zDB is designed to perform comparative genomics analyses and to integrate the results in a Django web-application.
Several analyses are currently supported, with more to come:
- Orthology inference
- Phylogenetic reconstructions
- COG annotation
- KEGG orthologs annotation and pathway completion analysis
- PFAM domains annotation
- Virulence factors
- Antimicrobial resistance genes
- Swissprot homologs search
- RefSeq homologs search: implemented, but significantly slows down the analysis. You'll also have to download and prepare the database for diamond search, as this was not included in the database setup script.
All the results are stored either in a SQLite database or directly as files and displayed in the web application. Interactive visualizations facilitates the comparison of gene content and static figure can be downloaded for publication.
Here is an overview of its architecture:
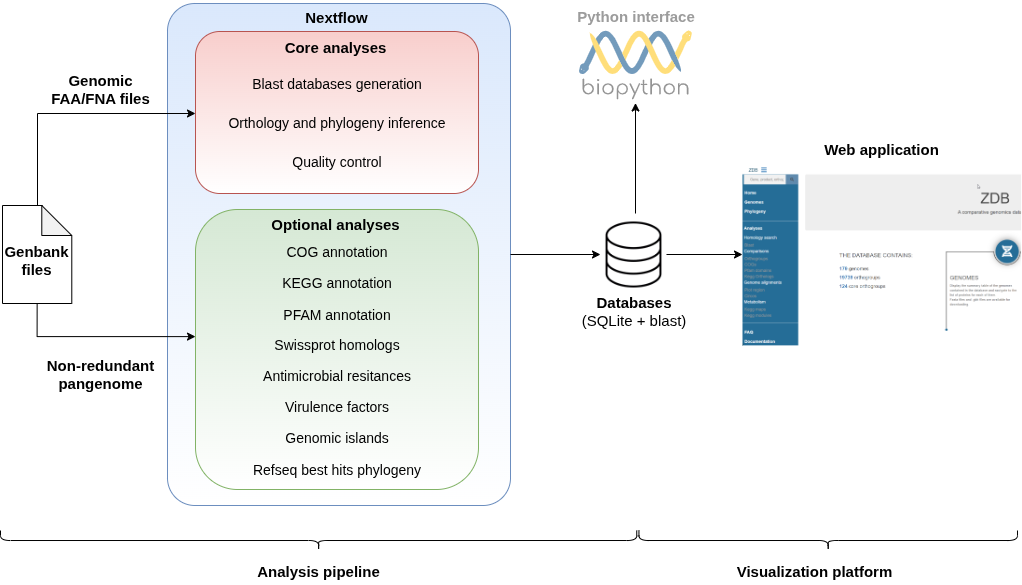
If you are not setting up your own database, but instead simply want to use the webapplication of an existing installation, you can directly refer to the website tutorial of the documentation.
If you are setting up your own database, you will need to install zDB, setup some reference databases, run the analysis pipeline and finally launch the web server. An overview of the workflow is shown below:
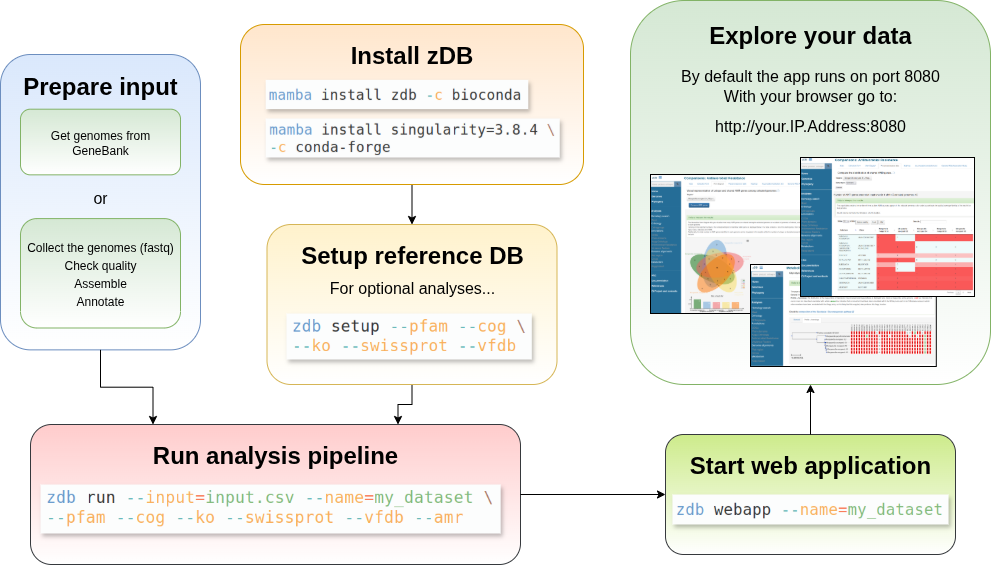
zDB can be installed from bioconda with the following command
conda install zdb -c conda-forge -c bioconda
Once zDB is installed, we advise you to run the analysis and/or webapp in containers, especially for MacOSX users. For this, you'll need to install either docker or singularity. Both the analyses and the webapp can also be run in conda environments, but this comes with several drawbacks:
- django will be run in native mode, without nginx and gunicorn and should not be used to set up a web-facing database (it is fine for a local access)
- containers allow us to have a precise control of the environment where the webapp is run; it is less the case for conda environment. Despite our best care, running the webapp in conda might not work due to local differences.
- some conda environments have numerous dependencies: to speed the installation, we strongly recommend the use a recent version of conda, which has included the solver from mamba and is much faster.
- Xvfb should be installed on your machine. The ete3 rendering engine unfortunately relies on Qt, which requires an X server running in headless mode. If you can't install Xvfb, please consider using singularity containers. We plan on developing our own Javascript tree rendering code to get rid of this dependency.
Of note, zDB has been tested on singularity v3.8.3 and v3.8.4 but should work on more recent versions. If you opt to use singularity, it can be installed with the following command:
conda install singularity=3.8.4 -c conda-forge
Or you could create a new environment containing zDB and singularity with:
conda create --name zdb -c conda-forge -c bioconda zdb singularity
For the installation of docker, please have a look here.
You can also install zdb directly from the github repository. This is particularly useful if you want to make modifications or if you want to have a direct access to Nextflow config file for a better control of the execution.
Check out the project (git clone [email protected]:metagenlab/zDB.git) or download and unpack a release, then edit this line of the bin/zdb bash script:
NEXTFLOW_DIR="${CONDA}/share/zdb-${VERSION}/"
and replace it by the directory where you downloaded the project (this should point to the directory where zdb's nextflow.config is located). Add zdb's bin directory to PATH and voila, zdb should run smoothly.
Note that zDB depends on nextflow (version 22.10 or lower) and singularity, so you'll need to install these packages, e.g. with conda:
conda env create -p ./env -f conda/main.yamlconda activate ./env
Several subcommands are available:
setup - download and prepare the reference databases
webapp - start the webapp
run - run the analysis pipeline
export - exports the results of a previous run in an archive
import - unpack an archive that was prepared with the export command in the current directory so that the results can be used to start the webapp
list_runs - lists the completed runs available to start the website in a given directory
Here are a few examples of workflows with a test dataset available from the git repository. To get the test dataset:
wget https://github.com/metagenlab/zDB/raw/master/test_dataset.tar.gz
tar xvf test_dataset.tar.gz
For a minimal database (assuming that singularity is installed):
conda install zdb -c conda-forge -c bioconda
zdb run --input=input.csv --name=simple_run # runs the analysis
zdb webapp --name=simple_run # Launches the webapp on simple run
The minimal database should take around 5 minutes to complete in a recent Desktop machine.
To do the same in conda environments:
conda install zdb -c conda-forge -c bioconda
zdb run --input=input.csv --name=simple_run_conda --conda # runs the analysis
zdb webapp --conda --name=simple_run_conda # Launches the webapp on the latest run
To have a more complete set of analyses (includes cog and pfam annotation):
conda install zdb -c conda-forge -c bioconda
zdb setup --pfam --cog --conda
zdb run --input=input.csv --name=more_complete_run --conda --cog --pfam # runs the analysis
zdb webapp --conda --name=more_complete_run # Launches the webapp on the latest run
For troubleshooting, please first read the more detailed sections below on how to set up reference databases, running the analysis and starting the webserver.
Depending on which analysis are to be run, reference databases will need to be downloaded and set up.
This is done using the zdb setup command.
You'll need to specifiy which databases are to be downloaded.
Of note, in minimal mode, zdb does not require any database to run.
The following databases can be downloaded:
--cog: downloads the CDD profiles used for COG annotations
--ko: downloads and sets up the hmm profiles of the ko database
--pfam: downloads and sets up the hmm profiles of the PFAM protein domains
--vfdb: downloads and sets up the virulence factor database (VFDB)
--swissprot: downloads and indexes the swissprot database
The database setup can be run either in singularity containers (by default), in conda environment (if the --conda flag is set) or in docker (if the --docker flag is set).
In addition, you can specify the directory where you want the databases to be installed with the --dir option: zdb setup --swissprot --dir=foobardir.
Downloading the HMM files from the KEGG server can take a bit of time (but you'll only need to do this once).
Example commmand, setting up all the databases in the current directory, using conda environments:
zdb setup --pfam --swissprot --cog --ko --conda
Once you have the reference databases set up, the genomes ready, just run the zdb run command. The run command expects a csv file as input. The csv should look like:
name, file
,foo/bar.gbk
,baz/bazz.gbk
foobar,foobar/baz.gbff
The name column is optional and can be omitted from the input csv file. By default, zdb will use the organism's name as defined in the genbank file to identify genomes in the web application. Specifying a name for a genome will tell zdb to use that name instead of the organism name from the genbank file. This is practical when working with assembled genomes that haven't been named yet or when working with genomes of different strains of a same species. If the same name is used in different files, zdb will just add a numbering suffix to make the names unique.
Any number of groups can also be specified in the input file, which will be available for selecting genomes easily in the web-application. They are specified as additional columns with headers of the form group:groupname, where groupname can be any string of your choice, and presence or absence is signified with 1 and 0 or other markers such as yes, no, e.g.:
name, file, group:first group, group: another one, group:third
,foo/bar.gbk,yes,1,no
,baz/bazz.gbk,0,1,1
foobar,foobar/baz.gbff,no,yes,0
Before launching the analysis, zdb will also check for the uniqueness of locus tags and generate new ones if necessary. This is usually not necessary for genomes downloaded from RefSeq or other databases, but if genomes were annotated with automated tools, name collisions might happen.
Several options are available and allow you to customize the run.
By default, the analysis are run in singularity containers, but you can change this by using the --conda or --docker flags to have them run in conda environments or docker containers, respectively. If singularity is enabled, the containers will have to be downloaded. By default, they are stored in the singularity folder of the current directory, but this can be changed using the --singularity_dir option. This might be useful if you want to share containers between analyses.
If the databases were set up, additional analyses can also be enabled with the --ko, --cog, --pfam, --vfdb and --swissprot flags. The --amr flag will add annotations of antimicrobial resistance genes (no database needed). The directory (by default zdb_ref in the current directory) where the database were installed can be specified with the --ref_dir option.
Other options include:
--resume: wrapper for nextflow resume, allows to restart a run that crashed without redoing all the computations
--out: directory where the files necessary for the webapp will be stored
--input: CSV file containing the path to the genbank files to include in the analysis
--name: custom run name (defaults to the name given by nextflow). The latest completed run is also named latest.
--cpu: number of parallel processes allowed (default 8)
--mem: max memory usage allowed (default 8GB)
--singularity_dir: the directory where the singularity images are downloaded (default singularity in current directory)
The --name option is optional and can be used to replace nextflow's randomly generated run names by more meaningful ones.
We noticed that undeterministic bugs sometimes happen when downloading a singularity container or when running long analysis. To resume the analysis when this happens, just add the --resume flag to the previous command. For example, if the run launched with the command
zdb run --input=input.csv --ko --cog
crashed after 5 hours of analysis, you can resume the run to where it was before crashing with the command:
zdb run --input=input.csv --ko --cog --resume
This can also be used if you want to add another analysis to a previous run:
zdb run --input=input.csv --ko --cog --pfam --resume
in this case, only the pfam annotations will be performed as the other analysis have already completed.
Once the analysis is complete, the web application can be run with the zdb webapp command. If the port 8080 is not in use, you can simply run the zdb webapp script without any parameters. zDB will launch the webapp on the last run of analysis.
The following options can be used:
--port=PORT_NUMBER the port the application will be listening to, 8080 by default.
--name=RUN_NAME when nextflow runs. If not specified, will default to the last successful run (latest).
--allowed_host=HOSTS the name of the host or the ip address of the server. If not specified, will default to the ip addresses of the current host.
By default, the webserver will be run in a singularity container. It can also be run in a conda environment by setting the --conda flag or in docker by setting the docker flag.
For MacOSX users, we advise to run the webapp in docker containers, setting --allowed_host=0.0.0.0 or 127.0.0.1, for the webapp to correctly display in your browser.
zdb webapp --docker --name=simple_run_docker # Launches the webapp using docker
Once the webserver has started, you'll be able to access the webpages with a web browser. You'll typically see a line of the sort:
Starting web server. The application will be accessible @155.105.138.249 172.17.0.1 on port 8080
To access the webapp, type either 155.105.138.249:8080 or 172.17.0.1:8080 on your web browser.
If you do not remember which runs are available, you can list them with the zdb list_runs command.
As the analysis may be run on a server or on an HPC cluster, the results may need to be exported to start a web application on a different machine.
This can be done with the zdb export command with a run name as parameter. This will create a compressed archive containing all the necessary results. The archive can then be transferred to a different machine and unpacked, either manually or with the zdb import command.
The web server can then be started as if the analysis had been run locally.
Suggestion and bug reports are very welcome here.
We already have several idea to improve the tool:
- make it possible to add or remove genomes in an existing database
- use d3.js to draw interactive phylogenetic trees
- add new annotations, in particular, we've already received some requests for antibiotic resistance and virulence
But we're definitely open for suggestions and contributions.
Running several instances
When trying to run several instances of the webapp on the same machine, you'll need to modify two files to avoid interferences between the instances.
In zdb/gunicorn/gunicorn.py:
bind = "0.0.0.0:8000"
modify the 8000 number to another, yet unused port. Same in the zdb/nginx/nginx.config file:
proxy_pass http://localhost:8000;
Modify the 8000 to the same number you attributed to the port number of gunicorn.
Running the webapp on a MacOSX
Please run the webapp in docker containers, setting --allowed_host=0.0.0.0 or 127.0.0.1, for the webapp to correctly display in your browser.
First you'll need to install zDB from source. You can then edit the different source files. Once you're done, use the zdb commands as you would for a normal use. Of note, if you want to modify the webapp code (the Javascript/python/HTML, etc), you can do so on the fly, while the webapp is running. We advise to use the --debug and (the hidden) --dev_server flags when testing changes in the webapp :
zdb webapp --debug --dev_server
The changes you make in the web server code will then reflect directly in the web page, with more log available in the console.
The nextflow pipeline is tested using a python integration of nextflow nextflow.py and standard unittests.
To run the tests you need a python environment with the required dependencies:
conda env create -f conda/testing.yaml
conda activate testing
You can then simply call
python -m unittest discover -s testing/pipelines/
Careful though with the nextflow pipeline tests:
- The test_db_setup module will download large volumes of data (tens of GBs), as the tests actually setup the zDB reference databases.
- The test_annotation_pipeline module expects you to have setup the reference databases.
The webapp is tested using the django testing tools. To run the tests you need a python environment with the required dependencies:
conda env create -f conda/webapp.yaml
conda activate webapp
You can then run the tests:
python webapp/manage.py test --settings=settings.testing_settings testing.webapp
Note that these tests will use the database created by the pipeline test TestAnnotationPipeline.test_full_pipeline, which therefore needs to have been executed first.
If you want to contribute, feel free to open a PR describing your changes and make sure the tests still pass and request a review from one of the developers (tpillone, bkm or njohner)
- Adapt CHANGELOG.md with release number and date.
- Run the tests (also checking that all conda envs build correctly).
- Create docker containers:
- Containers are defined in https://github.com/metagenlab/docker-images/
- Update
requirements.txtandDockerfiles forzdbandannotation-pipeline - Build and push the images, e.g.
docker build -t metagenlab/annotation-pipeline:1.4.1 annotation-pipeline/anddocker push metagenlab/annotation-pipeline:1.4.1 - Update zdb
annotation_pipeline.nfandbin/zdbaccordingly
- Test the bioconda release:
- To test the release locally we use bioconda-utils
- Activate the corresponding environment
- Clone
bioconda-recipes - Modify the zdb recipe (
recipes/zdb/meta.yaml) to download the tar file from master (set url to https://github.com/metagenlab/zDB/archive/refs/heads/master.tar.gz) - Run linting check (
bioconda-utils lint --packages zdb) - Build the package
bioconda-utils build --docker --mulled-test --packages zdb - Create a conda environment with the build package:
conda create -n test_zdb -c conda-forge -c /home/njohner/bin/miniconda3/envs/bioconda/conda-bld/ -c bioconda zdb singularity - Activate the environment and test zdb
- Make a release (can be done directly on github: https://github.com/metagenlab/zDB/releases)
- Release on bioconda:
- A pull request should be opened automatically on https://github.com/bioconda/bioconda-recipes. Check that the tests pass and that it gets merged
- Update documentation on readthedocs
- Demo of the website: https://zdb.metagenlab.ch/
- Documentation: https://zdb.readthedocs.io
- github repository: https://github.com/metagenlab/zDB
If you use zDB in your work, please cite the following paper:
Marquis B, Pillonel T, Carrara A, Bertelli C. 0. zDB: bacterial comparative genomics made easy. mSystems 0:e00473-24. https://doi.org/10.1128/msystems.00473-24