - 40px);overflow:hidden;-ms-overflow-y:visible}.modal-card-head,.modal-card-foot{align-items:center;background-color:#f5f5f5;display:flex;flex-shrink:0;justify-content:flex-start;padding:20px;position:relative}.modal-card-head{border-bottom:1px solid #dbdbdb;border-top-left-radius:6px;border-top-right-radius:6px}.modal-card-title{color:#222;flex-grow:1;flex-shrink:0;font-size:1.5rem;line-height:1}.modal-card-foot{border-bottom-left-radius:6px;border-bottom-right-radius:6px;border-top:1px solid #dbdbdb}.modal-card-foot .button:not(:last-child){margin-right:.5em}.modal-card-body{-webkit-overflow-scrolling:touch;background-color:#fff;flex-grow:1;flex-shrink:1;overflow:auto;padding:20px}.navbar{background-color:#fff;min-height:3.25rem;position:relative;z-index:30}.navbar.is-white{background-color:#fff;color:#0a0a0a}.navbar.is-white .navbar-brand>.navbar-item,.navbar.is-white .navbar-brand .navbar-link{color:#0a0a0a}.navbar.is-white .navbar-brand>a.navbar-item:focus,.navbar.is-white .navbar-brand>a.navbar-item:hover,.navbar.is-white .navbar-brand>a.navbar-item.is-active,.navbar.is-white .navbar-brand .navbar-link:focus,.navbar.is-white .navbar-brand .navbar-link:hover,.navbar.is-white .navbar-brand .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}.navbar.is-white .navbar-brand .navbar-link::after{border-color:#0a0a0a}.navbar.is-white .navbar-burger{color:#0a0a0a}@media screen and (min-width: 1056px){.navbar.is-white .navbar-start>.navbar-item,.navbar.is-white .navbar-start .navbar-link,.navbar.is-white .navbar-end>.navbar-item,.navbar.is-white .navbar-end .navbar-link{color:#0a0a0a}.navbar.is-white .navbar-start>a.navbar-item:focus,.navbar.is-white .navbar-start>a.navbar-item:hover,.navbar.is-white .navbar-start>a.navbar-item.is-active,.navbar.is-white .navbar-start .navbar-link:focus,.navbar.is-white .navbar-start .navbar-link:hover,.navbar.is-white .navbar-start .navbar-link.is-active,.navbar.is-white .navbar-end>a.navbar-item:focus,.navbar.is-white .navbar-end>a.navbar-item:hover,.navbar.is-white .navbar-end>a.navbar-item.is-active,.navbar.is-white .navbar-end .navbar-link:focus,.navbar.is-white .navbar-end .navbar-link:hover,.navbar.is-white .navbar-end .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}.navbar.is-white .navbar-start .navbar-link::after,.navbar.is-white .navbar-end .navbar-link::after{border-color:#0a0a0a}.navbar.is-white .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-white .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-white .navbar-item.has-dropdown.is-active .navbar-link{background-color:#f2f2f2;color:#0a0a0a}.navbar.is-white .navbar-dropdown a.navbar-item.is-active{background-color:#fff;color:#0a0a0a}}.navbar.is-black{background-color:#0a0a0a;color:#fff}.navbar.is-black .navbar-brand>.navbar-item,.navbar.is-black .navbar-brand .navbar-link{color:#fff}.navbar.is-black .navbar-brand>a.navbar-item:focus,.navbar.is-black .navbar-brand>a.navbar-item:hover,.navbar.is-black .navbar-brand>a.navbar-item.is-active,.navbar.is-black .navbar-brand .navbar-link:focus,.navbar.is-black .navbar-brand .navbar-link:hover,.navbar.is-black .navbar-brand .navbar-link.is-active{background-color:#000;color:#fff}.navbar.is-black .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-black .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-black .navbar-start>.navbar-item,.navbar.is-black .navbar-start .navbar-link,.navbar.is-black .navbar-end>.navbar-item,.navbar.is-black .navbar-end .navbar-link{color:#fff}.navbar.is-black .navbar-start>a.navbar-item:focus,.navbar.is-black .navbar-start>a.navbar-item:hover,.navbar.is-black .navbar-start>a.navbar-item.is-active,.navbar.is-black .navbar-start .navbar-link:focus,.navbar.is-black .navbar-start .navbar-link:hover,.navbar.is-black .navbar-start .navbar-link.is-active,.navbar.is-black .navbar-end>a.navbar-item:focus,.navbar.is-black .navbar-end>a.navbar-item:hover,.navbar.is-black .navbar-end>a.navbar-item.is-active,.navbar.is-black .navbar-end .navbar-link:focus,.navbar.is-black .navbar-end .navbar-link:hover,.navbar.is-black .navbar-end .navbar-link.is-active{background-color:#000;color:#fff}.navbar.is-black .navbar-start .navbar-link::after,.navbar.is-black .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-black .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-black .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-black .navbar-item.has-dropdown.is-active .navbar-link{background-color:#000;color:#fff}.navbar.is-black .navbar-dropdown a.navbar-item.is-active{background-color:#0a0a0a;color:#fff}}.navbar.is-light{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-brand>.navbar-item,.navbar.is-light .navbar-brand .navbar-link{color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-brand>a.navbar-item:focus,.navbar.is-light .navbar-brand>a.navbar-item:hover,.navbar.is-light .navbar-brand>a.navbar-item.is-active,.navbar.is-light .navbar-brand .navbar-link:focus,.navbar.is-light .navbar-brand .navbar-link:hover,.navbar.is-light .navbar-brand .navbar-link.is-active{background-color:#e8e8e8;color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-brand .navbar-link::after{border-color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-burger{color:rgba(0,0,0,0.7)}@media screen and (min-width: 1056px){.navbar.is-light .navbar-start>.navbar-item,.navbar.is-light .navbar-start .navbar-link,.navbar.is-light .navbar-end>.navbar-item,.navbar.is-light .navbar-end .navbar-link{color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-start>a.navbar-item:focus,.navbar.is-light .navbar-start>a.navbar-item:hover,.navbar.is-light .navbar-start>a.navbar-item.is-active,.navbar.is-light .navbar-start .navbar-link:focus,.navbar.is-light .navbar-start .navbar-link:hover,.navbar.is-light .navbar-start .navbar-link.is-active,.navbar.is-light .navbar-end>a.navbar-item:focus,.navbar.is-light .navbar-end>a.navbar-item:hover,.navbar.is-light .navbar-end>a.navbar-item.is-active,.navbar.is-light .navbar-end .navbar-link:focus,.navbar.is-light .navbar-end .navbar-link:hover,.navbar.is-light .navbar-end .navbar-link.is-active{background-color:#e8e8e8;color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-start .navbar-link::after,.navbar.is-light .navbar-end .navbar-link::after{border-color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-light .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-light .navbar-item.has-dropdown.is-active .navbar-link{background-color:#e8e8e8;color:rgba(0,0,0,0.7)}.navbar.is-light .navbar-dropdown a.navbar-item.is-active{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}}.navbar.is-dark,.content kbd.navbar{background-color:#363636;color:#fff}.navbar.is-dark .navbar-brand>.navbar-item,.content kbd.navbar .navbar-brand>.navbar-item,.navbar.is-dark .navbar-brand .navbar-link,.content kbd.navbar .navbar-brand .navbar-link{color:#fff}.navbar.is-dark .navbar-brand>a.navbar-item:focus,.content kbd.navbar .navbar-brand>a.navbar-item:focus,.navbar.is-dark .navbar-brand>a.navbar-item:hover,.content kbd.navbar .navbar-brand>a.navbar-item:hover,.navbar.is-dark .navbar-brand>a.navbar-item.is-active,.content kbd.navbar .navbar-brand>a.navbar-item.is-active,.navbar.is-dark .navbar-brand .navbar-link:focus,.content kbd.navbar .navbar-brand .navbar-link:focus,.navbar.is-dark .navbar-brand .navbar-link:hover,.content kbd.navbar .navbar-brand .navbar-link:hover,.navbar.is-dark .navbar-brand .navbar-link.is-active,.content kbd.navbar .navbar-brand .navbar-link.is-active{background-color:#292929;color:#fff}.navbar.is-dark .navbar-brand .navbar-link::after,.content kbd.navbar .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-dark .navbar-burger,.content kbd.navbar .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-dark .navbar-start>.navbar-item,.content kbd.navbar .navbar-start>.navbar-item,.navbar.is-dark .navbar-start .navbar-link,.content kbd.navbar .navbar-start .navbar-link,.navbar.is-dark .navbar-end>.navbar-item,.content kbd.navbar .navbar-end>.navbar-item,.navbar.is-dark .navbar-end .navbar-link,.content kbd.navbar .navbar-end .navbar-link{color:#fff}.navbar.is-dark .navbar-start>a.navbar-item:focus,.content kbd.navbar .navbar-start>a.navbar-item:focus,.navbar.is-dark .navbar-start>a.navbar-item:hover,.content kbd.navbar .navbar-start>a.navbar-item:hover,.navbar.is-dark .navbar-start>a.navbar-item.is-active,.content kbd.navbar .navbar-start>a.navbar-item.is-active,.navbar.is-dark .navbar-start .navbar-link:focus,.content kbd.navbar .navbar-start .navbar-link:focus,.navbar.is-dark .navbar-start .navbar-link:hover,.content kbd.navbar .navbar-start .navbar-link:hover,.navbar.is-dark .navbar-start .navbar-link.is-active,.content kbd.navbar .navbar-start .navbar-link.is-active,.navbar.is-dark .navbar-end>a.navbar-item:focus,.content kbd.navbar .navbar-end>a.navbar-item:focus,.navbar.is-dark .navbar-end>a.navbar-item:hover,.content kbd.navbar .navbar-end>a.navbar-item:hover,.navbar.is-dark .navbar-end>a.navbar-item.is-active,.content kbd.navbar .navbar-end>a.navbar-item.is-active,.navbar.is-dark .navbar-end .navbar-link:focus,.content kbd.navbar .navbar-end .navbar-link:focus,.navbar.is-dark .navbar-end .navbar-link:hover,.content kbd.navbar .navbar-end .navbar-link:hover,.navbar.is-dark .navbar-end .navbar-link.is-active,.content kbd.navbar .navbar-end .navbar-link.is-active{background-color:#292929;color:#fff}.navbar.is-dark .navbar-start .navbar-link::after,.content kbd.navbar .navbar-start .navbar-link::after,.navbar.is-dark .navbar-end .navbar-link::after,.content kbd.navbar .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-dark .navbar-item.has-dropdown:focus .navbar-link,.content kbd.navbar .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-dark .navbar-item.has-dropdown:hover .navbar-link,.content kbd.navbar .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-dark .navbar-item.has-dropdown.is-active .navbar-link,.content kbd.navbar .navbar-item.has-dropdown.is-active .navbar-link{background-color:#292929;color:#fff}.navbar.is-dark .navbar-dropdown a.navbar-item.is-active,.content kbd.navbar .navbar-dropdown a.navbar-item.is-active{background-color:#363636;color:#fff}}.navbar.is-primary,.docstring>section>a.navbar.docs-sourcelink{background-color:#4eb5de;color:#fff}.navbar.is-primary .navbar-brand>.navbar-item,.docstring>section>a.navbar.docs-sourcelink .navbar-brand>.navbar-item,.navbar.is-primary .navbar-brand .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link{color:#fff}.navbar.is-primary .navbar-brand>a.navbar-item:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item:focus,.navbar.is-primary .navbar-brand>a.navbar-item:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item:hover,.navbar.is-primary .navbar-brand>a.navbar-item.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-brand>a.navbar-item.is-active,.navbar.is-primary .navbar-brand .navbar-link:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link:focus,.navbar.is-primary .navbar-brand .navbar-link:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link:hover,.navbar.is-primary .navbar-brand .navbar-link.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link.is-active{background-color:#39acda;color:#fff}.navbar.is-primary .navbar-brand .navbar-link::after,.docstring>section>a.navbar.docs-sourcelink .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-primary .navbar-burger,.docstring>section>a.navbar.docs-sourcelink .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-primary .navbar-start>.navbar-item,.docstring>section>a.navbar.docs-sourcelink .navbar-start>.navbar-item,.navbar.is-primary .navbar-start .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link,.navbar.is-primary .navbar-end>.navbar-item,.docstring>section>a.navbar.docs-sourcelink .navbar-end>.navbar-item,.navbar.is-primary .navbar-end .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link{color:#fff}.navbar.is-primary .navbar-start>a.navbar-item:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item:focus,.navbar.is-primary .navbar-start>a.navbar-item:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item:hover,.navbar.is-primary .navbar-start>a.navbar-item.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-start>a.navbar-item.is-active,.navbar.is-primary .navbar-start .navbar-link:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link:focus,.navbar.is-primary .navbar-start .navbar-link:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link:hover,.navbar.is-primary .navbar-start .navbar-link.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link.is-active,.navbar.is-primary .navbar-end>a.navbar-item:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item:focus,.navbar.is-primary .navbar-end>a.navbar-item:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item:hover,.navbar.is-primary .navbar-end>a.navbar-item.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-end>a.navbar-item.is-active,.navbar.is-primary .navbar-end .navbar-link:focus,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link:focus,.navbar.is-primary .navbar-end .navbar-link:hover,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link:hover,.navbar.is-primary .navbar-end .navbar-link.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link.is-active{background-color:#39acda;color:#fff}.navbar.is-primary .navbar-start .navbar-link::after,.docstring>section>a.navbar.docs-sourcelink .navbar-start .navbar-link::after,.navbar.is-primary .navbar-end .navbar-link::after,.docstring>section>a.navbar.docs-sourcelink .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-primary .navbar-item.has-dropdown:focus .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-primary .navbar-item.has-dropdown:hover .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-primary .navbar-item.has-dropdown.is-active .navbar-link,.docstring>section>a.navbar.docs-sourcelink .navbar-item.has-dropdown.is-active .navbar-link{background-color:#39acda;color:#fff}.navbar.is-primary .navbar-dropdown a.navbar-item.is-active,.docstring>section>a.navbar.docs-sourcelink .navbar-dropdown a.navbar-item.is-active{background-color:#4eb5de;color:#fff}}.navbar.is-link{background-color:#2e63b8;color:#fff}.navbar.is-link .navbar-brand>.navbar-item,.navbar.is-link .navbar-brand .navbar-link{color:#fff}.navbar.is-link .navbar-brand>a.navbar-item:focus,.navbar.is-link .navbar-brand>a.navbar-item:hover,.navbar.is-link .navbar-brand>a.navbar-item.is-active,.navbar.is-link .navbar-brand .navbar-link:focus,.navbar.is-link .navbar-brand .navbar-link:hover,.navbar.is-link .navbar-brand .navbar-link.is-active{background-color:#2958a4;color:#fff}.navbar.is-link .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-link .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-link .navbar-start>.navbar-item,.navbar.is-link .navbar-start .navbar-link,.navbar.is-link .navbar-end>.navbar-item,.navbar.is-link .navbar-end .navbar-link{color:#fff}.navbar.is-link .navbar-start>a.navbar-item:focus,.navbar.is-link .navbar-start>a.navbar-item:hover,.navbar.is-link .navbar-start>a.navbar-item.is-active,.navbar.is-link .navbar-start .navbar-link:focus,.navbar.is-link .navbar-start .navbar-link:hover,.navbar.is-link .navbar-start .navbar-link.is-active,.navbar.is-link .navbar-end>a.navbar-item:focus,.navbar.is-link .navbar-end>a.navbar-item:hover,.navbar.is-link .navbar-end>a.navbar-item.is-active,.navbar.is-link .navbar-end .navbar-link:focus,.navbar.is-link .navbar-end .navbar-link:hover,.navbar.is-link .navbar-end .navbar-link.is-active{background-color:#2958a4;color:#fff}.navbar.is-link .navbar-start .navbar-link::after,.navbar.is-link .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-link .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-link .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-link .navbar-item.has-dropdown.is-active .navbar-link{background-color:#2958a4;color:#fff}.navbar.is-link .navbar-dropdown a.navbar-item.is-active{background-color:#2e63b8;color:#fff}}.navbar.is-info{background-color:#209cee;color:#fff}.navbar.is-info .navbar-brand>.navbar-item,.navbar.is-info .navbar-brand .navbar-link{color:#fff}.navbar.is-info .navbar-brand>a.navbar-item:focus,.navbar.is-info .navbar-brand>a.navbar-item:hover,.navbar.is-info .navbar-brand>a.navbar-item.is-active,.navbar.is-info .navbar-brand .navbar-link:focus,.navbar.is-info .navbar-brand .navbar-link:hover,.navbar.is-info .navbar-brand .navbar-link.is-active{background-color:#1190e3;color:#fff}.navbar.is-info .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-info .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-info .navbar-start>.navbar-item,.navbar.is-info .navbar-start .navbar-link,.navbar.is-info .navbar-end>.navbar-item,.navbar.is-info .navbar-end .navbar-link{color:#fff}.navbar.is-info .navbar-start>a.navbar-item:focus,.navbar.is-info .navbar-start>a.navbar-item:hover,.navbar.is-info .navbar-start>a.navbar-item.is-active,.navbar.is-info .navbar-start .navbar-link:focus,.navbar.is-info .navbar-start .navbar-link:hover,.navbar.is-info .navbar-start .navbar-link.is-active,.navbar.is-info .navbar-end>a.navbar-item:focus,.navbar.is-info .navbar-end>a.navbar-item:hover,.navbar.is-info .navbar-end>a.navbar-item.is-active,.navbar.is-info .navbar-end .navbar-link:focus,.navbar.is-info .navbar-end .navbar-link:hover,.navbar.is-info .navbar-end .navbar-link.is-active{background-color:#1190e3;color:#fff}.navbar.is-info .navbar-start .navbar-link::after,.navbar.is-info .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-info .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-info .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-info .navbar-item.has-dropdown.is-active .navbar-link{background-color:#1190e3;color:#fff}.navbar.is-info .navbar-dropdown a.navbar-item.is-active{background-color:#209cee;color:#fff}}.navbar.is-success{background-color:#22c35b;color:#fff}.navbar.is-success .navbar-brand>.navbar-item,.navbar.is-success .navbar-brand .navbar-link{color:#fff}.navbar.is-success .navbar-brand>a.navbar-item:focus,.navbar.is-success .navbar-brand>a.navbar-item:hover,.navbar.is-success .navbar-brand>a.navbar-item.is-active,.navbar.is-success .navbar-brand .navbar-link:focus,.navbar.is-success .navbar-brand .navbar-link:hover,.navbar.is-success .navbar-brand .navbar-link.is-active{background-color:#1ead51;color:#fff}.navbar.is-success .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-success .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-success .navbar-start>.navbar-item,.navbar.is-success .navbar-start .navbar-link,.navbar.is-success .navbar-end>.navbar-item,.navbar.is-success .navbar-end .navbar-link{color:#fff}.navbar.is-success .navbar-start>a.navbar-item:focus,.navbar.is-success .navbar-start>a.navbar-item:hover,.navbar.is-success .navbar-start>a.navbar-item.is-active,.navbar.is-success .navbar-start .navbar-link:focus,.navbar.is-success .navbar-start .navbar-link:hover,.navbar.is-success .navbar-start .navbar-link.is-active,.navbar.is-success .navbar-end>a.navbar-item:focus,.navbar.is-success .navbar-end>a.navbar-item:hover,.navbar.is-success .navbar-end>a.navbar-item.is-active,.navbar.is-success .navbar-end .navbar-link:focus,.navbar.is-success .navbar-end .navbar-link:hover,.navbar.is-success .navbar-end .navbar-link.is-active{background-color:#1ead51;color:#fff}.navbar.is-success .navbar-start .navbar-link::after,.navbar.is-success .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-success .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-success .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-success .navbar-item.has-dropdown.is-active .navbar-link{background-color:#1ead51;color:#fff}.navbar.is-success .navbar-dropdown a.navbar-item.is-active{background-color:#22c35b;color:#fff}}.navbar.is-warning{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-brand>.navbar-item,.navbar.is-warning .navbar-brand .navbar-link{color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-brand>a.navbar-item:focus,.navbar.is-warning .navbar-brand>a.navbar-item:hover,.navbar.is-warning .navbar-brand>a.navbar-item.is-active,.navbar.is-warning .navbar-brand .navbar-link:focus,.navbar.is-warning .navbar-brand .navbar-link:hover,.navbar.is-warning .navbar-brand .navbar-link.is-active{background-color:#ffd83e;color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-brand .navbar-link::after{border-color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-burger{color:rgba(0,0,0,0.7)}@media screen and (min-width: 1056px){.navbar.is-warning .navbar-start>.navbar-item,.navbar.is-warning .navbar-start .navbar-link,.navbar.is-warning .navbar-end>.navbar-item,.navbar.is-warning .navbar-end .navbar-link{color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-start>a.navbar-item:focus,.navbar.is-warning .navbar-start>a.navbar-item:hover,.navbar.is-warning .navbar-start>a.navbar-item.is-active,.navbar.is-warning .navbar-start .navbar-link:focus,.navbar.is-warning .navbar-start .navbar-link:hover,.navbar.is-warning .navbar-start .navbar-link.is-active,.navbar.is-warning .navbar-end>a.navbar-item:focus,.navbar.is-warning .navbar-end>a.navbar-item:hover,.navbar.is-warning .navbar-end>a.navbar-item.is-active,.navbar.is-warning .navbar-end .navbar-link:focus,.navbar.is-warning .navbar-end .navbar-link:hover,.navbar.is-warning .navbar-end .navbar-link.is-active{background-color:#ffd83e;color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-start .navbar-link::after,.navbar.is-warning .navbar-end .navbar-link::after{border-color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-warning .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-warning .navbar-item.has-dropdown.is-active .navbar-link{background-color:#ffd83e;color:rgba(0,0,0,0.7)}.navbar.is-warning .navbar-dropdown a.navbar-item.is-active{background-color:#ffdd57;color:rgba(0,0,0,0.7)}}.navbar.is-danger{background-color:#da0b00;color:#fff}.navbar.is-danger .navbar-brand>.navbar-item,.navbar.is-danger .navbar-brand .navbar-link{color:#fff}.navbar.is-danger .navbar-brand>a.navbar-item:focus,.navbar.is-danger .navbar-brand>a.navbar-item:hover,.navbar.is-danger .navbar-brand>a.navbar-item.is-active,.navbar.is-danger .navbar-brand .navbar-link:focus,.navbar.is-danger .navbar-brand .navbar-link:hover,.navbar.is-danger .navbar-brand .navbar-link.is-active{background-color:#c10a00;color:#fff}.navbar.is-danger .navbar-brand .navbar-link::after{border-color:#fff}.navbar.is-danger .navbar-burger{color:#fff}@media screen and (min-width: 1056px){.navbar.is-danger .navbar-start>.navbar-item,.navbar.is-danger .navbar-start .navbar-link,.navbar.is-danger .navbar-end>.navbar-item,.navbar.is-danger .navbar-end .navbar-link{color:#fff}.navbar.is-danger .navbar-start>a.navbar-item:focus,.navbar.is-danger .navbar-start>a.navbar-item:hover,.navbar.is-danger .navbar-start>a.navbar-item.is-active,.navbar.is-danger .navbar-start .navbar-link:focus,.navbar.is-danger .navbar-start .navbar-link:hover,.navbar.is-danger .navbar-start .navbar-link.is-active,.navbar.is-danger .navbar-end>a.navbar-item:focus,.navbar.is-danger .navbar-end>a.navbar-item:hover,.navbar.is-danger .navbar-end>a.navbar-item.is-active,.navbar.is-danger .navbar-end .navbar-link:focus,.navbar.is-danger .navbar-end .navbar-link:hover,.navbar.is-danger .navbar-end .navbar-link.is-active{background-color:#c10a00;color:#fff}.navbar.is-danger .navbar-start .navbar-link::after,.navbar.is-danger .navbar-end .navbar-link::after{border-color:#fff}.navbar.is-danger .navbar-item.has-dropdown:focus .navbar-link,.navbar.is-danger .navbar-item.has-dropdown:hover .navbar-link,.navbar.is-danger .navbar-item.has-dropdown.is-active .navbar-link{background-color:#c10a00;color:#fff}.navbar.is-danger .navbar-dropdown a.navbar-item.is-active{background-color:#da0b00;color:#fff}}.navbar>.container{align-items:stretch;display:flex;min-height:3.25rem;width:100%}.navbar.has-shadow{box-shadow:0 2px 0 0 #f5f5f5}.navbar.is-fixed-bottom,.navbar.is-fixed-top{left:0;position:fixed;right:0;z-index:30}.navbar.is-fixed-bottom{bottom:0}.navbar.is-fixed-bottom.has-shadow{box-shadow:0 -2px 0 0 #f5f5f5}.navbar.is-fixed-top{top:0}html.has-navbar-fixed-top,body.has-navbar-fixed-top{padding-top:3.25rem}html.has-navbar-fixed-bottom,body.has-navbar-fixed-bottom{padding-bottom:3.25rem}.navbar-brand,.navbar-tabs{align-items:stretch;display:flex;flex-shrink:0;min-height:3.25rem}.navbar-brand a.navbar-item:focus,.navbar-brand a.navbar-item:hover{background-color:transparent}.navbar-tabs{-webkit-overflow-scrolling:touch;max-width:100vw;overflow-x:auto;overflow-y:hidden}.navbar-burger{color:#222;-moz-appearance:none;-webkit-appearance:none;appearance:none;background:none;border:none;cursor:pointer;display:block;height:3.25rem;position:relative;width:3.25rem;margin-left:auto}.navbar-burger span{background-color:currentColor;display:block;height:1px;left:calc(50% - 8px);position:absolute;transform-origin:center;transition-duration:86ms;transition-property:background-color, opacity, transform;transition-timing-function:ease-out;width:16px}.navbar-burger span:nth-child(1){top:calc(50% - 6px)}.navbar-burger span:nth-child(2){top:calc(50% - 1px)}.navbar-burger span:nth-child(3){top:calc(50% + 4px)}.navbar-burger:hover{background-color:rgba(0,0,0,0.05)}.navbar-burger.is-active span:nth-child(1){transform:translateY(5px) rotate(45deg)}.navbar-burger.is-active span:nth-child(2){opacity:0}.navbar-burger.is-active span:nth-child(3){transform:translateY(-5px) rotate(-45deg)}.navbar-menu{display:none}.navbar-item,.navbar-link{color:#222;display:block;line-height:1.5;padding:0.5rem 0.75rem;position:relative}.navbar-item .icon:only-child,.navbar-link .icon:only-child{margin-left:-0.25rem;margin-right:-0.25rem}a.navbar-item,.navbar-link{cursor:pointer}a.navbar-item:focus,a.navbar-item:focus-within,a.navbar-item:hover,a.navbar-item.is-active,.navbar-link:focus,.navbar-link:focus-within,.navbar-link:hover,.navbar-link.is-active{background-color:#fafafa;color:#2e63b8}.navbar-item{flex-grow:0;flex-shrink:0}.navbar-item img{max-height:1.75rem}.navbar-item.has-dropdown{padding:0}.navbar-item.is-expanded{flex-grow:1;flex-shrink:1}.navbar-item.is-tab{border-bottom:1px solid transparent;min-height:3.25rem;padding-bottom:calc(0.5rem - 1px)}.navbar-item.is-tab:focus,.navbar-item.is-tab:hover{background-color:rgba(0,0,0,0);border-bottom-color:#2e63b8}.navbar-item.is-tab.is-active{background-color:rgba(0,0,0,0);border-bottom-color:#2e63b8;border-bottom-style:solid;border-bottom-width:3px;color:#2e63b8;padding-bottom:calc(0.5rem - 3px)}.navbar-content{flex-grow:1;flex-shrink:1}.navbar-link:not(.is-arrowless){padding-right:2.5em}.navbar-link:not(.is-arrowless)::after{border-color:#2e63b8;margin-top:-0.375em;right:1.125em}.navbar-dropdown{font-size:0.875rem;padding-bottom:0.5rem;padding-top:0.5rem}.navbar-dropdown .navbar-item{padding-left:1.5rem;padding-right:1.5rem}.navbar-divider{background-color:#f5f5f5;border:none;display:none;height:2px;margin:0.5rem 0}@media screen and (max-width: 1055px){.navbar>.container{display:block}.navbar-brand .navbar-item,.navbar-tabs .navbar-item{align-items:center;display:flex}.navbar-link::after{display:none}.navbar-menu{background-color:#fff;box-shadow:0 8px 16px rgba(10,10,10,0.1);padding:0.5rem 0}.navbar-menu.is-active{display:block}.navbar.is-fixed-bottom-touch,.navbar.is-fixed-top-touch{left:0;position:fixed;right:0;z-index:30}.navbar.is-fixed-bottom-touch{bottom:0}.navbar.is-fixed-bottom-touch.has-shadow{box-shadow:0 -2px 3px rgba(10,10,10,0.1)}.navbar.is-fixed-top-touch{top:0}.navbar.is-fixed-top .navbar-menu,.navbar.is-fixed-top-touch .navbar-menu{-webkit-overflow-scrolling:touch;max-height:calc(100vh - 3.25rem);overflow:auto}html.has-navbar-fixed-top-touch,body.has-navbar-fixed-top-touch{padding-top:3.25rem}html.has-navbar-fixed-bottom-touch,body.has-navbar-fixed-bottom-touch{padding-bottom:3.25rem}}@media screen and (min-width: 1056px){.navbar,.navbar-menu,.navbar-start,.navbar-end{align-items:stretch;display:flex}.navbar{min-height:3.25rem}.navbar.is-spaced{padding:1rem 2rem}.navbar.is-spaced .navbar-start,.navbar.is-spaced .navbar-end{align-items:center}.navbar.is-spaced a.navbar-item,.navbar.is-spaced .navbar-link{border-radius:4px}.navbar.is-transparent a.navbar-item:focus,.navbar.is-transparent a.navbar-item:hover,.navbar.is-transparent a.navbar-item.is-active,.navbar.is-transparent .navbar-link:focus,.navbar.is-transparent .navbar-link:hover,.navbar.is-transparent .navbar-link.is-active{background-color:transparent !important}.navbar.is-transparent .navbar-item.has-dropdown.is-active .navbar-link,.navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:focus .navbar-link,.navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:focus-within .navbar-link,.navbar.is-transparent .navbar-item.has-dropdown.is-hoverable:hover .navbar-link{background-color:transparent !important}.navbar.is-transparent .navbar-dropdown a.navbar-item:focus,.navbar.is-transparent .navbar-dropdown a.navbar-item:hover{background-color:#f5f5f5;color:#0a0a0a}.navbar.is-transparent .navbar-dropdown a.navbar-item.is-active{background-color:#f5f5f5;color:#2e63b8}.navbar-burger{display:none}.navbar-item,.navbar-link{align-items:center;display:flex}.navbar-item.has-dropdown{align-items:stretch}.navbar-item.has-dropdown-up .navbar-link::after{transform:rotate(135deg) translate(0.25em, -0.25em)}.navbar-item.has-dropdown-up .navbar-dropdown{border-bottom:2px solid #dbdbdb;border-radius:6px 6px 0 0;border-top:none;bottom:100%;box-shadow:0 -8px 8px rgba(10,10,10,0.1);top:auto}.navbar-item.is-active .navbar-dropdown,.navbar-item.is-hoverable:focus .navbar-dropdown,.navbar-item.is-hoverable:focus-within .navbar-dropdown,.navbar-item.is-hoverable:hover .navbar-dropdown{display:block}.navbar.is-spaced .navbar-item.is-active .navbar-dropdown,.navbar-item.is-active .navbar-dropdown.is-boxed,.navbar.is-spaced .navbar-item.is-hoverable:focus .navbar-dropdown,.navbar-item.is-hoverable:focus .navbar-dropdown.is-boxed,.navbar.is-spaced .navbar-item.is-hoverable:focus-within .navbar-dropdown,.navbar-item.is-hoverable:focus-within .navbar-dropdown.is-boxed,.navbar.is-spaced .navbar-item.is-hoverable:hover .navbar-dropdown,.navbar-item.is-hoverable:hover .navbar-dropdown.is-boxed{opacity:1;pointer-events:auto;transform:translateY(0)}.navbar-menu{flex-grow:1;flex-shrink:0}.navbar-start{justify-content:flex-start;margin-right:auto}.navbar-end{justify-content:flex-end;margin-left:auto}.navbar-dropdown{background-color:#fff;border-bottom-left-radius:6px;border-bottom-right-radius:6px;border-top:2px solid #dbdbdb;box-shadow:0 8px 8px rgba(10,10,10,0.1);display:none;font-size:0.875rem;left:0;min-width:100%;position:absolute;top:100%;z-index:20}.navbar-dropdown .navbar-item{padding:0.375rem 1rem;white-space:nowrap}.navbar-dropdown a.navbar-item{padding-right:3rem}.navbar-dropdown a.navbar-item:focus,.navbar-dropdown a.navbar-item:hover{background-color:#f5f5f5;color:#0a0a0a}.navbar-dropdown a.navbar-item.is-active{background-color:#f5f5f5;color:#2e63b8}.navbar.is-spaced .navbar-dropdown,.navbar-dropdown.is-boxed{border-radius:6px;border-top:none;box-shadow:0 8px 8px rgba(10,10,10,0.1), 0 0 0 1px rgba(10,10,10,0.1);display:block;opacity:0;pointer-events:none;top:calc(100% + (-4px));transform:translateY(-5px);transition-duration:86ms;transition-property:opacity, transform}.navbar-dropdown.is-right{left:auto;right:0}.navbar-divider{display:block}.navbar>.container .navbar-brand,.container>.navbar .navbar-brand{margin-left:-.75rem}.navbar>.container .navbar-menu,.container>.navbar .navbar-menu{margin-right:-.75rem}.navbar.is-fixed-bottom-desktop,.navbar.is-fixed-top-desktop{left:0;position:fixed;right:0;z-index:30}.navbar.is-fixed-bottom-desktop{bottom:0}.navbar.is-fixed-bottom-desktop.has-shadow{box-shadow:0 -2px 3px rgba(10,10,10,0.1)}.navbar.is-fixed-top-desktop{top:0}html.has-navbar-fixed-top-desktop,body.has-navbar-fixed-top-desktop{padding-top:3.25rem}html.has-navbar-fixed-bottom-desktop,body.has-navbar-fixed-bottom-desktop{padding-bottom:3.25rem}html.has-spaced-navbar-fixed-top,body.has-spaced-navbar-fixed-top{padding-top:5.25rem}html.has-spaced-navbar-fixed-bottom,body.has-spaced-navbar-fixed-bottom{padding-bottom:5.25rem}a.navbar-item.is-active,.navbar-link.is-active{color:#0a0a0a}a.navbar-item.is-active:not(:focus):not(:hover),.navbar-link.is-active:not(:focus):not(:hover){background-color:rgba(0,0,0,0)}.navbar-item.has-dropdown:focus .navbar-link,.navbar-item.has-dropdown:hover .navbar-link,.navbar-item.has-dropdown.is-active .navbar-link{background-color:#fafafa}}.hero.is-fullheight-with-navbar{min-height:calc(100vh - 3.25rem)}.pagination{font-size:1rem;margin:-.25rem}.pagination.is-small,#documenter .docs-sidebar form.docs-search>input.pagination{font-size:.75rem}.pagination.is-medium{font-size:1.25rem}.pagination.is-large{font-size:1.5rem}.pagination.is-rounded .pagination-previous,#documenter .docs-sidebar form.docs-search>input.pagination .pagination-previous,.pagination.is-rounded .pagination-next,#documenter .docs-sidebar form.docs-search>input.pagination .pagination-next{padding-left:1em;padding-right:1em;border-radius:9999px}.pagination.is-rounded .pagination-link,#documenter .docs-sidebar form.docs-search>input.pagination .pagination-link{border-radius:9999px}.pagination,.pagination-list{align-items:center;display:flex;justify-content:center;text-align:center}.pagination-previous,.pagination-next,.pagination-link,.pagination-ellipsis{font-size:1em;justify-content:center;margin:.25rem;padding-left:.5em;padding-right:.5em;text-align:center}.pagination-previous,.pagination-next,.pagination-link{border-color:#dbdbdb;color:#222;min-width:2.5em}.pagination-previous:hover,.pagination-next:hover,.pagination-link:hover{border-color:#b5b5b5;color:#363636}.pagination-previous:focus,.pagination-next:focus,.pagination-link:focus{border-color:#3c5dcd}.pagination-previous:active,.pagination-next:active,.pagination-link:active{box-shadow:inset 0 1px 2px rgba(10,10,10,0.2)}.pagination-previous[disabled],.pagination-previous.is-disabled,.pagination-next[disabled],.pagination-next.is-disabled,.pagination-link[disabled],.pagination-link.is-disabled{background-color:#dbdbdb;border-color:#dbdbdb;box-shadow:none;color:#6b6b6b;opacity:0.5}.pagination-previous,.pagination-next{padding-left:.75em;padding-right:.75em;white-space:nowrap}.pagination-link.is-current{background-color:#2e63b8;border-color:#2e63b8;color:#fff}.pagination-ellipsis{color:#b5b5b5;pointer-events:none}.pagination-list{flex-wrap:wrap}.pagination-list li{list-style:none}@media screen and (max-width: 768px){.pagination{flex-wrap:wrap}.pagination-previous,.pagination-next{flex-grow:1;flex-shrink:1}.pagination-list li{flex-grow:1;flex-shrink:1}}@media screen and (min-width: 769px),print{.pagination-list{flex-grow:1;flex-shrink:1;justify-content:flex-start;order:1}.pagination-previous,.pagination-next,.pagination-link,.pagination-ellipsis{margin-bottom:0;margin-top:0}.pagination-previous{order:2}.pagination-next{order:3}.pagination{justify-content:space-between;margin-bottom:0;margin-top:0}.pagination.is-centered .pagination-previous{order:1}.pagination.is-centered .pagination-list{justify-content:center;order:2}.pagination.is-centered .pagination-next{order:3}.pagination.is-right .pagination-previous{order:1}.pagination.is-right .pagination-next{order:2}.pagination.is-right .pagination-list{justify-content:flex-end;order:3}}.panel{border-radius:6px;box-shadow:#bbb;font-size:1rem}.panel:not(:last-child){margin-bottom:1.5rem}.panel.is-white .panel-heading{background-color:#fff;color:#0a0a0a}.panel.is-white .panel-tabs a.is-active{border-bottom-color:#fff}.panel.is-white .panel-block.is-active .panel-icon{color:#fff}.panel.is-black .panel-heading{background-color:#0a0a0a;color:#fff}.panel.is-black .panel-tabs a.is-active{border-bottom-color:#0a0a0a}.panel.is-black .panel-block.is-active .panel-icon{color:#0a0a0a}.panel.is-light .panel-heading{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.panel.is-light .panel-tabs a.is-active{border-bottom-color:#f5f5f5}.panel.is-light .panel-block.is-active .panel-icon{color:#f5f5f5}.panel.is-dark .panel-heading,.content kbd.panel .panel-heading{background-color:#363636;color:#fff}.panel.is-dark .panel-tabs a.is-active,.content kbd.panel .panel-tabs a.is-active{border-bottom-color:#363636}.panel.is-dark .panel-block.is-active .panel-icon,.content kbd.panel .panel-block.is-active .panel-icon{color:#363636}.panel.is-primary .panel-heading,.docstring>section>a.panel.docs-sourcelink .panel-heading{background-color:#4eb5de;color:#fff}.panel.is-primary .panel-tabs a.is-active,.docstring>section>a.panel.docs-sourcelink .panel-tabs a.is-active{border-bottom-color:#4eb5de}.panel.is-primary .panel-block.is-active .panel-icon,.docstring>section>a.panel.docs-sourcelink .panel-block.is-active .panel-icon{color:#4eb5de}.panel.is-link .panel-heading{background-color:#2e63b8;color:#fff}.panel.is-link .panel-tabs a.is-active{border-bottom-color:#2e63b8}.panel.is-link .panel-block.is-active .panel-icon{color:#2e63b8}.panel.is-info .panel-heading{background-color:#209cee;color:#fff}.panel.is-info .panel-tabs a.is-active{border-bottom-color:#209cee}.panel.is-info .panel-block.is-active .panel-icon{color:#209cee}.panel.is-success .panel-heading{background-color:#22c35b;color:#fff}.panel.is-success .panel-tabs a.is-active{border-bottom-color:#22c35b}.panel.is-success .panel-block.is-active .panel-icon{color:#22c35b}.panel.is-warning .panel-heading{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.panel.is-warning .panel-tabs a.is-active{border-bottom-color:#ffdd57}.panel.is-warning .panel-block.is-active .panel-icon{color:#ffdd57}.panel.is-danger .panel-heading{background-color:#da0b00;color:#fff}.panel.is-danger .panel-tabs a.is-active{border-bottom-color:#da0b00}.panel.is-danger .panel-block.is-active .panel-icon{color:#da0b00}.panel-tabs:not(:last-child),.panel-block:not(:last-child){border-bottom:1px solid #ededed}.panel-heading{background-color:#ededed;border-radius:6px 6px 0 0;color:#222;font-size:1.25em;font-weight:700;line-height:1.25;padding:0.75em 1em}.panel-tabs{align-items:flex-end;display:flex;font-size:.875em;justify-content:center}.panel-tabs a{border-bottom:1px solid #dbdbdb;margin-bottom:-1px;padding:0.5em}.panel-tabs a.is-active{border-bottom-color:#4a4a4a;color:#363636}.panel-list a{color:#222}.panel-list a:hover{color:#2e63b8}.panel-block{align-items:center;color:#222;display:flex;justify-content:flex-start;padding:0.5em 0.75em}.panel-block input[type="checkbox"]{margin-right:.75em}.panel-block>.control{flex-grow:1;flex-shrink:1;width:100%}.panel-block.is-wrapped{flex-wrap:wrap}.panel-block.is-active{border-left-color:#2e63b8;color:#363636}.panel-block.is-active .panel-icon{color:#2e63b8}.panel-block:last-child{border-bottom-left-radius:6px;border-bottom-right-radius:6px}a.panel-block,label.panel-block{cursor:pointer}a.panel-block:hover,label.panel-block:hover{background-color:#f5f5f5}.panel-icon{display:inline-block;font-size:14px;height:1em;line-height:1em;text-align:center;vertical-align:top;width:1em;color:#6b6b6b;margin-right:.75em}.panel-icon .fa{font-size:inherit;line-height:inherit}.tabs{-webkit-overflow-scrolling:touch;align-items:stretch;display:flex;font-size:1rem;justify-content:space-between;overflow:hidden;overflow-x:auto;white-space:nowrap}.tabs a{align-items:center;border-bottom-color:#dbdbdb;border-bottom-style:solid;border-bottom-width:1px;color:#222;display:flex;justify-content:center;margin-bottom:-1px;padding:0.5em 1em;vertical-align:top}.tabs a:hover{border-bottom-color:#222;color:#222}.tabs li{display:block}.tabs li.is-active a{border-bottom-color:#2e63b8;color:#2e63b8}.tabs ul{align-items:center;border-bottom-color:#dbdbdb;border-bottom-style:solid;border-bottom-width:1px;display:flex;flex-grow:1;flex-shrink:0;justify-content:flex-start}.tabs ul.is-left{padding-right:0.75em}.tabs ul.is-center{flex:none;justify-content:center;padding-left:0.75em;padding-right:0.75em}.tabs ul.is-right{justify-content:flex-end;padding-left:0.75em}.tabs .icon:first-child{margin-right:.5em}.tabs .icon:last-child{margin-left:.5em}.tabs.is-centered ul{justify-content:center}.tabs.is-right ul{justify-content:flex-end}.tabs.is-boxed a{border:1px solid transparent;border-radius:4px 4px 0 0}.tabs.is-boxed a:hover{background-color:#f5f5f5;border-bottom-color:#dbdbdb}.tabs.is-boxed li.is-active a{background-color:#fff;border-color:#dbdbdb;border-bottom-color:rgba(0,0,0,0) !important}.tabs.is-fullwidth li{flex-grow:1;flex-shrink:0}.tabs.is-toggle a{border-color:#dbdbdb;border-style:solid;border-width:1px;margin-bottom:0;position:relative}.tabs.is-toggle a:hover{background-color:#f5f5f5;border-color:#b5b5b5;z-index:2}.tabs.is-toggle li+li{margin-left:-1px}.tabs.is-toggle li:first-child a{border-top-left-radius:4px;border-bottom-left-radius:4px}.tabs.is-toggle li:last-child a{border-top-right-radius:4px;border-bottom-right-radius:4px}.tabs.is-toggle li.is-active a{background-color:#2e63b8;border-color:#2e63b8;color:#fff;z-index:1}.tabs.is-toggle ul{border-bottom:none}.tabs.is-toggle.is-toggle-rounded li:first-child a{border-bottom-left-radius:9999px;border-top-left-radius:9999px;padding-left:1.25em}.tabs.is-toggle.is-toggle-rounded li:last-child a{border-bottom-right-radius:9999px;border-top-right-radius:9999px;padding-right:1.25em}.tabs.is-small,#documenter .docs-sidebar form.docs-search>input.tabs{font-size:.75rem}.tabs.is-medium{font-size:1.25rem}.tabs.is-large{font-size:1.5rem}.column{display:block;flex-basis:0;flex-grow:1;flex-shrink:1;padding:.75rem}.columns.is-mobile>.column.is-narrow{flex:none;width:unset}.columns.is-mobile>.column.is-full{flex:none;width:100%}.columns.is-mobile>.column.is-three-quarters{flex:none;width:75%}.columns.is-mobile>.column.is-two-thirds{flex:none;width:66.6666%}.columns.is-mobile>.column.is-half{flex:none;width:50%}.columns.is-mobile>.column.is-one-third{flex:none;width:33.3333%}.columns.is-mobile>.column.is-one-quarter{flex:none;width:25%}.columns.is-mobile>.column.is-one-fifth{flex:none;width:20%}.columns.is-mobile>.column.is-two-fifths{flex:none;width:40%}.columns.is-mobile>.column.is-three-fifths{flex:none;width:60%}.columns.is-mobile>.column.is-four-fifths{flex:none;width:80%}.columns.is-mobile>.column.is-offset-three-quarters{margin-left:75%}.columns.is-mobile>.column.is-offset-two-thirds{margin-left:66.6666%}.columns.is-mobile>.column.is-offset-half{margin-left:50%}.columns.is-mobile>.column.is-offset-one-third{margin-left:33.3333%}.columns.is-mobile>.column.is-offset-one-quarter{margin-left:25%}.columns.is-mobile>.column.is-offset-one-fifth{margin-left:20%}.columns.is-mobile>.column.is-offset-two-fifths{margin-left:40%}.columns.is-mobile>.column.is-offset-three-fifths{margin-left:60%}.columns.is-mobile>.column.is-offset-four-fifths{margin-left:80%}.columns.is-mobile>.column.is-0{flex:none;width:0%}.columns.is-mobile>.column.is-offset-0{margin-left:0%}.columns.is-mobile>.column.is-1{flex:none;width:8.33333337%}.columns.is-mobile>.column.is-offset-1{margin-left:8.33333337%}.columns.is-mobile>.column.is-2{flex:none;width:16.66666674%}.columns.is-mobile>.column.is-offset-2{margin-left:16.66666674%}.columns.is-mobile>.column.is-3{flex:none;width:25%}.columns.is-mobile>.column.is-offset-3{margin-left:25%}.columns.is-mobile>.column.is-4{flex:none;width:33.33333337%}.columns.is-mobile>.column.is-offset-4{margin-left:33.33333337%}.columns.is-mobile>.column.is-5{flex:none;width:41.66666674%}.columns.is-mobile>.column.is-offset-5{margin-left:41.66666674%}.columns.is-mobile>.column.is-6{flex:none;width:50%}.columns.is-mobile>.column.is-offset-6{margin-left:50%}.columns.is-mobile>.column.is-7{flex:none;width:58.33333337%}.columns.is-mobile>.column.is-offset-7{margin-left:58.33333337%}.columns.is-mobile>.column.is-8{flex:none;width:66.66666674%}.columns.is-mobile>.column.is-offset-8{margin-left:66.66666674%}.columns.is-mobile>.column.is-9{flex:none;width:75%}.columns.is-mobile>.column.is-offset-9{margin-left:75%}.columns.is-mobile>.column.is-10{flex:none;width:83.33333337%}.columns.is-mobile>.column.is-offset-10{margin-left:83.33333337%}.columns.is-mobile>.column.is-11{flex:none;width:91.66666674%}.columns.is-mobile>.column.is-offset-11{margin-left:91.66666674%}.columns.is-mobile>.column.is-12{flex:none;width:100%}.columns.is-mobile>.column.is-offset-12{margin-left:100%}@media screen and (max-width: 768px){.column.is-narrow-mobile{flex:none;width:unset}.column.is-full-mobile{flex:none;width:100%}.column.is-three-quarters-mobile{flex:none;width:75%}.column.is-two-thirds-mobile{flex:none;width:66.6666%}.column.is-half-mobile{flex:none;width:50%}.column.is-one-third-mobile{flex:none;width:33.3333%}.column.is-one-quarter-mobile{flex:none;width:25%}.column.is-one-fifth-mobile{flex:none;width:20%}.column.is-two-fifths-mobile{flex:none;width:40%}.column.is-three-fifths-mobile{flex:none;width:60%}.column.is-four-fifths-mobile{flex:none;width:80%}.column.is-offset-three-quarters-mobile{margin-left:75%}.column.is-offset-two-thirds-mobile{margin-left:66.6666%}.column.is-offset-half-mobile{margin-left:50%}.column.is-offset-one-third-mobile{margin-left:33.3333%}.column.is-offset-one-quarter-mobile{margin-left:25%}.column.is-offset-one-fifth-mobile{margin-left:20%}.column.is-offset-two-fifths-mobile{margin-left:40%}.column.is-offset-three-fifths-mobile{margin-left:60%}.column.is-offset-four-fifths-mobile{margin-left:80%}.column.is-0-mobile{flex:none;width:0%}.column.is-offset-0-mobile{margin-left:0%}.column.is-1-mobile{flex:none;width:8.33333337%}.column.is-offset-1-mobile{margin-left:8.33333337%}.column.is-2-mobile{flex:none;width:16.66666674%}.column.is-offset-2-mobile{margin-left:16.66666674%}.column.is-3-mobile{flex:none;width:25%}.column.is-offset-3-mobile{margin-left:25%}.column.is-4-mobile{flex:none;width:33.33333337%}.column.is-offset-4-mobile{margin-left:33.33333337%}.column.is-5-mobile{flex:none;width:41.66666674%}.column.is-offset-5-mobile{margin-left:41.66666674%}.column.is-6-mobile{flex:none;width:50%}.column.is-offset-6-mobile{margin-left:50%}.column.is-7-mobile{flex:none;width:58.33333337%}.column.is-offset-7-mobile{margin-left:58.33333337%}.column.is-8-mobile{flex:none;width:66.66666674%}.column.is-offset-8-mobile{margin-left:66.66666674%}.column.is-9-mobile{flex:none;width:75%}.column.is-offset-9-mobile{margin-left:75%}.column.is-10-mobile{flex:none;width:83.33333337%}.column.is-offset-10-mobile{margin-left:83.33333337%}.column.is-11-mobile{flex:none;width:91.66666674%}.column.is-offset-11-mobile{margin-left:91.66666674%}.column.is-12-mobile{flex:none;width:100%}.column.is-offset-12-mobile{margin-left:100%}}@media screen and (min-width: 769px),print{.column.is-narrow,.column.is-narrow-tablet{flex:none;width:unset}.column.is-full,.column.is-full-tablet{flex:none;width:100%}.column.is-three-quarters,.column.is-three-quarters-tablet{flex:none;width:75%}.column.is-two-thirds,.column.is-two-thirds-tablet{flex:none;width:66.6666%}.column.is-half,.column.is-half-tablet{flex:none;width:50%}.column.is-one-third,.column.is-one-third-tablet{flex:none;width:33.3333%}.column.is-one-quarter,.column.is-one-quarter-tablet{flex:none;width:25%}.column.is-one-fifth,.column.is-one-fifth-tablet{flex:none;width:20%}.column.is-two-fifths,.column.is-two-fifths-tablet{flex:none;width:40%}.column.is-three-fifths,.column.is-three-fifths-tablet{flex:none;width:60%}.column.is-four-fifths,.column.is-four-fifths-tablet{flex:none;width:80%}.column.is-offset-three-quarters,.column.is-offset-three-quarters-tablet{margin-left:75%}.column.is-offset-two-thirds,.column.is-offset-two-thirds-tablet{margin-left:66.6666%}.column.is-offset-half,.column.is-offset-half-tablet{margin-left:50%}.column.is-offset-one-third,.column.is-offset-one-third-tablet{margin-left:33.3333%}.column.is-offset-one-quarter,.column.is-offset-one-quarter-tablet{margin-left:25%}.column.is-offset-one-fifth,.column.is-offset-one-fifth-tablet{margin-left:20%}.column.is-offset-two-fifths,.column.is-offset-two-fifths-tablet{margin-left:40%}.column.is-offset-three-fifths,.column.is-offset-three-fifths-tablet{margin-left:60%}.column.is-offset-four-fifths,.column.is-offset-four-fifths-tablet{margin-left:80%}.column.is-0,.column.is-0-tablet{flex:none;width:0%}.column.is-offset-0,.column.is-offset-0-tablet{margin-left:0%}.column.is-1,.column.is-1-tablet{flex:none;width:8.33333337%}.column.is-offset-1,.column.is-offset-1-tablet{margin-left:8.33333337%}.column.is-2,.column.is-2-tablet{flex:none;width:16.66666674%}.column.is-offset-2,.column.is-offset-2-tablet{margin-left:16.66666674%}.column.is-3,.column.is-3-tablet{flex:none;width:25%}.column.is-offset-3,.column.is-offset-3-tablet{margin-left:25%}.column.is-4,.column.is-4-tablet{flex:none;width:33.33333337%}.column.is-offset-4,.column.is-offset-4-tablet{margin-left:33.33333337%}.column.is-5,.column.is-5-tablet{flex:none;width:41.66666674%}.column.is-offset-5,.column.is-offset-5-tablet{margin-left:41.66666674%}.column.is-6,.column.is-6-tablet{flex:none;width:50%}.column.is-offset-6,.column.is-offset-6-tablet{margin-left:50%}.column.is-7,.column.is-7-tablet{flex:none;width:58.33333337%}.column.is-offset-7,.column.is-offset-7-tablet{margin-left:58.33333337%}.column.is-8,.column.is-8-tablet{flex:none;width:66.66666674%}.column.is-offset-8,.column.is-offset-8-tablet{margin-left:66.66666674%}.column.is-9,.column.is-9-tablet{flex:none;width:75%}.column.is-offset-9,.column.is-offset-9-tablet{margin-left:75%}.column.is-10,.column.is-10-tablet{flex:none;width:83.33333337%}.column.is-offset-10,.column.is-offset-10-tablet{margin-left:83.33333337%}.column.is-11,.column.is-11-tablet{flex:none;width:91.66666674%}.column.is-offset-11,.column.is-offset-11-tablet{margin-left:91.66666674%}.column.is-12,.column.is-12-tablet{flex:none;width:100%}.column.is-offset-12,.column.is-offset-12-tablet{margin-left:100%}}@media screen and (max-width: 1055px){.column.is-narrow-touch{flex:none;width:unset}.column.is-full-touch{flex:none;width:100%}.column.is-three-quarters-touch{flex:none;width:75%}.column.is-two-thirds-touch{flex:none;width:66.6666%}.column.is-half-touch{flex:none;width:50%}.column.is-one-third-touch{flex:none;width:33.3333%}.column.is-one-quarter-touch{flex:none;width:25%}.column.is-one-fifth-touch{flex:none;width:20%}.column.is-two-fifths-touch{flex:none;width:40%}.column.is-three-fifths-touch{flex:none;width:60%}.column.is-four-fifths-touch{flex:none;width:80%}.column.is-offset-three-quarters-touch{margin-left:75%}.column.is-offset-two-thirds-touch{margin-left:66.6666%}.column.is-offset-half-touch{margin-left:50%}.column.is-offset-one-third-touch{margin-left:33.3333%}.column.is-offset-one-quarter-touch{margin-left:25%}.column.is-offset-one-fifth-touch{margin-left:20%}.column.is-offset-two-fifths-touch{margin-left:40%}.column.is-offset-three-fifths-touch{margin-left:60%}.column.is-offset-four-fifths-touch{margin-left:80%}.column.is-0-touch{flex:none;width:0%}.column.is-offset-0-touch{margin-left:0%}.column.is-1-touch{flex:none;width:8.33333337%}.column.is-offset-1-touch{margin-left:8.33333337%}.column.is-2-touch{flex:none;width:16.66666674%}.column.is-offset-2-touch{margin-left:16.66666674%}.column.is-3-touch{flex:none;width:25%}.column.is-offset-3-touch{margin-left:25%}.column.is-4-touch{flex:none;width:33.33333337%}.column.is-offset-4-touch{margin-left:33.33333337%}.column.is-5-touch{flex:none;width:41.66666674%}.column.is-offset-5-touch{margin-left:41.66666674%}.column.is-6-touch{flex:none;width:50%}.column.is-offset-6-touch{margin-left:50%}.column.is-7-touch{flex:none;width:58.33333337%}.column.is-offset-7-touch{margin-left:58.33333337%}.column.is-8-touch{flex:none;width:66.66666674%}.column.is-offset-8-touch{margin-left:66.66666674%}.column.is-9-touch{flex:none;width:75%}.column.is-offset-9-touch{margin-left:75%}.column.is-10-touch{flex:none;width:83.33333337%}.column.is-offset-10-touch{margin-left:83.33333337%}.column.is-11-touch{flex:none;width:91.66666674%}.column.is-offset-11-touch{margin-left:91.66666674%}.column.is-12-touch{flex:none;width:100%}.column.is-offset-12-touch{margin-left:100%}}@media screen and (min-width: 1056px){.column.is-narrow-desktop{flex:none;width:unset}.column.is-full-desktop{flex:none;width:100%}.column.is-three-quarters-desktop{flex:none;width:75%}.column.is-two-thirds-desktop{flex:none;width:66.6666%}.column.is-half-desktop{flex:none;width:50%}.column.is-one-third-desktop{flex:none;width:33.3333%}.column.is-one-quarter-desktop{flex:none;width:25%}.column.is-one-fifth-desktop{flex:none;width:20%}.column.is-two-fifths-desktop{flex:none;width:40%}.column.is-three-fifths-desktop{flex:none;width:60%}.column.is-four-fifths-desktop{flex:none;width:80%}.column.is-offset-three-quarters-desktop{margin-left:75%}.column.is-offset-two-thirds-desktop{margin-left:66.6666%}.column.is-offset-half-desktop{margin-left:50%}.column.is-offset-one-third-desktop{margin-left:33.3333%}.column.is-offset-one-quarter-desktop{margin-left:25%}.column.is-offset-one-fifth-desktop{margin-left:20%}.column.is-offset-two-fifths-desktop{margin-left:40%}.column.is-offset-three-fifths-desktop{margin-left:60%}.column.is-offset-four-fifths-desktop{margin-left:80%}.column.is-0-desktop{flex:none;width:0%}.column.is-offset-0-desktop{margin-left:0%}.column.is-1-desktop{flex:none;width:8.33333337%}.column.is-offset-1-desktop{margin-left:8.33333337%}.column.is-2-desktop{flex:none;width:16.66666674%}.column.is-offset-2-desktop{margin-left:16.66666674%}.column.is-3-desktop{flex:none;width:25%}.column.is-offset-3-desktop{margin-left:25%}.column.is-4-desktop{flex:none;width:33.33333337%}.column.is-offset-4-desktop{margin-left:33.33333337%}.column.is-5-desktop{flex:none;width:41.66666674%}.column.is-offset-5-desktop{margin-left:41.66666674%}.column.is-6-desktop{flex:none;width:50%}.column.is-offset-6-desktop{margin-left:50%}.column.is-7-desktop{flex:none;width:58.33333337%}.column.is-offset-7-desktop{margin-left:58.33333337%}.column.is-8-desktop{flex:none;width:66.66666674%}.column.is-offset-8-desktop{margin-left:66.66666674%}.column.is-9-desktop{flex:none;width:75%}.column.is-offset-9-desktop{margin-left:75%}.column.is-10-desktop{flex:none;width:83.33333337%}.column.is-offset-10-desktop{margin-left:83.33333337%}.column.is-11-desktop{flex:none;width:91.66666674%}.column.is-offset-11-desktop{margin-left:91.66666674%}.column.is-12-desktop{flex:none;width:100%}.column.is-offset-12-desktop{margin-left:100%}}@media screen and (min-width: 1216px){.column.is-narrow-widescreen{flex:none;width:unset}.column.is-full-widescreen{flex:none;width:100%}.column.is-three-quarters-widescreen{flex:none;width:75%}.column.is-two-thirds-widescreen{flex:none;width:66.6666%}.column.is-half-widescreen{flex:none;width:50%}.column.is-one-third-widescreen{flex:none;width:33.3333%}.column.is-one-quarter-widescreen{flex:none;width:25%}.column.is-one-fifth-widescreen{flex:none;width:20%}.column.is-two-fifths-widescreen{flex:none;width:40%}.column.is-three-fifths-widescreen{flex:none;width:60%}.column.is-four-fifths-widescreen{flex:none;width:80%}.column.is-offset-three-quarters-widescreen{margin-left:75%}.column.is-offset-two-thirds-widescreen{margin-left:66.6666%}.column.is-offset-half-widescreen{margin-left:50%}.column.is-offset-one-third-widescreen{margin-left:33.3333%}.column.is-offset-one-quarter-widescreen{margin-left:25%}.column.is-offset-one-fifth-widescreen{margin-left:20%}.column.is-offset-two-fifths-widescreen{margin-left:40%}.column.is-offset-three-fifths-widescreen{margin-left:60%}.column.is-offset-four-fifths-widescreen{margin-left:80%}.column.is-0-widescreen{flex:none;width:0%}.column.is-offset-0-widescreen{margin-left:0%}.column.is-1-widescreen{flex:none;width:8.33333337%}.column.is-offset-1-widescreen{margin-left:8.33333337%}.column.is-2-widescreen{flex:none;width:16.66666674%}.column.is-offset-2-widescreen{margin-left:16.66666674%}.column.is-3-widescreen{flex:none;width:25%}.column.is-offset-3-widescreen{margin-left:25%}.column.is-4-widescreen{flex:none;width:33.33333337%}.column.is-offset-4-widescreen{margin-left:33.33333337%}.column.is-5-widescreen{flex:none;width:41.66666674%}.column.is-offset-5-widescreen{margin-left:41.66666674%}.column.is-6-widescreen{flex:none;width:50%}.column.is-offset-6-widescreen{margin-left:50%}.column.is-7-widescreen{flex:none;width:58.33333337%}.column.is-offset-7-widescreen{margin-left:58.33333337%}.column.is-8-widescreen{flex:none;width:66.66666674%}.column.is-offset-8-widescreen{margin-left:66.66666674%}.column.is-9-widescreen{flex:none;width:75%}.column.is-offset-9-widescreen{margin-left:75%}.column.is-10-widescreen{flex:none;width:83.33333337%}.column.is-offset-10-widescreen{margin-left:83.33333337%}.column.is-11-widescreen{flex:none;width:91.66666674%}.column.is-offset-11-widescreen{margin-left:91.66666674%}.column.is-12-widescreen{flex:none;width:100%}.column.is-offset-12-widescreen{margin-left:100%}}@media screen and (min-width: 1408px){.column.is-narrow-fullhd{flex:none;width:unset}.column.is-full-fullhd{flex:none;width:100%}.column.is-three-quarters-fullhd{flex:none;width:75%}.column.is-two-thirds-fullhd{flex:none;width:66.6666%}.column.is-half-fullhd{flex:none;width:50%}.column.is-one-third-fullhd{flex:none;width:33.3333%}.column.is-one-quarter-fullhd{flex:none;width:25%}.column.is-one-fifth-fullhd{flex:none;width:20%}.column.is-two-fifths-fullhd{flex:none;width:40%}.column.is-three-fifths-fullhd{flex:none;width:60%}.column.is-four-fifths-fullhd{flex:none;width:80%}.column.is-offset-three-quarters-fullhd{margin-left:75%}.column.is-offset-two-thirds-fullhd{margin-left:66.6666%}.column.is-offset-half-fullhd{margin-left:50%}.column.is-offset-one-third-fullhd{margin-left:33.3333%}.column.is-offset-one-quarter-fullhd{margin-left:25%}.column.is-offset-one-fifth-fullhd{margin-left:20%}.column.is-offset-two-fifths-fullhd{margin-left:40%}.column.is-offset-three-fifths-fullhd{margin-left:60%}.column.is-offset-four-fifths-fullhd{margin-left:80%}.column.is-0-fullhd{flex:none;width:0%}.column.is-offset-0-fullhd{margin-left:0%}.column.is-1-fullhd{flex:none;width:8.33333337%}.column.is-offset-1-fullhd{margin-left:8.33333337%}.column.is-2-fullhd{flex:none;width:16.66666674%}.column.is-offset-2-fullhd{margin-left:16.66666674%}.column.is-3-fullhd{flex:none;width:25%}.column.is-offset-3-fullhd{margin-left:25%}.column.is-4-fullhd{flex:none;width:33.33333337%}.column.is-offset-4-fullhd{margin-left:33.33333337%}.column.is-5-fullhd{flex:none;width:41.66666674%}.column.is-offset-5-fullhd{margin-left:41.66666674%}.column.is-6-fullhd{flex:none;width:50%}.column.is-offset-6-fullhd{margin-left:50%}.column.is-7-fullhd{flex:none;width:58.33333337%}.column.is-offset-7-fullhd{margin-left:58.33333337%}.column.is-8-fullhd{flex:none;width:66.66666674%}.column.is-offset-8-fullhd{margin-left:66.66666674%}.column.is-9-fullhd{flex:none;width:75%}.column.is-offset-9-fullhd{margin-left:75%}.column.is-10-fullhd{flex:none;width:83.33333337%}.column.is-offset-10-fullhd{margin-left:83.33333337%}.column.is-11-fullhd{flex:none;width:91.66666674%}.column.is-offset-11-fullhd{margin-left:91.66666674%}.column.is-12-fullhd{flex:none;width:100%}.column.is-offset-12-fullhd{margin-left:100%}}.columns{margin-left:-.75rem;margin-right:-.75rem;margin-top:-.75rem}.columns:last-child{margin-bottom:-.75rem}.columns:not(:last-child){margin-bottom:calc(1.5rem - .75rem)}.columns.is-centered{justify-content:center}.columns.is-gapless{margin-left:0;margin-right:0;margin-top:0}.columns.is-gapless>.column{margin:0;padding:0 !important}.columns.is-gapless:not(:last-child){margin-bottom:1.5rem}.columns.is-gapless:last-child{margin-bottom:0}.columns.is-mobile{display:flex}.columns.is-multiline{flex-wrap:wrap}.columns.is-vcentered{align-items:center}@media screen and (min-width: 769px),print{.columns:not(.is-desktop){display:flex}}@media screen and (min-width: 1056px){.columns.is-desktop{display:flex}}.columns.is-variable{--columnGap: 0.75rem;margin-left:calc(-1 * var(--columnGap));margin-right:calc(-1 * var(--columnGap))}.columns.is-variable>.column{padding-left:var(--columnGap);padding-right:var(--columnGap)}.columns.is-variable.is-0{--columnGap: 0rem}@media screen and (max-width: 768px){.columns.is-variable.is-0-mobile{--columnGap: 0rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-0-tablet{--columnGap: 0rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-0-tablet-only{--columnGap: 0rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-0-touch{--columnGap: 0rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-0-desktop{--columnGap: 0rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-0-desktop-only{--columnGap: 0rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-0-widescreen{--columnGap: 0rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-0-widescreen-only{--columnGap: 0rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-0-fullhd{--columnGap: 0rem}}.columns.is-variable.is-1{--columnGap: .25rem}@media screen and (max-width: 768px){.columns.is-variable.is-1-mobile{--columnGap: .25rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-1-tablet{--columnGap: .25rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-1-tablet-only{--columnGap: .25rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-1-touch{--columnGap: .25rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-1-desktop{--columnGap: .25rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-1-desktop-only{--columnGap: .25rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-1-widescreen{--columnGap: .25rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-1-widescreen-only{--columnGap: .25rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-1-fullhd{--columnGap: .25rem}}.columns.is-variable.is-2{--columnGap: .5rem}@media screen and (max-width: 768px){.columns.is-variable.is-2-mobile{--columnGap: .5rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-2-tablet{--columnGap: .5rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-2-tablet-only{--columnGap: .5rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-2-touch{--columnGap: .5rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-2-desktop{--columnGap: .5rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-2-desktop-only{--columnGap: .5rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-2-widescreen{--columnGap: .5rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-2-widescreen-only{--columnGap: .5rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-2-fullhd{--columnGap: .5rem}}.columns.is-variable.is-3{--columnGap: .75rem}@media screen and (max-width: 768px){.columns.is-variable.is-3-mobile{--columnGap: .75rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-3-tablet{--columnGap: .75rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-3-tablet-only{--columnGap: .75rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-3-touch{--columnGap: .75rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-3-desktop{--columnGap: .75rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-3-desktop-only{--columnGap: .75rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-3-widescreen{--columnGap: .75rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-3-widescreen-only{--columnGap: .75rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-3-fullhd{--columnGap: .75rem}}.columns.is-variable.is-4{--columnGap: 1rem}@media screen and (max-width: 768px){.columns.is-variable.is-4-mobile{--columnGap: 1rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-4-tablet{--columnGap: 1rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-4-tablet-only{--columnGap: 1rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-4-touch{--columnGap: 1rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-4-desktop{--columnGap: 1rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-4-desktop-only{--columnGap: 1rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-4-widescreen{--columnGap: 1rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-4-widescreen-only{--columnGap: 1rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-4-fullhd{--columnGap: 1rem}}.columns.is-variable.is-5{--columnGap: 1.25rem}@media screen and (max-width: 768px){.columns.is-variable.is-5-mobile{--columnGap: 1.25rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-5-tablet{--columnGap: 1.25rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-5-tablet-only{--columnGap: 1.25rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-5-touch{--columnGap: 1.25rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-5-desktop{--columnGap: 1.25rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-5-desktop-only{--columnGap: 1.25rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-5-widescreen{--columnGap: 1.25rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-5-widescreen-only{--columnGap: 1.25rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-5-fullhd{--columnGap: 1.25rem}}.columns.is-variable.is-6{--columnGap: 1.5rem}@media screen and (max-width: 768px){.columns.is-variable.is-6-mobile{--columnGap: 1.5rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-6-tablet{--columnGap: 1.5rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-6-tablet-only{--columnGap: 1.5rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-6-touch{--columnGap: 1.5rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-6-desktop{--columnGap: 1.5rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-6-desktop-only{--columnGap: 1.5rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-6-widescreen{--columnGap: 1.5rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-6-widescreen-only{--columnGap: 1.5rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-6-fullhd{--columnGap: 1.5rem}}.columns.is-variable.is-7{--columnGap: 1.75rem}@media screen and (max-width: 768px){.columns.is-variable.is-7-mobile{--columnGap: 1.75rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-7-tablet{--columnGap: 1.75rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-7-tablet-only{--columnGap: 1.75rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-7-touch{--columnGap: 1.75rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-7-desktop{--columnGap: 1.75rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-7-desktop-only{--columnGap: 1.75rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-7-widescreen{--columnGap: 1.75rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-7-widescreen-only{--columnGap: 1.75rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-7-fullhd{--columnGap: 1.75rem}}.columns.is-variable.is-8{--columnGap: 2rem}@media screen and (max-width: 768px){.columns.is-variable.is-8-mobile{--columnGap: 2rem}}@media screen and (min-width: 769px),print{.columns.is-variable.is-8-tablet{--columnGap: 2rem}}@media screen and (min-width: 769px) and (max-width: 1055px){.columns.is-variable.is-8-tablet-only{--columnGap: 2rem}}@media screen and (max-width: 1055px){.columns.is-variable.is-8-touch{--columnGap: 2rem}}@media screen and (min-width: 1056px){.columns.is-variable.is-8-desktop{--columnGap: 2rem}}@media screen and (min-width: 1056px) and (max-width: 1215px){.columns.is-variable.is-8-desktop-only{--columnGap: 2rem}}@media screen and (min-width: 1216px){.columns.is-variable.is-8-widescreen{--columnGap: 2rem}}@media screen and (min-width: 1216px) and (max-width: 1407px){.columns.is-variable.is-8-widescreen-only{--columnGap: 2rem}}@media screen and (min-width: 1408px){.columns.is-variable.is-8-fullhd{--columnGap: 2rem}}.tile{align-items:stretch;display:block;flex-basis:0;flex-grow:1;flex-shrink:1;min-height:min-content}.tile.is-ancestor{margin-left:-.75rem;margin-right:-.75rem;margin-top:-.75rem}.tile.is-ancestor:last-child{margin-bottom:-.75rem}.tile.is-ancestor:not(:last-child){margin-bottom:.75rem}.tile.is-child{margin:0 !important}.tile.is-parent{padding:.75rem}.tile.is-vertical{flex-direction:column}.tile.is-vertical>.tile.is-child:not(:last-child){margin-bottom:1.5rem !important}@media screen and (min-width: 769px),print{.tile:not(.is-child){display:flex}.tile.is-1{flex:none;width:8.33333337%}.tile.is-2{flex:none;width:16.66666674%}.tile.is-3{flex:none;width:25%}.tile.is-4{flex:none;width:33.33333337%}.tile.is-5{flex:none;width:41.66666674%}.tile.is-6{flex:none;width:50%}.tile.is-7{flex:none;width:58.33333337%}.tile.is-8{flex:none;width:66.66666674%}.tile.is-9{flex:none;width:75%}.tile.is-10{flex:none;width:83.33333337%}.tile.is-11{flex:none;width:91.66666674%}.tile.is-12{flex:none;width:100%}}.hero{align-items:stretch;display:flex;flex-direction:column;justify-content:space-between}.hero .navbar{background:none}.hero .tabs ul{border-bottom:none}.hero.is-white{background-color:#fff;color:#0a0a0a}.hero.is-white a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-white strong{color:inherit}.hero.is-white .title{color:#0a0a0a}.hero.is-white .subtitle{color:rgba(10,10,10,0.9)}.hero.is-white .subtitle a:not(.button),.hero.is-white .subtitle strong{color:#0a0a0a}@media screen and (max-width: 1055px){.hero.is-white .navbar-menu{background-color:#fff}}.hero.is-white .navbar-item,.hero.is-white .navbar-link{color:rgba(10,10,10,0.7)}.hero.is-white a.navbar-item:hover,.hero.is-white a.navbar-item.is-active,.hero.is-white .navbar-link:hover,.hero.is-white .navbar-link.is-active{background-color:#f2f2f2;color:#0a0a0a}.hero.is-white .tabs a{color:#0a0a0a;opacity:0.9}.hero.is-white .tabs a:hover{opacity:1}.hero.is-white .tabs li.is-active a{color:#fff !important;opacity:1}.hero.is-white .tabs.is-boxed a,.hero.is-white .tabs.is-toggle a{color:#0a0a0a}.hero.is-white .tabs.is-boxed a:hover,.hero.is-white .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-white .tabs.is-boxed li.is-active a,.hero.is-white .tabs.is-boxed li.is-active a:hover,.hero.is-white .tabs.is-toggle li.is-active a,.hero.is-white .tabs.is-toggle li.is-active a:hover{background-color:#0a0a0a;border-color:#0a0a0a;color:#fff}.hero.is-white.is-bold{background-image:linear-gradient(141deg, #e8e3e4 0%, #fff 71%, #fff 100%)}@media screen and (max-width: 768px){.hero.is-white.is-bold .navbar-menu{background-image:linear-gradient(141deg, #e8e3e4 0%, #fff 71%, #fff 100%)}}.hero.is-black{background-color:#0a0a0a;color:#fff}.hero.is-black a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-black strong{color:inherit}.hero.is-black .title{color:#fff}.hero.is-black .subtitle{color:rgba(255,255,255,0.9)}.hero.is-black .subtitle a:not(.button),.hero.is-black .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-black .navbar-menu{background-color:#0a0a0a}}.hero.is-black .navbar-item,.hero.is-black .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-black a.navbar-item:hover,.hero.is-black a.navbar-item.is-active,.hero.is-black .navbar-link:hover,.hero.is-black .navbar-link.is-active{background-color:#000;color:#fff}.hero.is-black .tabs a{color:#fff;opacity:0.9}.hero.is-black .tabs a:hover{opacity:1}.hero.is-black .tabs li.is-active a{color:#0a0a0a !important;opacity:1}.hero.is-black .tabs.is-boxed a,.hero.is-black .tabs.is-toggle a{color:#fff}.hero.is-black .tabs.is-boxed a:hover,.hero.is-black .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-black .tabs.is-boxed li.is-active a,.hero.is-black .tabs.is-boxed li.is-active a:hover,.hero.is-black .tabs.is-toggle li.is-active a,.hero.is-black .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#0a0a0a}.hero.is-black.is-bold{background-image:linear-gradient(141deg, #000 0%, #0a0a0a 71%, #181616 100%)}@media screen and (max-width: 768px){.hero.is-black.is-bold .navbar-menu{background-image:linear-gradient(141deg, #000 0%, #0a0a0a 71%, #181616 100%)}}.hero.is-light{background-color:#f5f5f5;color:rgba(0,0,0,0.7)}.hero.is-light a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-light strong{color:inherit}.hero.is-light .title{color:rgba(0,0,0,0.7)}.hero.is-light .subtitle{color:rgba(0,0,0,0.9)}.hero.is-light .subtitle a:not(.button),.hero.is-light .subtitle strong{color:rgba(0,0,0,0.7)}@media screen and (max-width: 1055px){.hero.is-light .navbar-menu{background-color:#f5f5f5}}.hero.is-light .navbar-item,.hero.is-light .navbar-link{color:rgba(0,0,0,0.7)}.hero.is-light a.navbar-item:hover,.hero.is-light a.navbar-item.is-active,.hero.is-light .navbar-link:hover,.hero.is-light .navbar-link.is-active{background-color:#e8e8e8;color:rgba(0,0,0,0.7)}.hero.is-light .tabs a{color:rgba(0,0,0,0.7);opacity:0.9}.hero.is-light .tabs a:hover{opacity:1}.hero.is-light .tabs li.is-active a{color:#f5f5f5 !important;opacity:1}.hero.is-light .tabs.is-boxed a,.hero.is-light .tabs.is-toggle a{color:rgba(0,0,0,0.7)}.hero.is-light .tabs.is-boxed a:hover,.hero.is-light .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-light .tabs.is-boxed li.is-active a,.hero.is-light .tabs.is-boxed li.is-active a:hover,.hero.is-light .tabs.is-toggle li.is-active a,.hero.is-light .tabs.is-toggle li.is-active a:hover{background-color:rgba(0,0,0,0.7);border-color:rgba(0,0,0,0.7);color:#f5f5f5}.hero.is-light.is-bold{background-image:linear-gradient(141deg, #dfd8d9 0%, #f5f5f5 71%, #fff 100%)}@media screen and (max-width: 768px){.hero.is-light.is-bold .navbar-menu{background-image:linear-gradient(141deg, #dfd8d9 0%, #f5f5f5 71%, #fff 100%)}}.hero.is-dark,.content kbd.hero{background-color:#363636;color:#fff}.hero.is-dark a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.content kbd.hero a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-dark strong,.content kbd.hero strong{color:inherit}.hero.is-dark .title,.content kbd.hero .title{color:#fff}.hero.is-dark .subtitle,.content kbd.hero .subtitle{color:rgba(255,255,255,0.9)}.hero.is-dark .subtitle a:not(.button),.content kbd.hero .subtitle a:not(.button),.hero.is-dark .subtitle strong,.content kbd.hero .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-dark .navbar-menu,.content kbd.hero .navbar-menu{background-color:#363636}}.hero.is-dark .navbar-item,.content kbd.hero .navbar-item,.hero.is-dark .navbar-link,.content kbd.hero .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-dark a.navbar-item:hover,.content kbd.hero a.navbar-item:hover,.hero.is-dark a.navbar-item.is-active,.content kbd.hero a.navbar-item.is-active,.hero.is-dark .navbar-link:hover,.content kbd.hero .navbar-link:hover,.hero.is-dark .navbar-link.is-active,.content kbd.hero .navbar-link.is-active{background-color:#292929;color:#fff}.hero.is-dark .tabs a,.content kbd.hero .tabs a{color:#fff;opacity:0.9}.hero.is-dark .tabs a:hover,.content kbd.hero .tabs a:hover{opacity:1}.hero.is-dark .tabs li.is-active a,.content kbd.hero .tabs li.is-active a{color:#363636 !important;opacity:1}.hero.is-dark .tabs.is-boxed a,.content kbd.hero .tabs.is-boxed a,.hero.is-dark .tabs.is-toggle a,.content kbd.hero .tabs.is-toggle a{color:#fff}.hero.is-dark .tabs.is-boxed a:hover,.content kbd.hero .tabs.is-boxed a:hover,.hero.is-dark .tabs.is-toggle a:hover,.content kbd.hero .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-dark .tabs.is-boxed li.is-active a,.content kbd.hero .tabs.is-boxed li.is-active a,.hero.is-dark .tabs.is-boxed li.is-active a:hover,.hero.is-dark .tabs.is-toggle li.is-active a,.content kbd.hero .tabs.is-toggle li.is-active a,.hero.is-dark .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#363636}.hero.is-dark.is-bold,.content kbd.hero.is-bold{background-image:linear-gradient(141deg, #1f191a 0%, #363636 71%, #46403f 100%)}@media screen and (max-width: 768px){.hero.is-dark.is-bold .navbar-menu,.content kbd.hero.is-bold .navbar-menu{background-image:linear-gradient(141deg, #1f191a 0%, #363636 71%, #46403f 100%)}}.hero.is-primary,.docstring>section>a.hero.docs-sourcelink{background-color:#4eb5de;color:#fff}.hero.is-primary a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.docstring>section>a.hero.docs-sourcelink a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-primary strong,.docstring>section>a.hero.docs-sourcelink strong{color:inherit}.hero.is-primary .title,.docstring>section>a.hero.docs-sourcelink .title{color:#fff}.hero.is-primary .subtitle,.docstring>section>a.hero.docs-sourcelink .subtitle{color:rgba(255,255,255,0.9)}.hero.is-primary .subtitle a:not(.button),.docstring>section>a.hero.docs-sourcelink .subtitle a:not(.button),.hero.is-primary .subtitle strong,.docstring>section>a.hero.docs-sourcelink .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-primary .navbar-menu,.docstring>section>a.hero.docs-sourcelink .navbar-menu{background-color:#4eb5de}}.hero.is-primary .navbar-item,.docstring>section>a.hero.docs-sourcelink .navbar-item,.hero.is-primary .navbar-link,.docstring>section>a.hero.docs-sourcelink .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-primary a.navbar-item:hover,.docstring>section>a.hero.docs-sourcelink a.navbar-item:hover,.hero.is-primary a.navbar-item.is-active,.docstring>section>a.hero.docs-sourcelink a.navbar-item.is-active,.hero.is-primary .navbar-link:hover,.docstring>section>a.hero.docs-sourcelink .navbar-link:hover,.hero.is-primary .navbar-link.is-active,.docstring>section>a.hero.docs-sourcelink .navbar-link.is-active{background-color:#39acda;color:#fff}.hero.is-primary .tabs a,.docstring>section>a.hero.docs-sourcelink .tabs a{color:#fff;opacity:0.9}.hero.is-primary .tabs a:hover,.docstring>section>a.hero.docs-sourcelink .tabs a:hover{opacity:1}.hero.is-primary .tabs li.is-active a,.docstring>section>a.hero.docs-sourcelink .tabs li.is-active a{color:#4eb5de !important;opacity:1}.hero.is-primary .tabs.is-boxed a,.docstring>section>a.hero.docs-sourcelink .tabs.is-boxed a,.hero.is-primary .tabs.is-toggle a,.docstring>section>a.hero.docs-sourcelink .tabs.is-toggle a{color:#fff}.hero.is-primary .tabs.is-boxed a:hover,.docstring>section>a.hero.docs-sourcelink .tabs.is-boxed a:hover,.hero.is-primary .tabs.is-toggle a:hover,.docstring>section>a.hero.docs-sourcelink .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-primary .tabs.is-boxed li.is-active a,.docstring>section>a.hero.docs-sourcelink .tabs.is-boxed li.is-active a,.hero.is-primary .tabs.is-boxed li.is-active a:hover,.hero.is-primary .tabs.is-toggle li.is-active a,.docstring>section>a.hero.docs-sourcelink .tabs.is-toggle li.is-active a,.hero.is-primary .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#4eb5de}.hero.is-primary.is-bold,.docstring>section>a.hero.is-bold.docs-sourcelink{background-image:linear-gradient(141deg, #1bc7de 0%, #4eb5de 71%, #5fa9e7 100%)}@media screen and (max-width: 768px){.hero.is-primary.is-bold .navbar-menu,.docstring>section>a.hero.is-bold.docs-sourcelink .navbar-menu{background-image:linear-gradient(141deg, #1bc7de 0%, #4eb5de 71%, #5fa9e7 100%)}}.hero.is-link{background-color:#2e63b8;color:#fff}.hero.is-link a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-link strong{color:inherit}.hero.is-link .title{color:#fff}.hero.is-link .subtitle{color:rgba(255,255,255,0.9)}.hero.is-link .subtitle a:not(.button),.hero.is-link .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-link .navbar-menu{background-color:#2e63b8}}.hero.is-link .navbar-item,.hero.is-link .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-link a.navbar-item:hover,.hero.is-link a.navbar-item.is-active,.hero.is-link .navbar-link:hover,.hero.is-link .navbar-link.is-active{background-color:#2958a4;color:#fff}.hero.is-link .tabs a{color:#fff;opacity:0.9}.hero.is-link .tabs a:hover{opacity:1}.hero.is-link .tabs li.is-active a{color:#2e63b8 !important;opacity:1}.hero.is-link .tabs.is-boxed a,.hero.is-link .tabs.is-toggle a{color:#fff}.hero.is-link .tabs.is-boxed a:hover,.hero.is-link .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-link .tabs.is-boxed li.is-active a,.hero.is-link .tabs.is-boxed li.is-active a:hover,.hero.is-link .tabs.is-toggle li.is-active a,.hero.is-link .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#2e63b8}.hero.is-link.is-bold{background-image:linear-gradient(141deg, #1b6098 0%, #2e63b8 71%, #2d51d2 100%)}@media screen and (max-width: 768px){.hero.is-link.is-bold .navbar-menu{background-image:linear-gradient(141deg, #1b6098 0%, #2e63b8 71%, #2d51d2 100%)}}.hero.is-info{background-color:#209cee;color:#fff}.hero.is-info a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-info strong{color:inherit}.hero.is-info .title{color:#fff}.hero.is-info .subtitle{color:rgba(255,255,255,0.9)}.hero.is-info .subtitle a:not(.button),.hero.is-info .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-info .navbar-menu{background-color:#209cee}}.hero.is-info .navbar-item,.hero.is-info .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-info a.navbar-item:hover,.hero.is-info a.navbar-item.is-active,.hero.is-info .navbar-link:hover,.hero.is-info .navbar-link.is-active{background-color:#1190e3;color:#fff}.hero.is-info .tabs a{color:#fff;opacity:0.9}.hero.is-info .tabs a:hover{opacity:1}.hero.is-info .tabs li.is-active a{color:#209cee !important;opacity:1}.hero.is-info .tabs.is-boxed a,.hero.is-info .tabs.is-toggle a{color:#fff}.hero.is-info .tabs.is-boxed a:hover,.hero.is-info .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-info .tabs.is-boxed li.is-active a,.hero.is-info .tabs.is-boxed li.is-active a:hover,.hero.is-info .tabs.is-toggle li.is-active a,.hero.is-info .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#209cee}.hero.is-info.is-bold{background-image:linear-gradient(141deg, #05a6d6 0%, #209cee 71%, #3287f5 100%)}@media screen and (max-width: 768px){.hero.is-info.is-bold .navbar-menu{background-image:linear-gradient(141deg, #05a6d6 0%, #209cee 71%, #3287f5 100%)}}.hero.is-success{background-color:#22c35b;color:#fff}.hero.is-success a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-success strong{color:inherit}.hero.is-success .title{color:#fff}.hero.is-success .subtitle{color:rgba(255,255,255,0.9)}.hero.is-success .subtitle a:not(.button),.hero.is-success .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-success .navbar-menu{background-color:#22c35b}}.hero.is-success .navbar-item,.hero.is-success .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-success a.navbar-item:hover,.hero.is-success a.navbar-item.is-active,.hero.is-success .navbar-link:hover,.hero.is-success .navbar-link.is-active{background-color:#1ead51;color:#fff}.hero.is-success .tabs a{color:#fff;opacity:0.9}.hero.is-success .tabs a:hover{opacity:1}.hero.is-success .tabs li.is-active a{color:#22c35b !important;opacity:1}.hero.is-success .tabs.is-boxed a,.hero.is-success .tabs.is-toggle a{color:#fff}.hero.is-success .tabs.is-boxed a:hover,.hero.is-success .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-success .tabs.is-boxed li.is-active a,.hero.is-success .tabs.is-boxed li.is-active a:hover,.hero.is-success .tabs.is-toggle li.is-active a,.hero.is-success .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#22c35b}.hero.is-success.is-bold{background-image:linear-gradient(141deg, #12a02c 0%, #22c35b 71%, #1fdf83 100%)}@media screen and (max-width: 768px){.hero.is-success.is-bold .navbar-menu{background-image:linear-gradient(141deg, #12a02c 0%, #22c35b 71%, #1fdf83 100%)}}.hero.is-warning{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.hero.is-warning a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-warning strong{color:inherit}.hero.is-warning .title{color:rgba(0,0,0,0.7)}.hero.is-warning .subtitle{color:rgba(0,0,0,0.9)}.hero.is-warning .subtitle a:not(.button),.hero.is-warning .subtitle strong{color:rgba(0,0,0,0.7)}@media screen and (max-width: 1055px){.hero.is-warning .navbar-menu{background-color:#ffdd57}}.hero.is-warning .navbar-item,.hero.is-warning .navbar-link{color:rgba(0,0,0,0.7)}.hero.is-warning a.navbar-item:hover,.hero.is-warning a.navbar-item.is-active,.hero.is-warning .navbar-link:hover,.hero.is-warning .navbar-link.is-active{background-color:#ffd83e;color:rgba(0,0,0,0.7)}.hero.is-warning .tabs a{color:rgba(0,0,0,0.7);opacity:0.9}.hero.is-warning .tabs a:hover{opacity:1}.hero.is-warning .tabs li.is-active a{color:#ffdd57 !important;opacity:1}.hero.is-warning .tabs.is-boxed a,.hero.is-warning .tabs.is-toggle a{color:rgba(0,0,0,0.7)}.hero.is-warning .tabs.is-boxed a:hover,.hero.is-warning .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-warning .tabs.is-boxed li.is-active a,.hero.is-warning .tabs.is-boxed li.is-active a:hover,.hero.is-warning .tabs.is-toggle li.is-active a,.hero.is-warning .tabs.is-toggle li.is-active a:hover{background-color:rgba(0,0,0,0.7);border-color:rgba(0,0,0,0.7);color:#ffdd57}.hero.is-warning.is-bold{background-image:linear-gradient(141deg, #ffae24 0%, #ffdd57 71%, #fffa71 100%)}@media screen and (max-width: 768px){.hero.is-warning.is-bold .navbar-menu{background-image:linear-gradient(141deg, #ffae24 0%, #ffdd57 71%, #fffa71 100%)}}.hero.is-danger{background-color:#da0b00;color:#fff}.hero.is-danger a:not(.button):not(.dropdown-item):not(.tag):not(.pagination-link.is-current),.hero.is-danger strong{color:inherit}.hero.is-danger .title{color:#fff}.hero.is-danger .subtitle{color:rgba(255,255,255,0.9)}.hero.is-danger .subtitle a:not(.button),.hero.is-danger .subtitle strong{color:#fff}@media screen and (max-width: 1055px){.hero.is-danger .navbar-menu{background-color:#da0b00}}.hero.is-danger .navbar-item,.hero.is-danger .navbar-link{color:rgba(255,255,255,0.7)}.hero.is-danger a.navbar-item:hover,.hero.is-danger a.navbar-item.is-active,.hero.is-danger .navbar-link:hover,.hero.is-danger .navbar-link.is-active{background-color:#c10a00;color:#fff}.hero.is-danger .tabs a{color:#fff;opacity:0.9}.hero.is-danger .tabs a:hover{opacity:1}.hero.is-danger .tabs li.is-active a{color:#da0b00 !important;opacity:1}.hero.is-danger .tabs.is-boxed a,.hero.is-danger .tabs.is-toggle a{color:#fff}.hero.is-danger .tabs.is-boxed a:hover,.hero.is-danger .tabs.is-toggle a:hover{background-color:rgba(10,10,10,0.1)}.hero.is-danger .tabs.is-boxed li.is-active a,.hero.is-danger .tabs.is-boxed li.is-active a:hover,.hero.is-danger .tabs.is-toggle li.is-active a,.hero.is-danger .tabs.is-toggle li.is-active a:hover{background-color:#fff;border-color:#fff;color:#da0b00}.hero.is-danger.is-bold{background-image:linear-gradient(141deg, #a70013 0%, #da0b00 71%, #f43500 100%)}@media screen and (max-width: 768px){.hero.is-danger.is-bold .navbar-menu{background-image:linear-gradient(141deg, #a70013 0%, #da0b00 71%, #f43500 100%)}}.hero.is-small .hero-body,#documenter .docs-sidebar form.docs-search>input.hero .hero-body{padding:1.5rem}@media screen and (min-width: 769px),print{.hero.is-medium .hero-body{padding:9rem 4.5rem}}@media screen and (min-width: 769px),print{.hero.is-large .hero-body{padding:18rem 6rem}}.hero.is-halfheight .hero-body,.hero.is-fullheight .hero-body,.hero.is-fullheight-with-navbar .hero-body{align-items:center;display:flex}.hero.is-halfheight .hero-body>.container,.hero.is-fullheight .hero-body>.container,.hero.is-fullheight-with-navbar .hero-body>.container{flex-grow:1;flex-shrink:1}.hero.is-halfheight{min-height:50vh}.hero.is-fullheight{min-height:100vh}.hero-video{overflow:hidden}.hero-video video{left:50%;min-height:100%;min-width:100%;position:absolute;top:50%;transform:translate3d(-50%, -50%, 0)}.hero-video.is-transparent{opacity:0.3}@media screen and (max-width: 768px){.hero-video{display:none}}.hero-buttons{margin-top:1.5rem}@media screen and (max-width: 768px){.hero-buttons .button{display:flex}.hero-buttons .button:not(:last-child){margin-bottom:0.75rem}}@media screen and (min-width: 769px),print{.hero-buttons{display:flex;justify-content:center}.hero-buttons .button:not(:last-child){margin-right:1.5rem}}.hero-head,.hero-foot{flex-grow:0;flex-shrink:0}.hero-body{flex-grow:1;flex-shrink:0;padding:3rem 1.5rem}@media screen and (min-width: 769px),print{.hero-body{padding:3rem 3rem}}.section{padding:3rem 1.5rem}@media screen and (min-width: 1056px){.section{padding:3rem 3rem}.section.is-medium{padding:9rem 4.5rem}.section.is-large{padding:18rem 6rem}}.footer{background-color:#fafafa;padding:3rem 1.5rem 6rem}h1 .docs-heading-anchor,h1 .docs-heading-anchor:hover,h1 .docs-heading-anchor:visited,h2 .docs-heading-anchor,h2 .docs-heading-anchor:hover,h2 .docs-heading-anchor:visited,h3 .docs-heading-anchor,h3 .docs-heading-anchor:hover,h3 .docs-heading-anchor:visited,h4 .docs-heading-anchor,h4 .docs-heading-anchor:hover,h4 .docs-heading-anchor:visited,h5 .docs-heading-anchor,h5 .docs-heading-anchor:hover,h5 .docs-heading-anchor:visited,h6 .docs-heading-anchor,h6 .docs-heading-anchor:hover,h6 .docs-heading-anchor:visited{color:#222}h1 .docs-heading-anchor-permalink,h2 .docs-heading-anchor-permalink,h3 .docs-heading-anchor-permalink,h4 .docs-heading-anchor-permalink,h5 .docs-heading-anchor-permalink,h6 .docs-heading-anchor-permalink{visibility:hidden;vertical-align:middle;margin-left:0.5em;font-size:0.7rem}h1 .docs-heading-anchor-permalink::before,h2 .docs-heading-anchor-permalink::before,h3 .docs-heading-anchor-permalink::before,h4 .docs-heading-anchor-permalink::before,h5 .docs-heading-anchor-permalink::before,h6 .docs-heading-anchor-permalink::before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f0c1"}h1:hover .docs-heading-anchor-permalink,h2:hover .docs-heading-anchor-permalink,h3:hover .docs-heading-anchor-permalink,h4:hover .docs-heading-anchor-permalink,h5:hover .docs-heading-anchor-permalink,h6:hover .docs-heading-anchor-permalink{visibility:visible}.docs-dark-only{display:none !important}pre{position:relative;overflow:hidden}pre code,pre code.hljs{padding:0 .75rem !important;overflow:auto;display:block}pre code:first-of-type,pre code.hljs:first-of-type{padding-top:0.5rem !important}pre code:last-of-type,pre code.hljs:last-of-type{padding-bottom:0.5rem !important}pre .copy-button{opacity:0.2;transition:opacity 0.2s;position:absolute;right:0em;top:0em;padding:0.5em;width:2.5em;height:2.5em;background:transparent;border:none;font-family:"Font Awesome 6 Free";color:#222;cursor:pointer;text-align:center}pre .copy-button:focus,pre .copy-button:hover{opacity:1;background:rgba(34,34,34,0.1);color:#2e63b8}pre .copy-button.success{color:#259a12;opacity:1}pre .copy-button.error{color:#cb3c33;opacity:1}pre:hover .copy-button{opacity:1}.admonition{background-color:#b5b5b5;border-style:solid;border-width:1px;border-color:#363636;border-radius:4px;font-size:1rem}.admonition strong{color:currentColor}.admonition.is-small,#documenter .docs-sidebar form.docs-search>input.admonition{font-size:.75rem}.admonition.is-medium{font-size:1.25rem}.admonition.is-large{font-size:1.5rem}.admonition.is-default{background-color:#b5b5b5;border-color:#363636}.admonition.is-default>.admonition-header{background-color:#363636;color:#fff}.admonition.is-default>.admonition-body{color:#fff}.admonition.is-info{background-color:#def0fc;border-color:#209cee}.admonition.is-info>.admonition-header{background-color:#209cee;color:#fff}.admonition.is-info>.admonition-body{color:rgba(0,0,0,0.7)}.admonition.is-success{background-color:#bdf4d1;border-color:#22c35b}.admonition.is-success>.admonition-header{background-color:#22c35b;color:#fff}.admonition.is-success>.admonition-body{color:rgba(0,0,0,0.7)}.admonition.is-warning{background-color:#fff3c5;border-color:#ffdd57}.admonition.is-warning>.admonition-header{background-color:#ffdd57;color:rgba(0,0,0,0.7)}.admonition.is-warning>.admonition-body{color:rgba(0,0,0,0.7)}.admonition.is-danger{background-color:#ffaba7;border-color:#da0b00}.admonition.is-danger>.admonition-header{background-color:#da0b00;color:#fff}.admonition.is-danger>.admonition-body{color:rgba(0,0,0,0.7)}.admonition.is-compat{background-color:#bdeff5;border-color:#1db5c9}.admonition.is-compat>.admonition-header{background-color:#1db5c9;color:#fff}.admonition.is-compat>.admonition-body{color:rgba(0,0,0,0.7)}.admonition-header{color:#fff;background-color:#363636;align-items:center;font-weight:700;justify-content:space-between;line-height:1.25;padding:0.5rem .75rem;position:relative}.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;margin-right:.75rem;content:"\f06a"}details.admonition.is-details>.admonition-header{list-style:none}details.admonition.is-details>.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f055"}details.admonition.is-details[open]>.admonition-header:before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f056"}.admonition-body{color:#222;padding:0.5rem .75rem}.admonition-body pre{background-color:#f5f5f5}.admonition-body code{background-color:rgba(0,0,0,0.05)}.docstring{margin-bottom:1em;background-color:rgba(0,0,0,0);border:1px solid #dbdbdb;box-shadow:2px 2px 3px rgba(10,10,10,0.1);max-width:100%}.docstring>header{cursor:pointer;display:flex;flex-grow:1;align-items:stretch;padding:0.5rem .75rem;background-color:#f5f5f5;box-shadow:0 0.125em 0.25em rgba(10,10,10,0.1);box-shadow:none;border-bottom:1px solid #dbdbdb}.docstring>header code{background-color:transparent}.docstring>header .docstring-article-toggle-button{min-width:1.1rem;padding:0.2rem 0.2rem 0.2rem 0}.docstring>header .docstring-binding{margin-right:0.3em}.docstring>header .docstring-category{margin-left:0.3em}.docstring>section{position:relative;padding:.75rem .75rem;border-bottom:1px solid #dbdbdb}.docstring>section:last-child{border-bottom:none}.docstring>section>a.docs-sourcelink{transition:opacity 0.3s;opacity:0;position:absolute;right:.375rem;bottom:.375rem}.docstring>section>a.docs-sourcelink:focus{opacity:1 !important}.docstring:hover>section>a.docs-sourcelink{opacity:0.2}.docstring:focus-within>section>a.docs-sourcelink{opacity:0.2}.docstring>section:hover a.docs-sourcelink{opacity:1}.documenter-example-output{background-color:#fff}.outdated-warning-overlay{position:fixed;top:0;left:0;right:0;box-shadow:0 0 10px rgba(0,0,0,0.3);z-index:999;background-color:#ffaba7;color:rgba(0,0,0,0.7);border-bottom:3px solid #da0b00;padding:10px 35px;text-align:center;font-size:15px}.outdated-warning-overlay .outdated-warning-closer{position:absolute;top:calc(50% - 10px);right:18px;cursor:pointer;width:12px}.outdated-warning-overlay a{color:#2e63b8}.outdated-warning-overlay a:hover{color:#363636}.content pre{border:1px solid #dbdbdb}.content code{font-weight:inherit}.content a code{color:#2e63b8}.content h1 code,.content h2 code,.content h3 code,.content h4 code,.content h5 code,.content h6 code{color:#222}.content table{display:block;width:initial;max-width:100%;overflow-x:auto}.content blockquote>ul:first-child,.content blockquote>ol:first-child,.content .admonition-body>ul:first-child,.content .admonition-body>ol:first-child{margin-top:0}pre,code{font-variant-ligatures:no-contextual}.breadcrumb a.is-disabled{cursor:default;pointer-events:none}.breadcrumb a.is-disabled,.breadcrumb a.is-disabled:hover{color:#222}.hljs{background:initial !important}.katex .katex-mathml{top:0;right:0}.katex-display,mjx-container,.MathJax_Display{margin:0.5em 0 !important}html{-moz-osx-font-smoothing:auto;-webkit-font-smoothing:auto}li.no-marker{list-style:none}#documenter .docs-main>article{overflow-wrap:break-word}#documenter .docs-main>article .math-container{overflow-x:auto;overflow-y:hidden}@media screen and (min-width: 1056px){#documenter .docs-main{max-width:52rem;margin-left:20rem;padding-right:1rem}}@media screen and (max-width: 1055px){#documenter .docs-main{width:100%}#documenter .docs-main>article{max-width:52rem;margin-left:auto;margin-right:auto;margin-bottom:1rem;padding:0 1rem}#documenter .docs-main>header,#documenter .docs-main>nav{max-width:100%;width:100%;margin:0}}#documenter .docs-main header.docs-navbar{background-color:#fff;border-bottom:1px solid #dbdbdb;z-index:2;min-height:4rem;margin-bottom:1rem;display:flex}#documenter .docs-main header.docs-navbar .breadcrumb{flex-grow:1}#documenter .docs-main header.docs-navbar .docs-sidebar-button{display:block;font-size:1.5rem;padding-bottom:0.1rem;margin-right:1rem}#documenter .docs-main header.docs-navbar .docs-right{display:flex;white-space:nowrap;gap:1rem;align-items:center}#documenter .docs-main header.docs-navbar .docs-right .docs-icon,#documenter .docs-main header.docs-navbar .docs-right .docs-label{display:inline-block}#documenter .docs-main header.docs-navbar .docs-right .docs-label{padding:0;margin-left:0.3em}@media screen and (max-width: 1055px){#documenter .docs-main header.docs-navbar .docs-right .docs-navbar-link{margin-left:0.4rem;margin-right:0.4rem}}#documenter .docs-main header.docs-navbar>*{margin:auto 0}@media screen and (max-width: 1055px){#documenter .docs-main header.docs-navbar{position:sticky;top:0;padding:0 1rem;transition-property:top, box-shadow;-webkit-transition-property:top, box-shadow;transition-duration:0.3s;-webkit-transition-duration:0.3s}#documenter .docs-main header.docs-navbar.headroom--not-top{box-shadow:.2rem 0rem .4rem #bbb;transition-duration:0.7s;-webkit-transition-duration:0.7s}#documenter .docs-main header.docs-navbar.headroom--unpinned.headroom--not-top.headroom--not-bottom{top:-4.5rem;transition-duration:0.7s;-webkit-transition-duration:0.7s}}#documenter .docs-main section.footnotes{border-top:1px solid #dbdbdb}#documenter .docs-main section.footnotes li .tag:first-child,#documenter .docs-main section.footnotes li .docstring>section>a.docs-sourcelink:first-child,#documenter .docs-main section.footnotes li .content kbd:first-child,.content #documenter .docs-main section.footnotes li kbd:first-child{margin-right:1em;margin-bottom:0.4em}#documenter .docs-main .docs-footer{display:flex;flex-wrap:wrap;margin-left:0;margin-right:0;border-top:1px solid #dbdbdb;padding-top:1rem;padding-bottom:1rem}@media screen and (max-width: 1055px){#documenter .docs-main .docs-footer{padding-left:1rem;padding-right:1rem}}#documenter .docs-main .docs-footer .docs-footer-nextpage,#documenter .docs-main .docs-footer .docs-footer-prevpage{flex-grow:1}#documenter .docs-main .docs-footer .docs-footer-nextpage{text-align:right}#documenter .docs-main .docs-footer .flexbox-break{flex-basis:100%;height:0}#documenter .docs-main .docs-footer .footer-message{font-size:0.8em;margin:0.5em auto 0 auto;text-align:center}#documenter .docs-sidebar{display:flex;flex-direction:column;color:#0a0a0a;background-color:#f5f5f5;border-right:1px solid #dbdbdb;padding:0;flex:0 0 18rem;z-index:5;font-size:1rem;position:fixed;left:-18rem;width:18rem;height:100%;transition:left 0.3s}#documenter .docs-sidebar.visible{left:0;box-shadow:.4rem 0rem .8rem #bbb}@media screen and (min-width: 1056px){#documenter .docs-sidebar.visible{box-shadow:none}}@media screen and (min-width: 1056px){#documenter .docs-sidebar{left:0;top:0}}#documenter .docs-sidebar .docs-logo{margin-top:1rem;padding:0 1rem}#documenter .docs-sidebar .docs-logo>img{max-height:6rem;margin:auto}#documenter .docs-sidebar .docs-package-name{flex-shrink:0;font-size:1.5rem;font-weight:700;text-align:center;white-space:nowrap;overflow:hidden;padding:0.5rem 0}#documenter .docs-sidebar .docs-package-name .docs-autofit{max-width:16.2rem}#documenter .docs-sidebar .docs-package-name a,#documenter .docs-sidebar .docs-package-name a:hover{color:#0a0a0a}#documenter .docs-sidebar .docs-version-selector{border-top:1px solid #dbdbdb;display:none;padding:0.5rem}#documenter .docs-sidebar .docs-version-selector.visible{display:flex}#documenter .docs-sidebar ul.docs-menu{flex-grow:1;user-select:none;border-top:1px solid #dbdbdb;padding-bottom:1.5rem}#documenter .docs-sidebar ul.docs-menu>li>.tocitem{font-weight:bold}#documenter .docs-sidebar ul.docs-menu>li li{font-size:.95rem;margin-left:1em;border-left:1px solid #dbdbdb}#documenter .docs-sidebar ul.docs-menu input.collapse-toggle{display:none}#documenter .docs-sidebar ul.docs-menu ul.collapsed{display:none}#documenter .docs-sidebar ul.docs-menu input:checked~ul.collapsed{display:block}#documenter .docs-sidebar ul.docs-menu label.tocitem{display:flex}#documenter .docs-sidebar ul.docs-menu label.tocitem .docs-label{flex-grow:2}#documenter .docs-sidebar ul.docs-menu label.tocitem .docs-chevron{display:inline-block;font-style:normal;font-variant:normal;text-rendering:auto;line-height:1;font-size:.75rem;margin-left:1rem;margin-top:auto;margin-bottom:auto}#documenter .docs-sidebar ul.docs-menu label.tocitem .docs-chevron::before{font-family:"Font Awesome 6 Free";font-weight:900;content:"\f054"}#documenter .docs-sidebar ul.docs-menu input:checked~label.tocitem .docs-chevron::before{content:"\f078"}#documenter .docs-sidebar ul.docs-menu .tocitem{display:block;padding:0.5rem 0.5rem}#documenter .docs-sidebar ul.docs-menu .tocitem,#documenter .docs-sidebar ul.docs-menu .tocitem:hover{color:#0a0a0a;background:#f5f5f5}#documenter .docs-sidebar ul.docs-menu a.tocitem:hover,#documenter .docs-sidebar ul.docs-menu label.tocitem:hover{color:#0a0a0a;background-color:#ebebeb}#documenter .docs-sidebar ul.docs-menu li.is-active{border-top:1px solid #dbdbdb;border-bottom:1px solid #dbdbdb;background-color:#fff}#documenter .docs-sidebar ul.docs-menu li.is-active .tocitem,#documenter .docs-sidebar ul.docs-menu li.is-active .tocitem:hover{background-color:#fff;color:#0a0a0a}#documenter .docs-sidebar ul.docs-menu li.is-active ul.internal .tocitem:hover{background-color:#ebebeb;color:#0a0a0a}#documenter .docs-sidebar ul.docs-menu>li.is-active:first-child{border-top:none}#documenter .docs-sidebar ul.docs-menu ul.internal{margin:0 0.5rem 0.5rem;border-top:1px solid #dbdbdb}#documenter .docs-sidebar ul.docs-menu ul.internal li{font-size:.85rem;border-left:none;margin-left:0;margin-top:0.5rem}#documenter .docs-sidebar ul.docs-menu ul.internal .tocitem{width:100%;padding:0}#documenter .docs-sidebar ul.docs-menu ul.internal .tocitem::before{content:"⚬";margin-right:0.4em}#documenter .docs-sidebar form.docs-search{margin:auto;margin-top:0.5rem;margin-bottom:0.5rem}#documenter .docs-sidebar form.docs-search>input{width:14.4rem}#documenter .docs-sidebar #documenter-search-query{color:#707070;width:14.4rem;box-shadow:inset 0 1px 2px rgba(10,10,10,0.1)}@media screen and (min-width: 1056px){#documenter .docs-sidebar ul.docs-menu{overflow-y:auto;-webkit-overflow-scroll:touch}#documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar{width:.3rem;background:none}#documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar-thumb{border-radius:5px 0px 0px 5px;background:#e0e0e0}#documenter .docs-sidebar ul.docs-menu::-webkit-scrollbar-thumb:hover{background:#ccc}}@media screen and (max-width: 1055px){#documenter .docs-sidebar{overflow-y:auto;-webkit-overflow-scroll:touch}#documenter .docs-sidebar::-webkit-scrollbar{width:.3rem;background:none}#documenter .docs-sidebar::-webkit-scrollbar-thumb{border-radius:5px 0px 0px 5px;background:#e0e0e0}#documenter .docs-sidebar::-webkit-scrollbar-thumb:hover{background:#ccc}}kbd.search-modal-key-hints{border-radius:0.25rem;border:1px solid rgba(0,0,0,0.6);box-shadow:0 2px 0 1px rgba(0,0,0,0.6);cursor:default;font-size:0.9rem;line-height:1.5;min-width:0.75rem;text-align:center;padding:0.1rem 0.3rem;position:relative;top:-1px}.search-min-width-50{min-width:50%}.search-min-height-100{min-height:100%}.search-modal-card-body{max-height:calc(100vh - 15rem)}.search-result-link{border-radius:0.7em;transition:all 300ms}.search-result-link:hover,.search-result-link:focus{background-color:rgba(0,128,128,0.1)}.search-result-link .property-search-result-badge,.search-result-link .search-filter{transition:all 300ms}.property-search-result-badge,.search-filter{padding:0.15em 0.5em;font-size:0.8em;font-style:italic;text-transform:none !important;line-height:1.5;color:#f5f5f5;background-color:rgba(51,65,85,0.501961);border-radius:0.6rem}.search-result-link:hover .property-search-result-badge,.search-result-link:hover .search-filter,.search-result-link:focus .property-search-result-badge,.search-result-link:focus .search-filter{color:#f1f5f9;background-color:#333}.search-filter{color:#333;background-color:#f5f5f5;transition:all 300ms}.search-filter:hover,.search-filter:focus{color:#333}.search-filter-selected{color:#f5f5f5;background-color:rgba(139,0,139,0.5)}.search-filter-selected:hover,.search-filter-selected:focus{color:#f5f5f5}.search-result-highlight{background-color:#ffdd57;color:black}.search-divider{border-bottom:1px solid #dbdbdb}.search-result-title{width:85%;color:#333}.search-result-code-title{font-size:0.875rem;font-family:"JuliaMono","SFMono-Regular","Menlo","Consolas","Liberation Mono","DejaVu Sans Mono",monospace}#search-modal .modal-card-body::-webkit-scrollbar,#search-modal .filter-tabs::-webkit-scrollbar{height:10px;width:10px;background-color:transparent}#search-modal .modal-card-body::-webkit-scrollbar-thumb,#search-modal .filter-tabs::-webkit-scrollbar-thumb{background-color:gray;border-radius:1rem}#search-modal .modal-card-body::-webkit-scrollbar-track,#search-modal .filter-tabs::-webkit-scrollbar-track{-webkit-box-shadow:inset 0 0 6px rgba(0,0,0,0.6);background-color:transparent}.w-100{width:100%}.gap-2{gap:0.5rem}.gap-4{gap:1rem}.gap-8{gap:2rem}.ansi span.sgr1{font-weight:bolder}.ansi span.sgr2{font-weight:lighter}.ansi span.sgr3{font-style:italic}.ansi span.sgr4{text-decoration:underline}.ansi span.sgr7{color:#fff;background-color:#222}.ansi span.sgr8{color:transparent}.ansi span.sgr8 span{color:transparent}.ansi span.sgr9{text-decoration:line-through}.ansi span.sgr30{color:#242424}.ansi span.sgr31{color:#a7201f}.ansi span.sgr32{color:#066f00}.ansi span.sgr33{color:#856b00}.ansi span.sgr34{color:#2149b0}.ansi span.sgr35{color:#7d4498}.ansi span.sgr36{color:#007989}.ansi span.sgr37{color:gray}.ansi span.sgr40{background-color:#242424}.ansi span.sgr41{background-color:#a7201f}.ansi span.sgr42{background-color:#066f00}.ansi span.sgr43{background-color:#856b00}.ansi span.sgr44{background-color:#2149b0}.ansi span.sgr45{background-color:#7d4498}.ansi span.sgr46{background-color:#007989}.ansi span.sgr47{background-color:gray}.ansi span.sgr90{color:#616161}.ansi span.sgr91{color:#cb3c33}.ansi span.sgr92{color:#0e8300}.ansi span.sgr93{color:#a98800}.ansi span.sgr94{color:#3c5dcd}.ansi span.sgr95{color:#9256af}.ansi span.sgr96{color:#008fa3}.ansi span.sgr97{color:#f5f5f5}.ansi span.sgr100{background-color:#616161}.ansi span.sgr101{background-color:#cb3c33}.ansi span.sgr102{background-color:#0e8300}.ansi span.sgr103{background-color:#a98800}.ansi span.sgr104{background-color:#3c5dcd}.ansi span.sgr105{background-color:#9256af}.ansi span.sgr106{background-color:#008fa3}.ansi span.sgr107{background-color:#f5f5f5}code.language-julia-repl>span.hljs-meta{color:#066f00;font-weight:bolder}/*!
+ Theme: Default
+ Description: Original highlight.js style
+ Author: (c) Ivan Sagalaev 


















 ","category":"page"},{"location":"writevtk.html#Simulation-data","page":"ParaView Rendering","title":"Simulation data","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"First, generate some correlation data in Sunny. We will use a 2D lattice, since the correlation data S(Q_xQ_yomega) is 3D and can be exported in its entirety. The following code sets up the system, thermalizes it, and records the correlation data in a SampledCorrelations called dsf.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"using Sunny\n\n# Single layer 12x12 periodic square lattice\nlatsize = (12,12,1)\n\nlatvecs = lattice_vectors(8.,8.,12.,90,100,90)\npositions = [[0,0,0]]\ntypes = [\"Cu\"]\nformfactors = [FormFactor(\"Cu2\")]\nxtal = Crystal(latvecs, positions; types)\n\nsys = System(xtal, latsize, [SpinInfo(1, S=1/2, g=2)], :SUN; seed=1)\n\nJ = 10.\nset_exchange!(sys, J, Bond(1,1,[1,0,0]))\nset_exchange!(sys, J, Bond(1,1,[0,1,0]))\n\nΔt = 0.01\nkT = 0.5\nlangevin = Langevin(Δt; λ=0.5, kT)\nrandomize_spins!(sys)\nfor i in 1:10_000 # Long enough to reach equilibrium\n step!(sys, langevin)\nend \n\nωmax=10.\n\ndsf = dynamical_correlations(sys\n ;Δt=Δt\n ,nω=48\n ,ωmax=ωmax\n ,process_trajectory=:symmetrize)\n\nnsamples = 10\nfor _ in 1:nsamples\n for _ in 1:1000 \n step!(sys, langevin)\n end\n add_sample!(dsf, sys)\nend","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The default histogram BinningParameters are already integrated over the z direction because the system is 2D:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"unit_resolution_binning_parameters(dsf)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 12 bins from -0.042 to +0.958 along [+1.27 dx] (Δ = 0.065)\n⊡ 12 bins from -0.042 to +0.958 along [+1.27 dy] (Δ = 0.065)\n∫ Integrated from +0.000 to +0.000 along [-0.33 dx +1.88 dz] (Δ = 0.524)\n⊡ 48 bins from -0.107 to +10.134 along [+1.00 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The histogram is very oblong; it's approximately 1x1x10. To make it a nicer shape, we will rescale the energy axis to be be fractions of ωmax:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"params = unit_resolution_binning_parameters(dsf)\nscale_factor = ωmax\nparams.binend[4] /= scale_factor\nparams.binstart[4] /= scale_factor\nparams.binwidth[4] /= scale_factor\nparams.covectors[4,:] ./= scale_factor","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Doing this changes the last axis of the histogram to fit in [0,1]:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 49 bins from -0.011 to +1.013 along [+0.10 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that our histogram is a cube, we compute the intensity in the histogram bins using the usual intensities_binned:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"formula = intensity_formula(dsf,:trace)\nsignal, counts = intensities_binned(dsf, params; formula)\nintensity = signal ./ counts","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that we have our intensity data and the binning parameters, we can export to VTK format using export_vtk and move to ParaView for the visualization.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"# Importing WriteVTK enables Sunny's export-to-VTK functions\nimport WriteVTK\n\n# [1,2,4] specifies that the (x,y,z) axes in ParaView are (Qx,Qy,ω)\nexport_vtk(\"square_lattice\", params, intensity; dims_kept = [1,2,4])\n# Writes a file square_lattice.vti in the current directory","category":"page"},{"location":"writevtk.html#Loading-in-ParaView","page":"ParaView Rendering","title":"Loading in ParaView","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In ParaView, use File > Open to open square_lattice.vti. This will add the file to the Pipeline Browser with a closed eye icon, indicating that the data is ready to be loaded.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In the Properties panel, both bin_centers and data will be selected for import by default. Uncheck bin_centers because we don't need that information for the visualization. Click the green Apply button to load the data.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"By default, only the outline of the data is shown in the 3D viewport. Since we adjusted the energy axis, the outline is a 1x1x1 cube. Optionally enable the axes grid under \"View\", and customize using the adjacent edit button.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"writevtk.html#Simulation-data","page":"ParaView Rendering","title":"Simulation data","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"First, generate some correlation data in Sunny. We will use a 2D lattice, since the correlation data S(Q_xQ_yomega) is 3D and can be exported in its entirety. The following code sets up the system, thermalizes it, and records the correlation data in a SampledCorrelations called dsf.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"using Sunny\n\n# Single layer 12x12 periodic square lattice\nlatsize = (12,12,1)\n\nlatvecs = lattice_vectors(8.,8.,12.,90,100,90)\npositions = [[0,0,0]]\ntypes = [\"Cu\"]\nformfactors = [FormFactor(\"Cu2\")]\nxtal = Crystal(latvecs, positions; types)\n\nsys = System(xtal, latsize, [SpinInfo(1, S=1/2, g=2)], :SUN; seed=1)\n\nJ = 10.\nset_exchange!(sys, J, Bond(1,1,[1,0,0]))\nset_exchange!(sys, J, Bond(1,1,[0,1,0]))\n\nΔt = 0.01\nkT = 0.5\nlangevin = Langevin(Δt; λ=0.5, kT)\nrandomize_spins!(sys)\nfor i in 1:10_000 # Long enough to reach equilibrium\n step!(sys, langevin)\nend \n\nωmax=10.\n\ndsf = dynamical_correlations(sys\n ;Δt=Δt\n ,nω=48\n ,ωmax=ωmax\n ,process_trajectory=:symmetrize)\n\nnsamples = 10\nfor _ in 1:nsamples\n for _ in 1:1000 \n step!(sys, langevin)\n end\n add_sample!(dsf, sys)\nend","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The default histogram BinningParameters are already integrated over the z direction because the system is 2D:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"unit_resolution_binning_parameters(dsf)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 12 bins from -0.042 to +0.958 along [+1.27 dx] (Δ = 0.065)\n⊡ 12 bins from -0.042 to +0.958 along [+1.27 dy] (Δ = 0.065)\n∫ Integrated from +0.000 to +0.000 along [-0.33 dx +1.88 dz] (Δ = 0.524)\n⊡ 48 bins from -0.107 to +10.134 along [+1.00 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"The histogram is very oblong; it's approximately 1x1x10. To make it a nicer shape, we will rescale the energy axis to be be fractions of ωmax:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"params = unit_resolution_binning_parameters(dsf)\nscale_factor = ωmax\nparams.binend[4] /= scale_factor\nparams.binstart[4] /= scale_factor\nparams.binwidth[4] /= scale_factor\nparams.covectors[4,:] ./= scale_factor","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Doing this changes the last axis of the histogram to fit in [0,1]:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"⊡ 49 bins from -0.011 to +1.013 along [+0.10 dE] (Δ = 0.213)","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that our histogram is a cube, we compute the intensity in the histogram bins using the usual intensities_binned:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"formula = intensity_formula(dsf,:trace)\nsignal, counts = intensities_binned(dsf, params; formula)\nintensity = signal ./ counts","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Now that we have our intensity data and the binning parameters, we can export to VTK format using export_vtk and move to ParaView for the visualization.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"# Importing WriteVTK enables Sunny's export-to-VTK functions\nimport WriteVTK\n\n# [1,2,4] specifies that the (x,y,z) axes in ParaView are (Qx,Qy,ω)\nexport_vtk(\"square_lattice\", params, intensity; dims_kept = [1,2,4])\n# Writes a file square_lattice.vti in the current directory","category":"page"},{"location":"writevtk.html#Loading-in-ParaView","page":"ParaView Rendering","title":"Loading in ParaView","text":"","category":"section"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In ParaView, use File > Open to open square_lattice.vti. This will add the file to the Pipeline Browser with a closed eye icon, indicating that the data is ready to be loaded.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"In the Properties panel, both bin_centers and data will be selected for import by default. Uncheck bin_centers because we don't need that information for the visualization. Click the green Apply button to load the data.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"By default, only the outline of the data is shown in the 3D viewport. Since we adjusted the energy axis, the outline is a 1x1x1 cube. Optionally enable the axes grid under \"View\", and customize using the adjacent edit button.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":" ","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"To enable the volumetric render:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Select \"Volume\" from the \"Representation\" drop-down menu under \"Display\".\nThe \"Coloring\" drop-down should automatically select data because it's the only data loaded.\nOpen the Color Map Editor to adjust the opacity of the fog, which may be too faint to see by default.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"To enable the volumetric render:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Select \"Volume\" from the \"Representation\" drop-down menu under \"Display\".\nThe \"Coloring\" drop-down should automatically select data because it's the only data loaded.\nOpen the Color Map Editor to adjust the opacity of the fog, which may be too faint to see by default.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":" ","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Depending on your computer and your dataset size, the volumetric rendering may be slow, but our dataset is relatively small, so the render should be fast.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"If nothing shows up at first, don't despair. Often, there are Bragg-like peaks in the correlation data which outshine everything else. To see this, enable Display Data Histogram in the Color Map Editor panel. To zoom in on the lower-intensity data, click and drag the right side handle of the opacity transfer function box to the middle a few times.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"Depending on your computer and your dataset size, the volumetric rendering may be slow, but our dataset is relatively small, so the render should be fast.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"If nothing shows up at first, don't despair. Often, there are Bragg-like peaks in the correlation data which outshine everything else. To see this, enable Display Data Histogram in the Color Map Editor panel. To zoom in on the lower-intensity data, click and drag the right side handle of the opacity transfer function box to the middle a few times.","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":" ","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"After suitable color mapping, the dispersion curve should become visible:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"
","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"After suitable color mapping, the dispersion curve should become visible:","category":"page"},{"location":"writevtk.html","page":"ParaView Rendering","title":"ParaView Rendering","text":"


