diff --git a/previews/PR345/.documenter-siteinfo.json b/previews/PR345/.documenter-siteinfo.json
index 04b8a9806..d92c02489 100644
--- a/previews/PR345/.documenter-siteinfo.json
+++ b/previews/PR345/.documenter-siteinfo.json
@@ -1 +1 @@
-{"documenter":{"julia_version":"1.10.7","generation_timestamp":"2024-12-17T16:53:12","documenter_version":"1.8.0"}}
\ No newline at end of file
+{"documenter":{"julia_version":"1.10.7","generation_timestamp":"2024-12-17T16:57:17","documenter_version":"1.8.0"}}
\ No newline at end of file
diff --git a/previews/PR345/examples/01_LSWT_CoRh2O4.html b/previews/PR345/examples/01_LSWT_CoRh2O4.html
index d7fce5495..1689330ec 100644
--- a/previews/PR345/examples/01_LSWT_CoRh2O4.html
+++ b/previews/PR345/examples/01_LSWT_CoRh2O4.html
@@ -41,4 +41,4 @@
res = powder_average(cryst, radii, 2000) do qs
intensities(swt, qs; energies, kernel)
end
-plot_intensities(res; units, saturation=1.0, title="CoRh₂O₄ Powder Average")
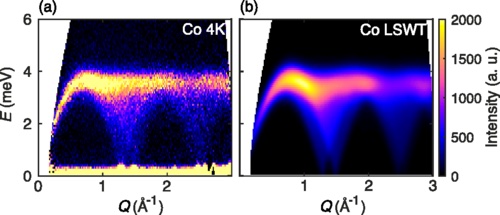
This result can be compared to experimental neutron scattering data from Fig. 5 of Ge et al.
Spin wave theory neglects thermal fluctuations of the magnetic order. The next CoRh₂O₄ tutorial demonstrates how to sample spins in thermal equilibrium, and measure dynamical correlations from the classical spin dynamics.
Sunny also offers features that go beyond the dipole approximation of a quantum spin via the theory of SU(N) coherent states. This can be especially useful for systems with strong single-ion anisotropy, as demonstrated in the FeI₂ tutorial.
Settings
This document was generated with Documenter.jl version 1.8.0 on Tuesday 17 December 2024. Using Julia version 1.10.7.